

Genomic and Precision Medicine Approaches in Atherosclerotic Cardiovascular Disease: From Risk Prediction to Therapy—A Review

 , ,
, ,  and
and
Abstract
1. Introduction
2. Search Strategy
3. Genomic Architecture of Atherosclerosis: Monogenic and Polygenic Contributions
3.1. Monogenic Forms of Atherosclerosis
3.2. Polygenic Architecture of Atherosclerosis
4. Clinical Application of Polygenic Risk Scores (PRS)
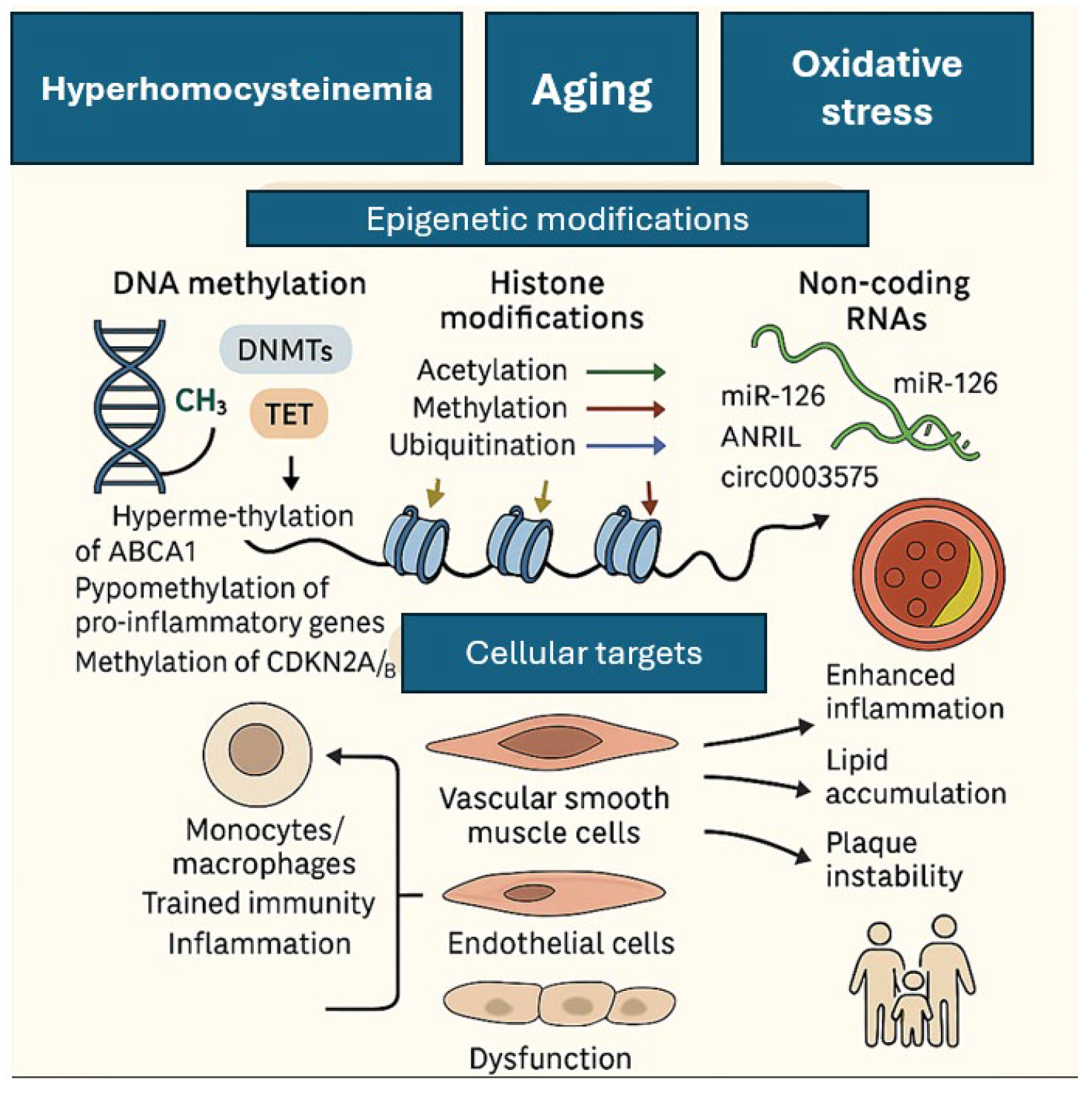
5. Epigenomics
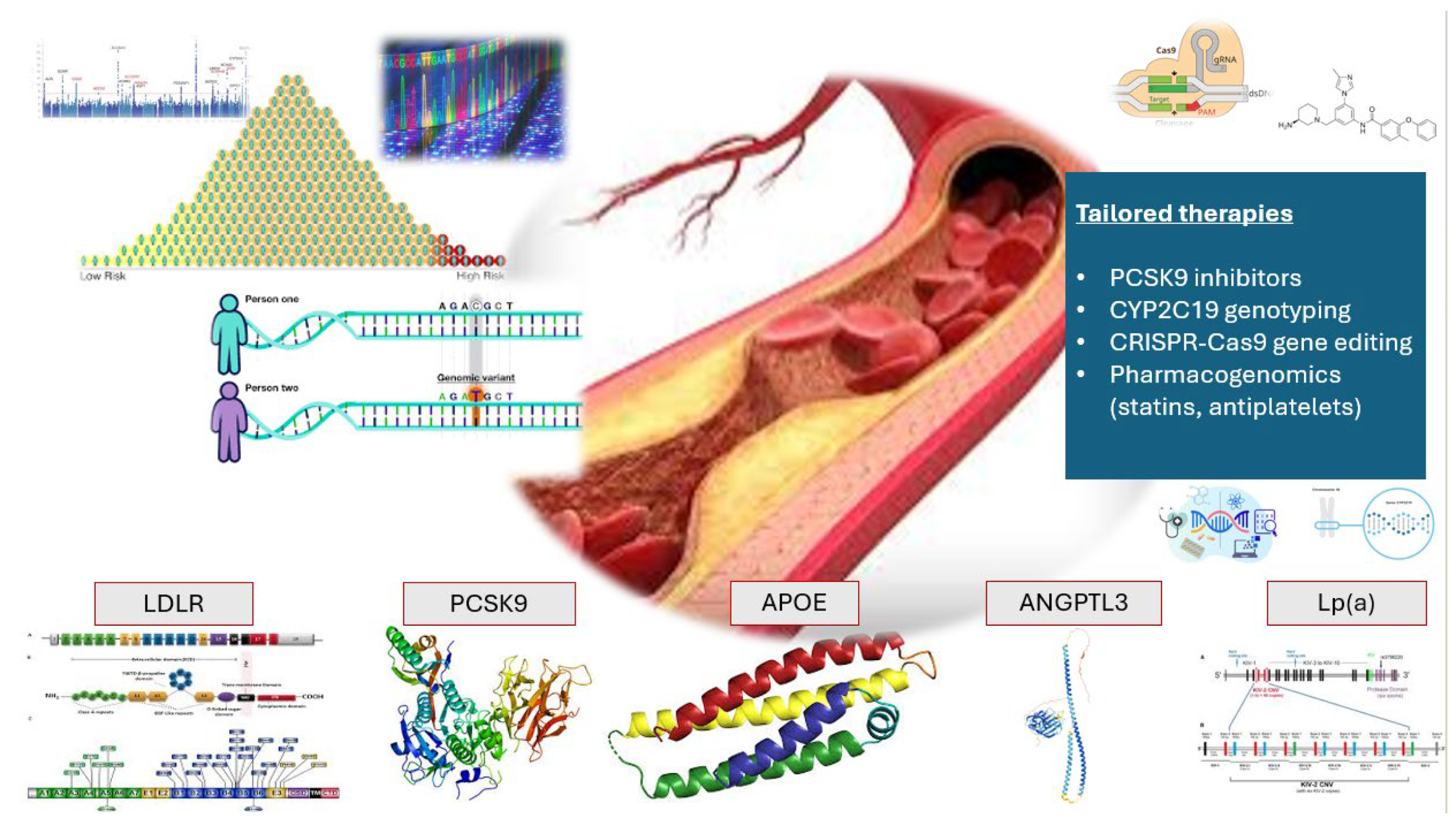
6. Precision Medicine and Tailored Drug Therapies in ASCVDs
6.1. PCSK9: From Genetic Discovery to Precision Therapeutics
6.2. Pharmacogenomics in Statin Therapy for ASCVD
6.3. Tailored Drug Therapies: Antiplatelet Therapy
6.4. Pharmacogenomics of Antihypertensive Therapy in ASCVD
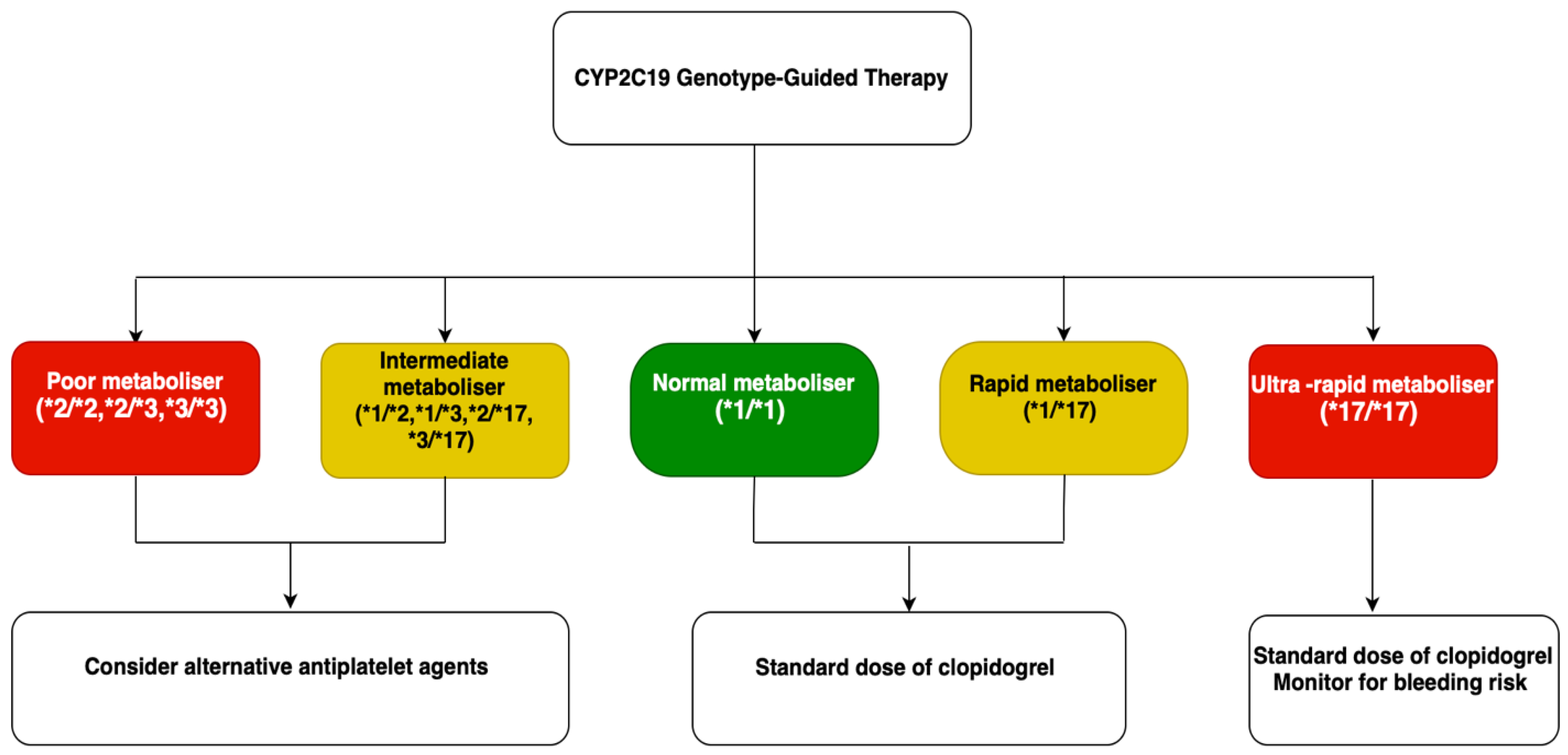
6.5. Current Guidelines for CYP2C19 Genotyping
6.6. Gene Editing and Therapy in Lipid Management
6.7. Precision Medicine in Polygenic Atherosclerosis
7. Technological Advances in Genomic Medicine: Next-Generation Sequencing (NGS) and Genomic Insights
8. Challenges and Future Directions
9. Conclusions
Funding
Conflicts of Interest
References
- Zhang, K.; Kan, C.; Chen, J.; Shi, J.; Ma, Y.; Wang, X.; Li, X.; Cai, W.; Pan, R.; Zhang, J.; et al. Epidemiology of 369 Diseases and Injuries Attributable to 84 Risk Factors: 1990–2019 with 2040 Projection. iScience 2024, 27, 109508. [Google Scholar] [CrossRef]
- Libby, P. The Changing Landscape of Atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Yang, R.; Yao, L.; Du, C.; Wu, Y. Identification of Key Pathways and Core Genes Involved in Atherosclerotic Plaque Progression. Ann. Transl. Med. 2021, 9, 267. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Y.; Zhao, Y.; Li, Y.; Zhang, X.; Bao, L.; Yan, R.; Yang, Y.; Zhou, H.; Zhang, J.; et al. Research Progress and Clinical Translation Potential of Coronary Atherosclerosis Diagnostic Markers from a Genomic Perspective. Genes 2025, 16, 98. [Google Scholar] [CrossRef]
- Currie, G.; Delles, C. Precision Medicine and Personalized Medicine in Cardiovascular Disease. Adv. Exp. Med. Biol. 2018, 1065, 589–605. [Google Scholar] [CrossRef]
- Usova, E.I.; Alieva, A.S.; Yakovlev, A.N.; Alieva, M.S.; Prokhorikhin, A.A.; Konradi, A.O.; Shlyakhto, E.V.; Magni, P.; Catapano, A.L.; Baragetti, A. Integrative Analysis of Multi-Omics and Genetic Approaches—A New Level in Atherosclerotic Cardiovascular Risk Prediction. Biomolecules 2021, 11, 1597. [Google Scholar] [CrossRef]
- Tuteja, S.; Rader, D.J. Genomic Medicine in the Prevention and Treatment of Atherosclerotic Cardiovascular Disease. Pers. Med. 2012, 9, 395–404. [Google Scholar] [CrossRef]
- Nayor, M.; Brown, K.J.; Vasan, R.S. The Molecular Basis of Predicting Atherosclerotic Cardiovascular Disease Risk. Circ. Res. 2021, 128, 287–303. [Google Scholar] [CrossRef]
- Mitsis, A.; Kadoglou, N.P.E.; Lambadiari, V.; Alexiou, S.; Theodoropoulos, K.C.; Avraamides, P.; Kassimis, G. Prognostic Role of Inflammatory Cytokines and Novel Adipokines in Acute Myocardial Infarction: An Updated and Comprehensive Review. Cytokine 2022, 153, 155848. [Google Scholar] [CrossRef]
- Cadby, G.; Giles, C.; Melton, P.E.; Huynh, K.; Mellett, N.A.; Duong, T.; Nguyen, A.; Cinel, M.; Smith, A.; Olshansky, G.; et al. Comprehensive Genetic Analysis of the Human Lipidome Identifies Loci Associated with Lipid Homeostasis with Links to Coronary Artery Disease. Nat. Commun. 2022, 13, 3124. [Google Scholar] [CrossRef]
- Aherrahrou, R.; Reinberger, T.; Hashmi, S.; Erdmann, J. GWAS Breakthroughs: Mapping the Journey from One Locus to 393 Significant Coronary Artery Disease Associations. Cardiovasc. Res. 2024, 120, 1508–1530. [Google Scholar] [CrossRef]
- Kaya, E.; Kayıkçıoğlu, M.; Tetik Vardarlı, A.; Eroğlu, Z.; Payzın, S.; Can, L. PCSK 9 Gain-of-Function Mutations (R496W and D374Y) and Clinical Cardiovascular Characteristics in a Cohort of Turkish Patients with Familial Hypercholesterolemia. Anatol. J. Cardiol. 2017, 18, 266–272. [Google Scholar] [CrossRef]
- McMaster, M.W.; Shah, A.; Kangarlu, J.; Cheikhali, R.; Frishman, W.H.; Aronow, W.S. The Impact of the Apolipoprotein E Genotype on Cardiovascular Disease and Cognitive Disorders. Cardiol. Rev. 2024, 10–1097. [Google Scholar] [CrossRef]
- Behbodikhah, J.; Ahmed, S.; Elyasi, A.; Kasselman, L.J.; de Leon, J.; Glass, A.D.; Reiss, A.B. Apolipoprotein B and Cardiovascular Disease: Biomarker and Potential Therapeutic Target. Metabolites 2021, 11, 690. [Google Scholar] [CrossRef]
- Gabel, B.R.; Koschinsky, M.L. Analysis of the Proteolytic Activity of a Recombinant Form of Apolipoprotein(a). Biochemistry 1995, 34, 15777–15784. [Google Scholar] [CrossRef]
- Lackner, C.; Cohen, J.C.; Hobbs, H.H. Molecular Definition of the Extreme Size Polymorphism in Apolipoprotein(a). Hum. Mol. Genet. 1993, 2, 933–940. [Google Scholar] [CrossRef]
- Clarke, R.; Peden, J.F.; Hopewell, J.C.; Kyriakou, T.; Goel, A.; Heath, S.C.; Parish, S.; Barlera, S.; Franzosi, M.G.; Rust, S.; et al. Genetic Variants Associated with Lp(a) Lipoprotein Level and Coronary Disease. N. Engl. J. Med. 2009, 361, 2518–2528. [Google Scholar] [CrossRef]
- Polisecki, E.; Muallem, H.; Maeda, N.; Peter, I.; Robertson, M.; McMahon, A.D.; Ford, I.; Packard, C.; Shepherd, J.; Jukema, J.W.; et al. Genetic Variation at the LDL Receptor and HMG-CoA Reductase Gene Loci, Lipid Levels, Statin Response, and Cardiovascular Disease Incidence in PROSPER. Atherosclerosis 2008, 200, 109–114. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. The LDL Receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef]
- Ølnes, Å.S.; Teigen, M.; Laerdahl, J.K.; Leren, T.P.; Strøm, T.B.; Bjune, K.; Rosak-Szyrocka, J. Variants in the CETP Gene Affect Levels of HDL Cholesterol by Reducing the Amount, and Not the Specific Lipid Transfer Activity, of Secreted CETP. PLoS ONE 2023, 18, e0294764. [Google Scholar] [CrossRef]
- Yu, C.; Bakshi, A.; Watts, G.F.; Renton, A.E.; Fulton-Howard, B.; Goate, A.M.; Natarajan, P.; Chasman, D.I.; Robman, L.; Woods, R.L.; et al. Genome-Wide Association Study of Cardiovascular Resilience Identifies Protective Variation in the CETP Gene. J. Am. Heart Assoc. 2023, 12, e031459. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, F.; Mansfield, B.S.; Raal, F.J. ANGPTL3 as a Drug Target in Hyperlipidemia and Atherosclerosis. Curr. Atheroscler. Rep. 2022, 24, 959–967. [Google Scholar] [CrossRef]
