

From Jumping Gene to Cancer: Revisiting the Role of JTB Protein
 ,
,  and
and
Abstract
1. Introduction
2. Jumping Translocations (JTs) in Cancer: Mechanisms and Clinical Implications
3. From Gene to Protein: Multifaceted Roles of JTB and Implications in Cancer
3.1. Discovery and Genomic Context of JTB
3.2. Structural Features and JTB Molecular Interactions
3.3. Functional Implications in Normal and Malignant Cells
3.4. JTB and Mitotic Regulation
3.5. JTB Contributes to Neoplastic Transformation by Disrupting Mitochondrial Function
3.6. JTB’s Role in Hematologic Malignancies
3.7. JTB in Prostate Cancer: Androgen Regulation and Therapeutic Implications
3.8. Proteomics-Based Characterization of JTB in Breast Cancer (BC)
3.9. JTB Expression Imbalance Promotes Malignant Phenotypes
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABL | Abelson murine leukemia viral oncogene homolog |
| AML | acute myeloid leukemia |
| AURKA | Aurora kinase A |
| AURKB | Aurora kinase B |
| BC | breast cancer |
| BOR | borealin |
| CPC | chromosomal passenger complex |
| CRC | colorectal cancer |
| EDC | epidermal differentiation complex |
| EMT | epithelial–mesenchymal transition |
| FISH | fluorescence in situ hybridization |
| GSEA | Gene Set Enrichment Analysis |
| HCC | hepatocellular carcinoma |
| HER2/neu | human epidermal growth factor receptor 2 |
| INCENP | inner centromere protein |
| JTB | jumping translocation breakpoint gene/protein |
| JT | jumping translocation |
| MDS | myelodysplastic syndrome |
| MM | multiple myeloma |
| MS | mass spectrometry |
| PAR | prostate androgen-regulated gene/protein |
| PCa | prostate cancer |
| PPI | protein–protein interaction |
| PTM | posttranslational modification |
| SJT | sequential jumping translocation |
References
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.; Bernard, O.A. Jumping translocations. Genes Chromosomes Cancer 2007, 46, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.R.; Stephens, D.; Ruppert, A.S.; Racke, F.; McFaddin, A.; Breidenbach, H.; Lin, H.-J.; Waller, K.; Bannerman, T.; Jones, J.A.; et al. Jumping translocations, a novel finding in chronic lymphocytic leukaemia. Br. J. Haematol. 2015, 170, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Sanford, D.; DiNardo, C.D.; Tang, G.; Cortes, J.E.; Verstovsek, S.; Jabbour, E.; Ravandi, F.; Kantarjian, H.; Garcia-Manero, G. Jumping Translocations in Myeloid Malignancies Associated with Treatment Resistance and Poor Survival. Clin. Lymphoma Myeloma Leuk. 2015, 15, 556–562. [Google Scholar] [CrossRef]
- Halper-Stromberg, E.; Stinnett, V.; Morsberger, L.; Pallavajjala, A.; Levis, M.J.; DeZern, A.E.; Lei, M.; Phan, B.; Xian, R.R.; Gocke, C.D.; et al. 1q jumping translocation as a biomarker in myeloid malignancy: Frequently mutated genes associated with bad prognosis and low survival. Exp. Hematol. Oncol. 2024, 13, 73. [Google Scholar] [CrossRef]
- Lee, I.; Gudipati, M.A.; Waters, E.; Duong, V.H.; Baer, M.R.; Zou, Y. Jumping translocations of chromosome 1q occurring by a multi-stage process in an acute myeloid leukemia progressed from myelodysplastic syndrome with a TET2 mutation. Mol. Cytogenet. 2019, 12, 47. [Google Scholar] [CrossRef]
- Kanome, T.; Itoh, N.; Ishikawa, F.; Mori, K.; Kim-Kaneyama, J.R.; Nose, K.; Shibanuma, M. Characterization of Jumping translocation breakpoint (JTB) gene product isolated as a TGF-β1-inducible clone involved in regulation of mitochondrial function, cell growth and cell death. Oncogene 2007, 26, 5991–6001. [Google Scholar] [CrossRef]
- Lema Fernandez, A.G.; Nardelli, C.; Pierini, V.; Crescenzi, B.; Pellanera, F.; Matteucci, C.; Crocioni, M.; Arniani, S.; Di Battista, V.; Quintini, M.; et al. Epigenetic Modeling of Jumping Translocations of 1q Heterochromatin in Acute Myeloid Leukemia After 5′-Azacytidine Treatment. Genes Chromosomes Cancer 2024, 63, e70013. [Google Scholar] [CrossRef]
- Platica, M.; Ivan, E.; Ionescu, A.; Holland, J.F.; Mora, G.; Tindall, D.J.; Mandeli, J.; Unger, P.D.; Platica, O. Transformation of NIH 3T3 cells by enhanced PAR expression. Biochem. Biophys. Res. Commun. 2004, 314, 891–896. [Google Scholar] [CrossRef]
- Platica, M.; Ionescu, A.; Ivan, E.; Holland, J.; Mandeli, J.; Platica, O. PAR, a protein involved in the cell cycle, is functionally related to chromosomal passenger proteins. Int. J. Oncol. 2011, 38, 777–785. [Google Scholar] [CrossRef]