- Kjolby, M.; Andersen, O.M.; Breiderhoff, T.; Fjorback, A.W.; Pedersen, K.M.; Madsen, P.; Jansen, P.; Heeren, J.; Willnow, T.E.; Nykjaer, A. Sort1, Encoded by the Cardiovascular Risk Locus 1p13.3, Is a Regulator of Hepatic Lipoprotein Export. Cell Metab. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Sivapalaratnam, S.; Motazacker, M.M.; Maiwald, S.; Hovingh, G.K.; Kastelein, J.J.P.; Levi, M.; Trip, M.D.; Dallinga-Thie, G.M. Genome-Wide Association Studies in Atherosclerosis. Curr. Atheroscler. Rep. 2011, 13, 225–232. [Google Scholar] [CrossRef]
- Zhong, J.; Chen, X.; Ye, H.; Wu, N.; Chen, X.; Duan, S. CDKN2A and CDKN2B Methylation in Coronary Heart Disease Cases and Controls. Exp. Ther. Med. 2017, 14, 6093–6098. [Google Scholar] [CrossRef]
- Newman, C.B.; Tobert, J.A. Targeting PCSK9 with Antibodies and Gene Silencing to Reduce LDL Cholesterol. J. Clin. Endocrinol. Metab. 2023, 108, 784–790. [Google Scholar] [CrossRef]
- Wang, Z.; Yao, H.; Lin, S.; Zhu, X.; Shen, Z.; Lu, G.; Poon, W.S.; Xie, D.; Lin, M.C.; Kung, H. Transcriptional and Epigenetic Regulation of Human microRNAs. Cancer Lett. 2013, 331, 1–10. [Google Scholar] [CrossRef]
- Elguindy, A.; Yacoub, M.H. The Discovery of PCSK9 Inhibitors: A Tale of Creativity and Multifaceted Translational Research. Glob. Cardiol. Sci. Pract. 2013, 2013, 343–347. [Google Scholar] [CrossRef]
- Cariou, B.; Le May, C.; Costet, P. Clinical Aspects of PCSK9. Atherosclerosis 2011, 216, 258–265. [Google Scholar] [CrossRef]
- Grejtakova, D.; Boronova, I.; Bernasovska, J.; Bellosta, S. PCSK9 and Lipid Metabolism: Genetic Variants, Current Therapies, and Cardiovascular Outcomes. Cardiovasc. Drugs Ther. 2024, 1–13. [Google Scholar] [CrossRef]
- Vrablik, M.; Dlouha, D.; Todorovova, V.; Stefler, D.; Hubacek, J.A. Genetics of Cardiovascular Disease: How Far Are We from Personalized CVD Risk Prediction and Management? Int. J. Mol. Sci. 2021, 22, 4182. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Ma, L.; Chen, Y.; Li, J.; Wang, Y.; You, W.; Yuan, H.; Tang, X.; Ouyang, H.; Pang, D. Large-Scale CRISPR Screen of LDLR Pathogenic Variants. Research 2023, 6, 0203. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Langsted, A.; Nordestgaard, B.G. Remnant Cholesterol, LDL Cholesterol, and apoB Absolute Mass Changes Explain Results of the PROMINENT Trial. Atherosclerosis 2024, 393, 117556. [Google Scholar] [CrossRef]
- Utermann, G.; Menzel, H.J.; Kraft, H.G.; Duba, H.C.; Kemmler, H.G.; Seitz, C. Lp(a) Glycoprotein Phenotypes. Inheritance and Relation to Lp(a)-Lipoprotein Concentrations in Plasma. J. Clin. Investig. 1987, 80, 458–465. [Google Scholar] [CrossRef]
- Tsimikas, S.; Hall, J.L. Lipoprotein(a) as a Potential Causal Genetic Risk Factor of Cardiovascular Disease. J. Am. Coll. Cardiol. 2012, 60, 716–721. [Google Scholar] [CrossRef]
- Trégouët, D.A.; König, I.R.; Erdmann, J.; Munteanu, A.; Braund, P.S.; Hall, A.S.; Großhennig, A.; Linsel-Nitschke, P.; Perret, C.; DeSuremain, M.; et al. Genome-Wide Haplotype Association Study Identifies the SLC22A3-LPAL2-LPA Gene Cluster as a Risk Locus for Coronary Artery Disease. Nat. Genet. 2009, 41, 283–285. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Tybjærg-Hansen, A.; Steffensen, R.; Nordestgaard, B.G. Genetically Elevated Lipoprotein(a) and Increased Risk of Myocardial Infarction. JAMA 2009, 301, 2331–2339. [Google Scholar] [CrossRef]
- Qi, L.; Parast, L.; Cai, T.; Powers, C.; Gervino, E.V.; Hauser, T.H.; Hu, F.B.; Doria, A. Genetic Susceptibility to Coronary Heart Disease in Type 2 Diabetes. J. Am. Coll. Cardiol. 2011, 58, 2675–2682. [Google Scholar] [CrossRef]
- Lettre, G.; Palmer, C.D.; Young, T.; Ejebe, K.G.; Allayee, H.; Benjamin, E.J.; Bennett, F.; Bowden, D.W.; Chakravarti, A.; Dreisbach, A.; et al. Genome-Wide Association Study of Coronary Heart Disease and Its Risk Factors in 8,090 African Americans: The NHLBI CARe Project. PLoS Genet. 2011, 7, e1001300. [Google Scholar] [CrossRef]
- Pott, J.; Burkhardt, R.; Beutner, F.; Horn, K.; Teren, A.; Kirsten, H.; Holdt, L.M.; Schuler, G.; Teupser, D.; Loeffler, M.; et al. Genome-Wide Meta-Analysis Identifies Novel Loci of Plaque Burden in Carotid Artery. Atherosclerosis 2017, 259, 32–40. [Google Scholar] [CrossRef]
- Trinder, M.; Vikulova, D.; Pimstone, S.; Mancini, G.B.J.; Brunham, L.R. Polygenic Architecture and Cardiovascular Risk of Familial Combined Hyperlipidemia. Atherosclerosis 2022, 340, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Maouche, S.; Schunkert, H. Strategies beyond Genome-Wide Association Studies for Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Kamiza, A.B.; Touré, S.M.; Zhou, F.; Soremekun, O.; Cissé, C.; Wélé, M.; Touré, A.M.; Nashiru, O.; Corpas, M.; Nyirenda, M.; et al. Multi-Trait Discovery and Fine-Mapping of Lipid Loci in 125,000 Individuals of African Ancestry. Nat. Commun. 2023, 14, 5403. [Google Scholar] [CrossRef] [PubMed]
- Dron, J.S.; Natarajan, P.; Peloso, G.M. The Breadth and Impact of the Global Lipids Genetics Consortium. Curr. Opin. Lipidol. 2025, 36, 61–70. [Google Scholar] [CrossRef]
- Ottensmann, L.; Tabassum, R.; Ruotsalainen, S.E.; Gerl, M.J.; Klose, C.; Widén, E.; Gen, F.; Simons, K.; Ripatti, S.; Pirinen, M. Genome-Wide Association Analysis of Plasma Lipidome Identifies 495 Genetic Associations. Nat. Commun. 2023, 14, 6934. [Google Scholar] [CrossRef]
- Leite, J.M.R.S.; Pereira, J.L.; de Souza, C.A.; Pavan Soler, J.M.; Mingroni-Netto, R.C.; Fisberg, R.M.; Rogero, M.M.; Sarti, F.M. Novel Loci Linked to Serum Lipid Traits Are Identified in a Genome-Wide Association Study of a Highly Admixed Brazilian Population–the 2015 ISA Nutrition. Lipids Health Dis. 2024, 23, 229. [Google Scholar] [CrossRef]
- Aragam, K.G.; Natarajan, P. Polygenic Scores to Assess Atherosclerotic Cardiovascular Disease Risk: Clinical Perspectives and Basic Implications. Circ. Res. 2020, 126, 1159–1177. [Google Scholar] [CrossRef]
- Cornelissen, A.; Gadhoke, N.V.; Ryan, K.; Hodonsky, C.J.; Mitchell, R.; Bihlmeyer, N.A.; Duong, T.; Chen, Z.; Dikongue, A.; Sakamoto, A.; et al. Polygenic Risk Score Associates with Atherosclerotic Plaque Characteristics at Autopsy. Arterioscler. Thromb. Vasc. Biol. 2024, 44, 300–313. [Google Scholar] [CrossRef]
- Lerga-Jaso, J.; Terpolovsky, A.; Novković, B.; Osama, A.; Manson, C.; Bohn, S.; De Marino, A.; Kunitomi, M.; Yazdi, P.G. Optimization of Multi-Ancestry Polygenic Risk Score Disease Prediction Models. Sci. Rep. 2025, 15, 17495. [Google Scholar] [CrossRef]
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.-M.; Capodanno, D.; et al. 2021 ESC Guidelines on Cardiovascular Disease Prevention in Clinical Practice. Eur. Heart J. 2021, 42, 3227–3337. [Google Scholar] [CrossRef]
- Muntner, P.; Colantonio, L.D.; Cushman, M.; Goff, D.C.; Howard, G.; Howard, V.J.; Kissela, B.; Levitan, E.B.; Lloyd-Jones, D.M.; Safford, M.M. Validation of the Atherosclerotic Cardiovascular Disease Pooled Cohort Risk Equations. JAMA 2014, 311, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Muse, E.D.; Chen, S.-F.; Torkamani, A. Monogenic and Polygenic Models of Coronary Artery Disease. Curr. Cardiol. Rep. 2021, 23, 107. [Google Scholar] [CrossRef] [PubMed]
- Kessler, T.; Vilne, B.; Schunkert, H. The Impact of Genome-Wide Association Studies on the Pathophysiology and Therapy of Cardiovascular Disease. EMBO Mol. Med. 2016, 8, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fahed, A.C. Breaking Binary in Cardiovascular Disease Risk Prediction. npj Cardiovasc. Health 2025, 2, 2. [Google Scholar] [CrossRef]
- Walsh, R.; Jurgens, S.J.; Erdmann, J.; Bezzina, C.R. Genome-Wide Association Studies of Cardiovascular Disease. Physiol. Rev. 2023, 103, 2039–2055. [Google Scholar] [CrossRef]
- O’Sullivan, J.W.; Raghavan, S.; Marquez-Luna, C.; Luzum, J.A.; Damrauer, S.M.; Ashley, E.A.; O’Donnell, C.J.; Willer, C.J.; Natarajan, P.; on behalf of the American Heart Association Council on Genomic and Precision Medicine; Council on Clinical Cardiology; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Radiology and Intervention; Council on Lifestyle and Cardiometabolic Health; and Council on Peripheral Vascular Disease Polygenic Risk Scores for Cardiovascular Disease. Polygenic Risk Scores for Cardiovascular Disease: A Scientific Statement from the American Heart Association. Circulation 2022, 146, E93–E118. [Google Scholar] [CrossRef]
- Agbaedeng, T.A.; Noubiap, J.J.; Mofo Mato, E.P.; Chew, D.P.; Figtree, G.A.; Said, M.A.; Van Der Harst, P. Polygenic Risk Score and Coronary Artery Disease: A Meta-Analysis of 979,286 Participant Data. Atherosclerosis 2021, 333, 48–55. [Google Scholar] [CrossRef]
- Parsons, R.E.; Liu, X.; Collister, J.A.; Clifton, D.A.; Cairns, B.J.; Clifton, L. Independent External Validation of the QRISK3 Cardiovascular Disease Risk Prediction Model Using UK Biobank. Heart 2023, 109, 1690–1697. [Google Scholar] [CrossRef]
- Vilhjálmsson, B.J.; Yang, J.; Finucane, H.K.; Gusev, A.; Lindström, S.; Ripke, S.; Genovese, G.; Loh, P.-R.; Bhatia, G.; Do, R.; et al. Modeling Linkage Disequilibrium Increases Accuracy of Polygenic Risk Scores. Am. J. Hum. Genet. 2015, 97, 576–592. [Google Scholar] [CrossRef]
- Tada, H.; Usui, S.; Sakata, K.; Takamura, M.; Kawashiri, M. Challenges of Precision Medicine for Atherosclerotic Cardiovascular Disease Based on Human Genome Information. J. Atheroscler. Thromb. 2021, 28, 305–313. [Google Scholar] [CrossRef]
- Inouye, M.; Abraham, G.; Nelson, C.P.; Wood, A.M.; Sweeting, M.J.; Dudbridge, F.; Lai, F.Y.; Kaptoge, S.; Brozynska, M.; Wang, T.; et al. Genomic Risk Prediction of Coronary Artery Disease in 480,000 Adults: Implications for Primary Prevention. J. Am. Coll. Cardiol. 2018, 72, 1883–1893. [Google Scholar] [CrossRef] [PubMed]
- Tikkanen, E.; Havulinna, A.S.; Palotie, A.; Salomaa, V.; Ripatti, S. Genetic Risk Prediction and a 2-Stage Risk Screening Strategy for Coronary Heart Disease. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2261–2266. [Google Scholar] [CrossRef] [PubMed]
- Wells, Q.S.; Bagheri, M.; Aday, A.W.; Gupta, D.K.; Shaffer, C.M.; Wei, W.-Q.; Vaitinadin, N.S.; Khan, S.S.; Greenland, P.; Wang, T.J.; et al. Polygenic Risk Score to Identify Subclinical Coronary Heart Disease Risk in Young Adults. Circ. Genom. Precis. Med. 2021, 14, e003341. [Google Scholar] [CrossRef] [PubMed]
- Marston, N.A.; Pirruccello, J.P.; Melloni, G.E.M.; Koyama, S.; Kamanu, F.K.; Weng, L.-C.; Roselli, C.; Kamatani, Y.; Komuro, I.; Aragam, K.G.; et al. Predictive Utility of a Coronary Artery Disease Polygenic Risk Score in Primary Prevention. JAMA Cardiol. 2023, 8, 130–137. [Google Scholar] [CrossRef]
- Gray, M.P.; Berman, Y.; Bottà, G.; Grieve, S.M.; Ho, A.; Hu, J.; Hyun, K.; Ingles, J.; Jennings, G.; Kilov, G.; et al. Incorporating a Polygenic Risk Score-Triaged Coronary Calcium Score into Cardiovascular Disease Examinations to Identify Subclinical Coronary Artery Disease (ESCALATE): Protocol for a Prospective, Nonrandomized Implementation Trial. Am. Heart J. 2023, 264, 163–173. [Google Scholar] [CrossRef]
- Fahed, A.C.; Natarajan, P. Clinical Applications of Polygenic Risk Score for Coronary Artery Disease through the Life Course. Atherosclerosis 2023, 386, 117356. [Google Scholar] [CrossRef]
- Barlevy, D.; Lencz, T.; Carmi, S.; Kostick, K.M.; Mukherjee, M.; Pereira, S.; Lázaro-Muñoz, G. Capacities and Limitations of Using Polygenic Risk Scores for Reproductive Decision Making. Am. J. Bioeth. 2022, 22, 42–45. [Google Scholar] [CrossRef]
- Damiani, I.; Solberg, E.H.; Iyer, M.; Cheng, P.; Weldy, C.S.; Kim, J.B. Environmental Pollutants and Atherosclerosis: Epigenetic Mechanisms Linking Genetic Risk and Disease. Atherosclerosis 2025, 404, 119131. [Google Scholar] [CrossRef]
- Dai, Y.; Chen, D.; Xu, T. DNA Methylation Aberrant in Atherosclerosis. Front. Pharmacol. 2022, 13, 815977. [Google Scholar] [CrossRef]
- Jiang, W.; Agrawal, D.; Boosani, C. Cell-specific Histone Modifications in Atherosclerosis (Review). Mol. Med. Rep. 2018, 18, 1215–1224. [Google Scholar] [CrossRef]
- Jeltsch, A.; Broche, J.; Bashtrykov, P. Molecular Processes Connecting DNA Methylation Patterns with DNA Methyltransferases and Histone Modifications in Mammalian Genomes. Genes 2018, 9, 566. [Google Scholar] [CrossRef] [PubMed]
- Lucas, T.; Bonauer, A.; Dimmeler, S. RNA Therapeutics in Cardiovascular Disease. Circ. Res. 2018, 123, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Holdt, L.M.; Stahringer, A.; Sass, K.; Pichler, G.; Kulak, N.A.; Wilfert, W.; Kohlmaier, A.; Herbst, A.; Northoff, B.H.; Nicolaou, A.; et al. Circular Non-Coding RNA ANRIL Modulates Ribosomal RNA Maturation and Atherosclerosis in Humans. Nat. Commun. 2016, 7, 12429. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.; Li, H.; Han, X.; Chen, S.; Yang, B.; Hu, Z.; Zhu, H.; Cai, C.; Chen, J.; et al. Characterization of LncRNA Expression Profile and Identification of Novel LncRNA Biomarkers to Diagnose Coronary Artery Disease. Atherosclerosis 2018, 275, 359–367. [Google Scholar] [CrossRef]