- Rousseau, F.; Pan, B.; Fairbrother, W.J.; Bazan, J.F.; Lingel, A. The Structure of the Extracellular Domain of the Jumping Translocation Breakpoint Protein Reveals a Variation of the Midkine Fold. J. Mol. Biol. 2012, 415, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Jayathirtha, M.; Jayaweera, T.; Whitham, D.; Petre, B.A.; Neagu, A.-N.; Darie, C.C. Two-Dimensional Polyacrylamide Gel Electrophoresis Coupled with Nanoliquid Chromatography–Tandem Mass Spectrometry-Based Identification of Differentially Expressed Proteins and Tumorigenic Pathways in the MCF7 Breast Cancer Cell Line Transfected for Jumping Translocation Breakpoint Protein Overexpression. Int. J. Mol. Sci. 2023, 24, 14714. [Google Scholar] [PubMed]
- Jayathirtha, M.; Neagu, A.-N.; Whitham, D.; Alwine, S.; Darie, C. Investigation of the effects of overexpression of jumping translocation breakpoint (JTB) protein in MCF7 cells for potential use as a biomarker in breast cancer. Am. J. Cancer Res. 2022, 12, 1784–1823. [Google Scholar] [PubMed]
- Jayathirtha, M.; Neagu, A.-N.; Whitham, D.; Alwine, S.; Darie, C.C. Investigation of the effects of downregulation of jumping translocation breakpoint (JTB) protein expression in MCF7 cells for potential use as a biomarker in breast cancer. Am. J. Cancer Res. 2022, 12, 4373–4398. [Google Scholar]
- Jayathirtha, M.; Whitham, D.; Alwine, S.; Donnelly, M.; Neagu, A.-N.; Darie, C.C. Investigating the Function of Human Jumping Translocation Breakpoint Protein (hJTB) and Its Interacting Partners through In-Solution Proteomics of MCF7 Cells. Molecules 2022, 27, 8301. [Google Scholar] [CrossRef]
- Jayathirtha, M.; Jayaweera, T.; Whitham, D.; Sullivan, I.; Petre, B.A.; Darie, C.C.; Neagu, A.-N. Two-Dimensional-PAGE Coupled with nLC-MS/MS-Based Identification of Differentially Expressed Proteins and Tumorigenic Pathways in MCF7 Breast Cancer Cells Transfected for JTB Protein Silencing. Molecules 2023, 28, 7501. [Google Scholar] [CrossRef]
- Jayathirtha, M.; Channaveerappa, D.; Darie, C. Investigation and Characterization of the Jumping Translocation Breakpoint (JTB) Protein using Mass Spectrometry based Proteomics. FASEB J. 2021, 35. [Google Scholar] [CrossRef]
- Padilla-Nash, H.M.; Heselmeyer-Haddad, K.; Wangsa, D.; Zhang, H.; Ghadimi, B.M.; Macville, M.; Augustus, M.; Schröck, E.; Hilgenfeld, E.; Ried, T. Jumping translocations are common in solid tumor cell lines and result in recurrent fusions of whole chromosome arms. Genes Chromosomes Cancer 2001, 30, 349–363. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, B.; Park, J.; Choi, E.; Oh, A.; Lee, S.; Ryu, H.; Kang, I.; Yang, K.; Park, S. Rarely Observed Jumping Translocation in Spontaneous Abortion. J. Genet. Med. 2010, 7, 82. [Google Scholar] [CrossRef]
- Couture, T.; Amato, K.; Diadamo, A.; Li, P. Jumping Translocations of 1q in Myelodysplastic Syndrome and Acute Myeloid Leukemia: Report of Three Cases and Review of Literature. Case Rep. Genet. 2018, 2018, 1–5. [Google Scholar] [CrossRef]
- Hatakeyama, S.; Osawa, M.; Omine, M.; Ishikawa, F. JTB: A novel membrane protein gene at 1q21 rearranged in a jumping translocation. Oncogene 1999, 18, 2085–2090. [Google Scholar] [CrossRef]
- Ma, N.-F.; Hu, L.; Fung, J.M.; Xie, D.; Zheng, B.-J.; Chen, L.; Tang, D.-J.; Fu, L.; Wu, Z.; Chen, M.; et al. Isolation and characterization of a novel oncogene, amplified in liver cancer 1, within a commonly amplified region at 1q21 in hepatocellular carcinoma. Hepatology 2008, 47, 503–510. [Google Scholar] [CrossRef]
- Le Baccon, P.; Leroux, D.; Dascalescu, C.; Duley, S.; Marais, D.; Esmenjaud, E.; Sotto, J.; Callanan, M. Novel evidence of a role for chromosome 1 pericentric heterochromatin in the pathogenesis of B-cell lymphoma and multiple myeloma. Genes Chromosomes Cancer 2001, 32, 250–264. [Google Scholar] [CrossRef]
- Liu, N.; Xie, Z.; Li, H.; Wang, L. The numerous facets of 1q21+ in multiple myeloma: Pathogenesis, clinicopathological features, prognosis and clinical progress (Review). Oncol. Lett. 2024, 27, 258. [Google Scholar] [CrossRef]
- Sawyer, J.R.; Tricot, G.; Mattox, S.; Jagannath, S.; Barlogie, B. Jumping Translocations of Chromosome 1q in Multiple Myeloma: Evidence for a Mechanism Involving Decondensation of Pericentromeric Heterochromatin. Blood 1998, 91, 1732–1741. [Google Scholar] [CrossRef]
- Platica, O.; Chen, S.; Ivan, E.; Lopingco, M.; Holland, J.; Platica, M. PAR, a novel androgen regulated gene, ubiquitously expressed in normal and malignant cells. Int. J. Oncol. 2000, 16, 1055–1061. [Google Scholar] [CrossRef]
- Carmena, M.; Wheelock, M.; Funabiki, H.; Earnshaw, W.C. The chromosomal passenger complex (CPC): From easy rider to the godfather of mitosis. Nat. Rev. Mol. Cell Biol. 2012, 13, 789–803. [Google Scholar] [CrossRef]
- GSEA. 2025. Available online: https://www.gsea-msigdb.org/gsea/msigdb/human/geneset/chr1q21.html (accessed on 23 April 2025).