- Bekkering, S.; Joosten, L.A.B.; Meer, J.W.M.V.D.; Netea, M.G.; Riksen, N.P. Trained Innate Immunity and Atherosclerosis. Curr. Opin. Lipidol. 2013, 24, 487–492. [Google Scholar] [CrossRef]
- Wang, B.; Qiu, J.; Lian, J.; Yang, X.; Zhou, J. Gut Metabolite Trimethylamine-N-Oxide in Atherosclerosis: From Mechanism to Therapy. Front. Cardiovasc. Med. 2021, 8, 723886. [Google Scholar] [CrossRef]
- DeGroat, W.; Abdelhalim, H.; Peker, E.; Sheth, N.; Narayanan, R.; Zeeshan, S.; Liang, B.T.; Ahmed, Z. Multimodal AI/ML for Discovering Novel Biomarkers and Predicting Disease Using Multi-Omics Profiles of Patients with Cardiovascular Diseases. Sci. Rep. 2024, 14, 26503. [Google Scholar] [CrossRef]
- Qian, Q.; Xue, R.; Xu, C.; Wang, F.; Zeng, J.; Xiao, J. CVD Atlas: A Multi-Omics Database of Cardiovascular Disease. Nucleic Acids Res. 2025, 53, D1348–D1355. [Google Scholar] [CrossRef]
- Leon-Mimila, P.; Wang, J.; Huertas-Vazquez, A. Relevance of Multi-Omics Studies in Cardiovascular Diseases. Front. Cardiovasc. Med. 2019, 6, 91. [Google Scholar] [CrossRef]
- Schuermans, A.; Pournamdari, A.B.; Lee, J.; Bhukar, R.; Ganesh, S.; Darosa, N.; Small, A.M.; Yu, Z.; Hornsby, W.; Koyama, S.; et al. Integrative Proteomic Analyses across Common Cardiac Diseases Yield Mechanistic Insights and Enhanced Prediction. Nat. Cardiovasc. Res. 2024, 3, 1516–1530. [Google Scholar] [CrossRef]
- Schiano, C.; Costa, V.; Aprile, M.; Grimaldi, V.; Maiello, C.; Esposito, R.; Soricelli, A.; Colantuoni, V.; Donatelli, F.; Ciccodicola, A.; et al. Heart Failure: Pilot Transcriptomic Analysis of Cardiac Tissue by RNA-Sequencing. Cardiol. J. 2017, 24, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H.; Hobbs, H.H. Sequence Variations in PCSK9, Low LDL, and Protection against Coronary Heart Disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J. Management of Severe Dyslipidaemia: Role of PCSK9 Inhibitors. Eur. Cardiol. Rev. 2018, 13, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.P.; Wiviott, S.D.; Raal, F.J.; Blom, D.J.; Robinson, J.; Ballantyne, C.M.; Somaratne, R.; Legg, J.; Wasserman, S.M.; et al. Efficacy and Safety of Evolocumab in Reducing Lipids and Cardiovascular Events. N. Engl. J. Med. 2015, 372, 1500–1509. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Giugliano, R.P.; Wiviott, S.D.; Atar, D.; Keech, A.; Kuder, J.F.; Im, K.; Murphy, S.A.; Flores-Arredondo, J.H.; López, J.A.G.; et al. Long-Term Evolocumab in Patients with Established Atherosclerotic Cardiovascular Disease. Circulation 2022, 146, 1109–1119. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Bessac, L.; Berdan, L.G.; Bhatt, D.L.; Bittner, V.; Diaz, R.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; Jukema, J.W.; et al. Effect of Alirocumab, a Monoclonal Antibody to PCSK9, on Long-Term Cardiovascular Outcomes Following Acute Coronary Syndromes: Rationale and Design of the ODYSSEY Outcomes Trial. Am. Heart J. 2014, 168, 682–689. [Google Scholar] [CrossRef]
- Goodman, S.G.; Steg, P.G.; Poulouin, Y.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Garon, G.; Harrington, R.A.; Jukema, J.W.; Manvelian, G.; et al. Long-Term Efficacy, Safety, and Tolerability of Alirocumab in 8242 Patients Eligible for 3 to 5 Years of Placebo-Controlled Observation in the ODYSSEY OUTCOMES Trial. J. Am. Heart Assoc. 2023, 12, e029216. [Google Scholar] [CrossRef]
- Räber, L.; Ueki, Y.; Otsuka, T.; Losdat, S.; Häner, J.D.; Lonborg, J.; Fahrni, G.; Iglesias, J.F.; van Geuns, R.-J.; Ondracek, A.S.; et al. Effect of Alirocumab Added to High-Intensity Statin Therapy on Coronary Atherosclerosis in Patients with Acute Myocardial Infarction: The PACMAN-AMI Randomized Clinical Trial. JAMA 2022, 327, 1771–1781. [Google Scholar] [CrossRef]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Ray, K.K.; Troquay, R.P.T.; Visseren, F.L.J.; Leiter, L.A.; Scott Wright, R.; Vikarunnessa, S.; Talloczy, Z.; Zang, X.; Maheux, P.; Lesogor, A.; et al. Long-Term Efficacy and Safety of Inclisiran in Patients with High Cardiovascular Risk and Elevated LDL Cholesterol (ORION-3): Results from the 4-Year Open-Label Extension of the ORION-1 Trial. Lancet Diabetes Endocrinol. 2023, 11, 109–119. [Google Scholar] [CrossRef]
- Wright, R.S.; Raal, F.J.; Koenig, W.; Landmesser, U.; Leiter, L.A.; Vikarunnessa, S.; Lesogor, A.; Maheux, P.; Talloczy, Z.; Zang, X.; et al. Inclisiran Administration Potently and Durably Lowers LDL-C over an Extended-Term Follow-up: The ORION-8 Trial. Cardiovasc. Res. 2024, 120, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Landmesser, U.; Leiter, L.A.; Kallend, D.; Dufour, R.; Karakas, M.; Hall, T.; Troquay, R.P.T.; Turner, T.; Visseren, F.L.J.; et al. Inclisiran in Patients at High Cardiovascular Risk with Elevated LDL Cholesterol. N. Engl. J. Med. 2017, 376, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Hajar, R. PCSK 9 Inhibitors: A Short History and a New Era of Lipid-Lowering Therapy. Heart Views 2019, 20, 74–75. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Jeswani, B.M.; Sharma, S.; Rathore, S.S.; Nazir, A.; Bhatheja, R.; Kapoor, K. PCSK9 Inhibitors: The Evolving Future. Health Sci. Rep. 2024, 7, e70174. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, R.; Zhu, J.; Yang, X.; Luo, H.; Wang, H.; Luo, X. Failure of Lipid Control by PCSK9 Inhibitors in Compound Heterozygous Familial Hypercholesterolemia Complicated with Premature Myocardial Infarction: A Case Report. Clin. Case Rep. 2024, 12, e8498. [Google Scholar] [CrossRef] [PubMed]
- Arca, M.; Celant, S.; Olimpieri, P.P.; Colatrella, A.; Tomassini, L.; D’Erasmo, L.; Averna, M.; Zambon, A.; Catapano, A.L.; Russo, P. Real-World Effectiveness of PCSK9 Inhibitors in Reducing LDL-C in Patients with Familial Hypercholesterolemia in Italy: A Retrospective Cohort Study Based on the AIFA Monitoring Registries. J. Am. Heart Assoc. 2023, 12, e026550. [Google Scholar] [CrossRef]
- Istvan, E.S.; Deisenhofer, J. Structural Mechanism for Statin Inhibition of HMG-CoA Reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef]
- Zheng, E.; Madura, P.; Grandos, J.; Broncel, M.; Pawlos, A.; Woźniak, E.; Gorzelak-Pabiś, P. When the Same Treatment Has Different Response: The Role of Pharmacogenomics in Statin Therapy. Biomed. Pharmacother. 2024, 170, 115966. [Google Scholar] [CrossRef]
- Klyushova, L.S.; Perepechaeva, M.L.; Grishanova, A.Y. The Role of CYP3A in Health and Disease. Biomedicines 2022, 10, 2686. [Google Scholar] [CrossRef]
- Patel, K.A.; Bhatt, M.H.; Hirani, R.V.; Patel, V.A.; Patel, V.N.; Shah, G.B.; Chorawala, M.R. Assessment of Potential Drug-Drug Interactions among Outpatients in a Tertiary Care Hospital: Focusing on the Role of P-Glycoprotein and CYP3A4 (Retrospective Observational Study). Heliyon 2022, 8, e11278. [Google Scholar] [CrossRef] [PubMed]
- Tsamandouras, N.; Dickinson, G.; Guo, Y.; Hall, S.; Rostami-Hodjegan, A.; Galetin, A.; Aarons, L. Identification of the Effect of Multiple Polymorphisms on the Pharmacokinetics of Simvastatin and Simvastatin Acid Using a Population-Modeling Approach. Clin. Pharmacol. Ther. 2014, 96, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Elens, L.; Becker, M.L.; Haufroid, V.; Hofman, A.; Visser, L.E.; Uitterlinden, A.G.; Stricker, B.C.; van Schaik, R.H.N. Novel CYP3A4 Intron 6 Single Nucleotide Polymorphism Is Associated with Simvastatin-Mediated Cholesterol Reduction in The Rotterdam Study. Pharmacogenet. Genom. 2011, 21, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Klein, K.; Thomas, M.; Winter, S.; Nussler, A.K.; Niemi, M.; Schwab, M.; Zanger, U.M. PPARA: A Novel Genetic Determinant of CYP3A4 In Vitro and In Vivo. Clin. Pharmacol. Ther. 2012, 91, 1044–1052. [Google Scholar] [CrossRef]
- Zhou, X.-Y.; Hu, X.-X.; Wang, C.-C.; Lu, X.-R.; Chen, Z.; Liu, Q.; Hu, G.-X.; Cai, J.-P. Enzymatic Activities of CYP3A4 Allelic Variants on Quinine 3-Hydroxylation In Vitro. Front. Pharmacol. 2019, 10, 591. [Google Scholar] [CrossRef]
- Sprowl, J.A.; Ong, S.S.; Gibson, A.A.; Hu, S.; Du, G.; Lin, W.; Li, L.; Bharill, S.; Ness, R.A.; Stecula, A.; et al. A Phosphotyrosine Switch Regulates Organic Cation Transporters. Nat. Commun. 2016, 7, 10880. [Google Scholar] [CrossRef]
- Perland, E.; Fredriksson, R. Classification Systems of Secondary Active Transporters. Trends Pharmacol. Sci. 2017, 38, 305–315. [Google Scholar] [CrossRef]
- Postmus, I.; Trompet, S.; Deshmukh, H.A.; Barnes, M.R.; Li, X.; Warren, H.R.; Chasman, D.I.; Zhou, K.; Arsenault, B.J.; Donnelly, L.A.; et al. Pharmacogenetic Meta-Analysis of Genome-Wide Association Studies of LDL Cholesterol Response to Statins. Nat. Commun. 2014, 5, 5068. [Google Scholar] [CrossRef]
- Ramsey, L.B.; Johnson, S.G.; Caudle, K.E.; Haidar, C.E.; Voora, D.; Wilke, R.A.; Maxwell, W.D.; McLeod, H.L.; Krauss, R.M.; Roden, D.M.; et al. The Clinical Pharmacogenetics Implementation Consortium Guideline for SLCO1B1 and Simvastatin-Induced Myopathy: 2014 Update. Clin. Pharmacol. Ther. 2014, 96, 423–428. [Google Scholar] [CrossRef]
- SEARCH Collaborative Group; Link, E.; Parish, S.; Armitage, J.; Bowman, L.; Heath, S.; Matsuda, F.; Gut, I.; Lathrop, M.; Collins, R. SLCO1B1 Variants and Statin-Induced Myopathy—A Genomewide Study. N. Engl. J. Med. 2008, 359, 789–799. [Google Scholar] [CrossRef]
- Chasman, D.I.; Giulianini, F.; MacFadyen, J.; Barratt, B.J.; Nyberg, F.; Ridker, P.M. Genetic Determinants of Statin-Induced Low-Density Lipoprotein Cholesterol Reduction: The Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) Trial. Circ. Cardiovasc. Genet. 2012, 5, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Sortica, V.A.; Fiegenbaum, M.; Lima, L.O.; Van Der Sand, C.R.; Van Der Sand, L.C.; Ferreira, M.E.W.; Pires, R.C.; Hutz, M.H. SLCO1B1 Gene Variability Influences Lipid-Lowering Efficacy on Simvastatin Therapy in Southern Brazilians. Clin. Chem. Lab. Med. 2012, 50, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.C.; Perin, P.M.S.; Purim, S.G.; Silbiger, V.N.; Genvigir, F.D.V.; Willrich, M.A.V.; Arazi, S.S.; Luchessi, A.D.; Hirata, M.H.; Bernik, M.M.S.; et al. Pharmacogenetics of OATP Transporters Reveals That SLCO1B1 c.388A>G Variant Is Determinant of Increased Atorvastatin Response. Int. J. Mol. Sci. 2011, 12, 5815–5827. [Google Scholar] [CrossRef]
- Moon, J.Y.; Franchi, F.; Rollini, F.; Rivas Rios, J.R.; Kureti, M.; Cavallari, L.H.; Angiolillo, D.J. Role of Genetic Testing in Patients Undergoing Percutaneous Coronary Intervention. Expert Rev. Clin. Pharmacol. 2018, 11, 151–164. [Google Scholar] [CrossRef]
- Angiolillo, D.J.; Fernandez-Ortiz, A.; Bernardo, E.; Alfonso, F.; Macaya, C.; Bass, T.A.; Costa, M.A. Variability in Individual Responsiveness to Clopidogrel: Clinical Implications, Management, and Future Perspectives. J. Am. Coll. Cardiol. 2007, 49, 1505–1516. [Google Scholar] [CrossRef] [PubMed]
- Dean, L.; Kane, M. Clopidogrel Therapy and CYP2C19 Genotype. In Medical Genetics Summaries; Pratt, V.M., Scott, S.A., Pirmohamed, M., Esquivel, B., Kattman, B.L., Malheiro, A.J., Eds.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012. [Google Scholar]
- Shuldiner, A.R.; O’connell, J.R.; Bliden, K.P.; Gandhi, A.; Ryan, K.; Horenstein, R.B.; Damcott, C.M.; Pakyz, R.; Tantry, U.S.; Gibson, Q.; et al. Association of Cytochrome P450 2C19 Genotype with the Antiplatelet Effect and Clinical Efficacy of Clopidogrel Therapy. JAMA 2009, 302, 849–857. [Google Scholar] [CrossRef]
- Scott, S.A.; Sangkuhl, K.; Shuldiner, A.R.; Hulot, J.-S.; Thorn, C.F.; Altman, R.B.; Klein, T.E. PharmGKB Summary: Very Important Pharmacogene Information for Cytochrome P450, Family 2, Subfamily C, Polypeptide 19. Pharmacogenet. Genom. 2012, 22, 159–165. [Google Scholar] [CrossRef]
- Frére, C.; Cuisset, T.; Gaborit, B.; Alessi, M.C.; Hulot, J.S. The CYP2C19*17 Allele Is Associated with Better Platelet Response to Clopidogrel in Patients Admitted for Non-ST Acute Coronary Syndrome. J. Thromb. Haemost. 2009, 7, 1409–1411. [Google Scholar] [CrossRef]
- Lee, C.R.; Luzum, J.A.; Sangkuhl, K.; Gammal, R.S.; Sabatine, M.S.; Stein, C.M.; Kisor, D.F.; Limdi, N.A.; Lee, Y.M.; Scott, S.A.; et al. Clinical Pharmacogenetics Implementation Consortium Guideline for CYP2C19 Genotype and Clopidogrel Therapy: 2022 Update. Clin. Pharmacol. Ther. 2022, 112, 959–967. [Google Scholar] [CrossRef]
- Mega, J.L.; Close, S.L.; Wiviott, S.D.; Shen, L.; Hockett, R.D.; Brandt, J.T.; Walker, J.R.; Antman, E.M.; Macias, W.; Braunwald, E.; et al. Cytochrome P-450 Polymorphisms and Response to Clopidogrel. N. Engl. J. Med. 2009, 360, 354–362. [Google Scholar] [CrossRef]
- Mega, J.L.; Simon, T.; Collet, J.-P.; Anderson, J.L.; Antman, E.M.; Bliden, K.; Cannon, C.P.; Danchin, N.; Giusti, B.; Gurbel, P.; et al. Reduced-Function CYP2C19 Genotype and Risk of Adverse Clinical Outcomes among Patients Treated with Clopidogrel Predominantly for PCI: A Meta-Analysis. JAMA 2010, 304, 1821–1830. [Google Scholar] [CrossRef]
- Siller-Matula, J.M.; Trenk, D.; Schrör, K.; Gawaz, M.; Kristensen, S.D.; Storey, R.F.; Huber, K.; European Platelet Academy. How to Improve the Concept of Individualised Antiplatelet Therapy with P2Y12 Receptor Inhibitors–Is an Algorithm the Answer? Thromb. Haemost. 2015, 113, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xiao, T.; Chen, L.; Xie, S.; Deng, M.; Wu, D. The Association of ADRB1 and CYP2D6 Polymorphisms with Antihypertensive Effects and Analysis of Their Contribution to Hypertension Risk. Am. J. Med. Sci. 2018, 355, 235–239. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Z.Q.; Tan, Z.R.; Chen, X.P.; Wang, L.S.; Zhou, G.; Zhou, H.H. Gly389Arg Polymorphism of β1-Adrenergic Receptor Is Associated with the Cardiovascular Response to Metoprolol. Clin. Pharmacol. Ther. 2003, 74, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Petrović, J.; Pešić, V.; Lauschke, V.M. Frequencies of Clinically Important CYP2C19 and CYP2D6 Alleles Are Graded across Europe. Eur. J. Hum. Genet. 2020, 28, 88–94. [Google Scholar] [CrossRef]
- Cavallari, L.H.; Obeng, A.O. Genetic Determinants of P2Y12 Inhibitors and Clinical Implications. Interv. Cardiol. Clin. 2017, 6, 141–149. [Google Scholar] [CrossRef]
- Rysz, J.; Franczyk, B.; Rysz-Górzyńska, M.; Gluba-Brzózka, A. Pharmacogenomics of Hypertension Treatment. Int. J. Mol. Sci. 2020, 21, 4709. [Google Scholar] [CrossRef]
- Authors/Task Force members; Windecker, S.; Kolh, P.; Alfonso, F.; Collet, J.-P.; Cremer, J.; Falk, V.; Filippatos, G.; Hamm, C.; Head, S.J. 2014 ESC/EACTS Guidelines on Myocardial Revascularization: The Task Force on Myocardial Revascularization of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS)Developed with the Special Contribution of the European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur. Heart J. 2014, 35, 2541–2619. [Google Scholar] [CrossRef]
- Levine, G.N.; Bates, E.R.; Bittl, J.A.; Brindis, R.G.; Fihn, S.D.; Fleisher, L.A.; Granger, C.B.; Lange, R.A.; Mack, M.J.; Mauri, L.; et al. 2016 ACC/AHA Guideline Focused Update on Duration of Dual Antiplatelet Therapy in Patients with Coronary Artery Disease. Circulation 2016, 68, e1082–e1115. [Google Scholar] [CrossRef]
- Hochholzer, W.; Trenk, D.; Fromm, M.F.; Valina, C.M.; Stratz, C.; Bestehorn, H.-P.; Büttner, H.J.; Neumann, F.-J. Impact of Cytochrome P450 2C19 Loss-of-Function Polymorphism and of Major Demographic Characteristics on Residual Platelet Function After Loading and Maintenance Treatment with Clopidogrel in Patients Undergoing Elective Coronary Stent Placement. J. Am. Coll. Cardiol. 2010, 55, 2427–2434. [Google Scholar] [CrossRef]
- Alexopoulos, D.; Dimitropoulos, G.; Davlouros, P.; Xanthopoulou, I.; Kassimis, G.; Stavrou, E.F.; Hahalis, G.; Athanassiadou, A. Prasugrel Overcomes High On-Clopidogrel Platelet Reactivity Post-Stenting More Effectively Than High-Dose (150-Mg) Clopidogrel. JACC Cardiovasc. Interv. 2011, 4, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Malinin, A.; Pokov, A.; Spergling, M.; Defranco, A.; Schwartz, K.; Schwartz, D.; Mahmud, E.; Atar, D.; Serebruany, V. Monitoring Platelet Inhibition after Clopidogrel with the VerifyNow-P2Y12® Rapid Analyzer: The VERIfy Thrombosis Risk ASsessment (VERITAS) Study. Thromb. Res. 2007, 119, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.D.; Wells, G.A.; Le May, M.R.; Labinaz, M.; Glover, C.; Froeschl, M.; Dick, A.; Marquis, J.-F.; O’Brien, E.; Goncalves, S.; et al. Point-of-Care Genetic Testing for Personalisation of Antiplatelet Treatment (RAPID GENE): A Prospective, Randomised, Proof-of-Concept Trial. Lancet 2012, 379, 1705–1711. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, A.C.; Musunuru, K. CRISPR-Cas9 Genome Editing for Treatment of Atherogenic Dyslipidemia. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 12–18. [Google Scholar] [CrossRef]
- Lee, R.G.; Mazzola, A.M.; Braun, M.C.; Platt, C.; Vafai, S.B.; Kathiresan, S.; Rohde, E.; Bellinger, A.M.; Khera, A.V. Efficacy and Safety of an Investigational Single-Course CRISPR Base-Editing Therapy Targeting PCSK9 in Nonhuman Primate and Mouse Models. Circulation 2023, 147, 242–253. [Google Scholar] [CrossRef]
- Zuo, Y.; Zhang, C.; Zhou, Y.; Li, H.; Xiao, W.; Herzog, R.W.; Xu, J.; Zhang, J.; Chen, Y.E.; Han, R. Liver-Specific in Vivo Base Editing of Angptl3 via AAV Delivery Efficiently Lowers Blood Lipid Levels in Mice. Cell Biosci. 2023, 13, 109. [Google Scholar] [CrossRef]
- Li, Y.; Liu, H.; Shen, C.; Li, J.; Liu, F.; Huang, K.; Gu, D.; Li, Y.; Lu, X. Association of Genetic Variants Related to Combined Lipid-Lowering and Antihypertensive Therapies with Risk of Cardiovascular Disease: 2 × 2 Factorial Mendelian Randomization Analyses. BMC Med. 2024, 22, 201. [Google Scholar] [CrossRef]
- Shan, H.; Fei, T. CRISPR Screening in Cardiovascular Research. Front. Cell Dev. Biol. 2023, 11, 1175849. [Google Scholar] [CrossRef]
- Shinwari, Z.K.; Tanveer, F.; Khalil, A.T. Ethical Issues Regarding CRISPR Mediated Genome Editing. Curr. Issues Mol. Biol. 2018, 26, 103–110. [Google Scholar] [CrossRef]
- Hooper, A.J.; Tang, X.L.; Burnett, J.R. VERVE-101, a CRISPR Base-Editing Therapy Designed to Permanently Inactivate Hepatic PCSK9 and Reduce LDL-Cholesterol. Expert Opin. Investig. Drugs 2024, 33, 753–756. [Google Scholar] [CrossRef]
- Coltell, O.; Asensio, E.M.; Sorlí, J.V.; Ortega-Azorín, C.; Fernández-Carrión, R.; Pascual, E.C.; Barragán, R.; González, J.I.; Estruch, R.; Alzate, J.F.; et al. Associations between the New DNA-Methylation-Based Telomere Length Estimator, the Mediterranean Diet and Genetics in a Spanish Population at High Cardiovascular Risk. Antioxidants 2023, 12, 2004. [Google Scholar] [CrossRef] [PubMed]
- Salata, E.; Fotis, T.; Ntoumou, E.; Sagonas, A.; Panagiotou, N. Adjusted Polygenic Risk Score: A Novel Biomarker for the Prevention of Cardiovascular Diseases. World Acad. Sci. J. 2025, 7, 32. [Google Scholar] [CrossRef]
- Mathew, J.; Ragesh, K.; Kunhunni, D.G.P.; Sreekumar, S.; Chandrababu, A. Advancements in Precision Medicine for Imaging and Pharmacotherapy in Atherosclerosis: Recent Discoveries and Emerging Technologies. J. Young Pharm. 2024, 16, 660–666. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of Age: Ten Years of next-Generation Sequencing Technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Lu, Y.; Li, M.; Gao, Z.; Ma, H.; Chong, Y.; Hong, J.; Wu, J.; Wu, D.; Xi, D.; Deng, W. Advances in Whole Genome Sequencing: Methods, Tools, and Applications in Population Genomics. Int. J. Mol. Sci. 2025, 26, 372. [Google Scholar] [CrossRef]
- Bartha, Á.; Győrffy, B. Comprehensive Outline of Whole Exome Sequencing Data Analysis Tools Available in Clinical Oncology. Cancers 2019, 11, 1725. [Google Scholar] [CrossRef]
- Pei, X.M.; Yeung, M.H.Y.; Wong, A.N.N.; Tsang, H.F.; Yu, A.C.S.; Yim, A.K.Y.; Wong, S.C.C. Targeted Sequencing Approach and Its Clinical Applications for the Molecular Diagnosis of Human Diseases. Cells 2023, 12, 493. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-Omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14, 117793221989905. [Google Scholar] [CrossRef]
- He, B.; Zhu, R.; Yang, H.; Lu, Q.; Wang, W.; Song, L.; Sun, X.; Zhang, G.; Li, S.; Yang, J.; et al. Assessing the Impact of Data Preprocessing on Analyzing Next Generation Sequencing Data. Front. Bioeng. Biotechnol. 2020, 8, 817. [Google Scholar] [CrossRef]
- Lischer, H.E.L.; Shimizu, K.K. Reference-Guided de Novo Assembly Approach Improves Genome Reconstruction for Related Species. BMC Bioinform. 2017, 18, 474. [Google Scholar] [CrossRef]
- Zhao, B.H.; Ruze, A.; Zhao, L.; Li, Q.L.; Tang, J.; Xiefukaiti, N.; Gai, M.T.; Deng, A.X.; Shan, X.F.; Gao, X.M. The Role and Mechanisms of Microvascular Damage in the Ischemic Myocardium. Cell. Mol. Life Sci. 2023, 80, 341. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Baxter, S.L.; Xu, J.; Xu, J.; Zhou, X.; Zhang, K. The Practical Implementation of Artificial Intelligence Technologies in Medicine. Nat. Med. 2019, 25, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Lysenko, A.; Jia, S.; Boroevich, K.A.; Tsunoda, T. Advances in AI and Machine Learning for Predictive Medicine. J. Hum. Genet. 2024, 69, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Sopic, M.; Vilne, B.; Gerdts, E.; Trindade, F.; Uchida, S.; Khatib, S.; Wettinger, S.B.; Devaux, Y.; Magni, P.; EU-AtheroNET COST Action CA21153. Multiomics Tools for Improved Atherosclerotic Cardiovascular Disease Management. Trends Mol. Med. 2023, 29, 983–995. [Google Scholar] [CrossRef]
- Mitsis, A.; Myrianthefs, M.; Sokratous, S.; Karmioti, G.; Kyriakou, M.; Drakomathioulakis, M.; Tzikas, S.; Kadoglou, N.P.E.; Karagiannidis, E.; Nasoufidou, A.; et al. Emerging Therapeutic Targets for Acute Coronary Syndromes: Novel Advancements and Future Directions. Biomedicines 2024, 12, 1670. [Google Scholar] [CrossRef]
- Kerr, M.; Pears, R.; Miedzybrodzka, Z.; Haralambos, K.; Cather, M.; Watson, M.; Humphries, S.E. Cost Effectiveness of Cascade Testing for Familial Hypercholesterolaemia, Based on Data from Familial Hypercholesterolaemia Services in the UK. Eur. Heart J. 2017, 38, 1832–1839. [Google Scholar] [CrossRef]
- Bowdin, S.; Gilbert, A.; Bedoukian, E.; Carew, C.; Adam, M.P.; Belmont, J.; Bernhardt, B.; Biesecker, L.; Bjornsson, H.T.; Blitzer, M.; et al. Recommendations for the Integration of Genomics into Clinical Practice. Genet. Med. 2016, 18, 1075–1084. [Google Scholar] [CrossRef]
- Wang, R.S.; Maron, B.A.; Loscalzo, J. Multi-Omics Network Medicine Approaches to Precision Medicine and Therapeutics in Cardiovascular Diseases. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 493–503. [Google Scholar] [CrossRef]
- Bozyel, S.; Şimşek, E.; Koçyiğit, D.; Güler, A.; Korkmaz, Y.; Şeker, M.; Ertürk, M.; Keser, N. Artificial Intelligence-Based Clinical Decision Support Systems in Cardiovascular Diseases. Anatol. J. Cardiol. 2024, 28, 74–86. [Google Scholar] [CrossRef]
- Cuevas-Chávez, A.; Hernández, Y.; Ortiz-Hernandez, J.; Sánchez-Jiménez, E.; Ochoa-Ruiz, G.; Pérez, J.; González-Serna, G. A Systematic Review of Machine Learning and IoT Applied to the Prediction and Monitoring of Cardiovascular Diseases. Healthcare 2023, 11, 2240. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Gene | Chromosomal Location | Function | Impact on ASCVD | Clinical Implications | References+9 |
|---|---|---|---|---|---|
| PCSK9 | 1p32 | Promotes LDLR degradation | ↑ LDL-C (GOF) ↓ LDL-C (LOF) | PCSK9 inhibitors reduce LDL-C ↑ ASCVD risk | Aherrahrou R. [11] Kaya E. [12] |
| APOE | 19q13.32 | Cholesterol transport | E4 isoform raises ASCVD risk | Screening helps assess genetic risk | McMaster M.W. [13] |
| APOB | 2p24.1 | LDL binding to LDLR | Impaired binding increases ASCVD risk | Reflects atherogenic particle count ↑ ASCVD risk | Behbodikhah J. [14] |
| LPA | 6q2.6–2.7 | Forms Lp(a) particles | Elevated Lp(a) ↑ ASCVD risk | ↑ ASCVD risk | Gabel B.R. [15] Lackner C. [16] Clarke R. [17] |
| LDLR | 19p13.2 | LDL-C clearance | FH | Targeted by statins and PCSK9 inhibitors ↑ ASCVD risk | Polisecki E. [18] Goldstein J.L. [19] |
| CETP | 16q21 | Transfers cholesteryl esters | Favorable variants reduce ASCVD risk | ↓ ASCVD risk | Ølnes Å.S. [20] Yu C. [21] |
| ANGPTL3 | 1p31.3 | Inhibits lipid clearance enzymes | LOF mutations reduce ASCVD risk | Targeted for lipid-lowering therapies | Mohamed, F. [22] |
| SORT1 | 1p31 | Regulates lipid metabolism | Variants affect LDL-C and ASCVD | Potential therapeutic target | Kjolby M [23] |
| CELSR2 | 1p31 | Alters hepatic expression | Affects LDL-C metabolism | Biomarker for ASCVD risk | Sivapalaratnam S. [24] |
| PSRC1 | 1p31 | Modulates hepatic gene expression | Impacts cholesterol levels | Role in ASCVD under investigation | Sivapalaratnam S. [24] |
| CDKN2A | 9p21 | Regulates the cell cycle | ↑ ASCVD risk | Potential predictive biomarker | Zhong J. [25] |
| Drug Class | Example | Key Gene(s) Involved | Genetic Impact on Drug Response | Clinical Application | References |
|---|---|---|---|---|---|
| Statin | Simvastatin Atorvastatin | CYP3A4 | LoF: *22(rs35599367): ↑ bioavailability of simvastatin and ↓ concentration of atorvastatin metabolites. → Variability in plasma levels. | Be aware of CYP3A4 inhibitors and inducers to avoid drug-drug interactions. | Zheng E. [99] Patel K.A. [101] Tsamandouras N. [102] Elens L. [103] Sprowl J.A. [106] |
| Statin | Fluvastatin | CYP2C9 | LoF: *3 ↑ plasma concentration of fluvastatin. ↑ risk of concentration-dependent side effects. | Patients carrying the CYP2C9*3 allele may require dose adjustment or consideration of alternative statins due to reduced metabolic clearance of fluvastatin. Monitoring for concentration-dependent adverse effects, such as myopathy or elevated liver enzymes, is advised. | Perland, E. [107] |
| Statin | All statins (notably simvastatin, atorvastatin, rosuvastatin | SLCO1B1 | LoF: c.521T>C, rs4149056 ↓ OATP1B1 transporter function. ↑ plasma statin levels → increased risk of statin-induced myopathy. ↓ hepatic uptake → ↓ efficacy of statin therapy GoF: c.388A>G, rs2306283 ↑ OATP1B1 transporter function. ↑ hepatic uptake → ↑ efficacy of statin therapy | SLCO1B1 genotyping (e.g., testing for c.521T>C) is recommended to guide statin selection and dosing, particularly to reduce myopathy risk [114]. For patients with reduced function alleles, lower doses or alternative statins may be preferred to reduce adverse effects and maintain therapeutic efficacy | Ramsey, L.B. [109] Sortica, V.A. [112] Rodrigues, A.C. [113] |
| PCSK9 INH | Alirocumab Evolocumab | LDLR | HoFH: Few or non-functional LDL receptors → poor response to PCSK9 inhibitors. HeFH: Some functional LDLRs → good response. | Approved for patients with HeFH or HoFH. In clinical ASCVD, when further LDL-C reduction is needed beyond statins and ezetimibe. | Sabatine M.S. [84] Schwartz G.G. [86] O’Donoghue [85] Goodman S.G. [87] |
| PCSK9 INH | Alirocumab Evolocumab | ApoB | Defective ApoB: Impaired LDL binding despite the presence of LDLRs → reduced response. Normal ApoB: Functional binding → good response due to effective LDL clearance. | Beneficial for patients with statin intolerance. Can be used as an adjunct to lifestyle modifications and dietary therapy for high-risk patients. | Sabatine M.S. [84] Schwartz G.G. [86] O’Donoghue [85] Goodman S.G. [87] |
| PCSK9 INH | Alirocumab Evolocumab Inclisiran | PCSK9 | GoF: ↑ PCSK9 activity → enhanced response to inhibitors as they block excess PCSK9. LoF: ↓ PCSK9 activity → limited additional benefit since endogenous PCSK9 is already low Lower LDL-C levels. Protective effect against ASCVD. | Inclisiran offers the advantage of twice-yearly dosing, improving adherence in long-term lipid management Effective in patients with PCSK9 gain-of-function variants, where endogenous PCSK9 levels are elevated. | Ray K.K. [89] Ray K.K. [90] Wright R.S. [91] |
| Antiplatelets | Clopidogrel | CYP2C19 | Normal alleles (e.g., *1/*1): Normal CYP2C19 activity. Adequate clopidogrel activation. Normal/Rapid Metabolizers (NM/RM). LoF: e.g., *2, *3: ↓ CYP2C19 enzymatic activity. ↓ conversion of clopidogrel to the active metabolite. ↓ antiplatelet effect. ↑ risk of thrombotic events. Intermediate/Poor Metabolizers (IM/PM). GoF: e.g., *17: ↑ CYP2C19 activity. ↑ clopidogrel activation. Potentially increased bleeding risk Ultra-rapid Metabolizers (UM). | Pharmacogenetic Testing: Recommended assays include CYP2C19 genotyping or platelet function tests (e.g., VerifyNow). Therapeutic Recommendations: IM/PM: Consider alternative antiplatelet agents (e.g., Ticagrelor, Prasugrel) due to reduced efficacy of clopidogrel. NM/RM: Standard clopidogrel dosing is appropriate. UM: Standard dosing is generally acceptable; monitor for bleeding risk. | Angiolillo D.J. [115] Dean L. [116] Shuldiner, A.R. [117] Scott S.A. [118] Frére C. [119] Lee C.R. [120] Mega J.L. [121] Mega J.L. [122] Siller-Matula J.M. [123] |
| Antihypertensives | B–blockers | ADRB1 | Affects receptor sensitivity The Arg389Arg genotype often shows enhanced response to beta-blockers compared to Gly389 carriers. | Genotyping may help predict BP response and risk of side effects. | Chen L. [124] |
| Antihypertensives | B–blockers | CYP2D6 | CYP2D6 polymorphisms alter drug metabolism. PM of CYP2D6 substrates may experience elevated drug levels and increased risk of side effects. | Use lower doses or alternative β-blockers in CYP2D6 poor metabolizers to reduce risk of adverse effects. Monitor closely. | Liu J. [125] Petrović J. [126] |
| Challenge | Description | Potential Solutions |
|---|---|---|
| Cost and Accessibility | Genetic testing and advanced therapies remain expensive and are not widely available | Expansion of insurance coverage, government-funded research initiatives |
| Limited Clinical Guidelines | Lack of standardized protocols for integrating genomics into cardiovascular care | Development of consensus guidelines by major cardiology societies |
| Ethical and Privacy Concerns | Genetic data privacy and potential discrimination in insurance/employment | Implementation of strong regulatory frameworks (e.g., GDPR, HIPAA) |
| Physician Awareness and Training | Many clinicians lack formal training in genetic risk assessment | Increased medical education and integration into cardiology fellowships |
| Data Interpretation and Integration | Challenges in translating genetic risk scores into actionable clinical decisions | AI-driven decision support tools and multi-omics approaches |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitsis, A.; Khattab, E.; Kyriakou, M.; Sokratous, S.; Sakellaropoulos, S.G.; Tzikas, S.; Kadogou, N.P.E.; Kassimis, G. Genomic and Precision Medicine Approaches in Atherosclerotic Cardiovascular Disease: From Risk Prediction to Therapy—A Review. Biomedicines 2025, 13, 1723. https://doi.org/10.3390/biomedicines13071723
Mitsis A, Khattab E, Kyriakou M, Sokratous S, Sakellaropoulos SG, Tzikas S, Kadogou NPE, Kassimis G. Genomic and Precision Medicine Approaches in Atherosclerotic Cardiovascular Disease: From Risk Prediction to Therapy—A Review. Biomedicines. 2025; 13(7):1723. https://doi.org/10.3390/biomedicines13071723
Chicago/Turabian StyleMitsis, Andreas, Elina Khattab, Michaella Kyriakou, Stefanos Sokratous, Stefanos G. Sakellaropoulos, Stergios Tzikas, Nikolaos P. E. Kadogou, and George Kassimis. 2025. "Genomic and Precision Medicine Approaches in Atherosclerotic Cardiovascular Disease: From Risk Prediction to Therapy—A Review" Biomedicines 13, no. 7: 1723. https://doi.org/10.3390/biomedicines13071723
APA StyleMitsis, A., Khattab, E., Kyriakou, M., Sokratous, S., Sakellaropoulos, S. G., Tzikas, S., Kadogou, N. P. E., & Kassimis, G. (2025). Genomic and Precision Medicine Approaches in Atherosclerotic Cardiovascular Disease: From Risk Prediction to Therapy—A Review. Biomedicines, 13(7), 1723. https://doi.org/10.3390/biomedicines13071723