- Bisht, K.; Walker, B.; Kumar, S.K.; Spicka, I.; Moreau, P.; Martin, T.; Costa, L.J.; Richter, J.; Taro, F.; Sandrine, M.; et al. Chromosomal 1q21 abnormalities in multiple myeloma: A review of translational, clinical research, and therapeutic strategies. Expert Rev. Hematol. 2021, 14, 1099–1114. [Google Scholar] [CrossRef]
- Nagai, S.; Nannya, Y.; Takahashi, T.; Kurokawa, M. Jumping translocation involving 1q21 during long-term complete remission of acute myeloid leukemia. Ann. Hematol. 2010, 89, 741–742. [Google Scholar] [CrossRef]
- Tyszkiewicz, T.; Jarząb, M.; Szymczyk, C.; Kowal, M.; Krajewska, J.; Jaworska, M.; Fraczek, M.; Krajewska, A.; Hadas, E.; Swierniak, M.; et al. Epidermal differentiation complex (locus 1q21) gene expression in head and neck cancer and normal mucosa. Folia Histochem. et Cytobiol. 2014, 52, 79–89. [Google Scholar] [CrossRef]
- Abhishek, S.; Palamadai Krishnan, S. Epidermal Differentiation Complex: A Review on Its Epigenetic Regulation and Potential Drug Targets. Cell J. 2016, 18, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Henry, J. Update on the epidermal differentiation complex. Front. Biosci. 2012, 17, 1517. [Google Scholar] [CrossRef] [PubMed]
- Qin, D.; Ma, L.; Qin, L. Potential Role of the Epidermal Differentiation Complex in the Pathogenesis of Psoriasis. FBL 2022, 27, 325. [Google Scholar] [CrossRef] [PubMed]
- Holthaus, K.B.; Eckhart, L. Development-Associated Genes of the Epidermal Differentiation Complex (EDC). J. Dev. Biol. 2024, 12, 4. [Google Scholar] [CrossRef]
- Sun, M.; Veschi, V.; Bagchi, S.; Xu, M.; Mendoza, A.; Liu, Z.; Thiele, C.J. Targeting the Chromosomal Passenger Complex Subunit INCENP Induces Polyploidization, Apoptosis, and Senescence in Neuroblastoma. Cancer Res. 2019, 79, 4937–4950. [Google Scholar] [CrossRef]
- D’Avino, P.P.; Capalbo, L. New Auroras on the Roles of the Chromosomal Passenger Complex in Cytokinesis: Implications for Cancer Therapies. Front. Oncol. 2015, 5, 221. [Google Scholar] [CrossRef]
- Hindriksen, S.; Meppelink, A.; Lens, S.M.A. Functionality of the chromosomal passenger complex in cancer. Biochem. Soc. Trans. 2015, 43, 23–32. [Google Scholar] [CrossRef]
- Alphafold. GSEA. 2025. Available online: https://www.uniprot.org/uniprotkb/O76095/entry#sequences (accessed on 23 April 2025).
- Platica, M.; Ivan, E.; Chen, S.; Holland, J.F.; Gil, J.; Mandeli, J.; Platica, O. Stable lower PAR expression decreased DU145 prostate cancer cell growth in SCID mice. Prostate 2001, 49, 200–207. [Google Scholar] [CrossRef]
- Lin, X.; Xiang, X.; Hao, L.; Wang, T.; Lai, Y.; Abudoureyimu, M.; Zhou, H.; Feng-Bing, M.; Chu, X.; Wang, R. The role of Aurora-A in human cancers and future therapeutics. Am. J. Cancer Res. 2020, 10, 2705–2729. [Google Scholar]
- Baba, A.B.; Rah, B.; Bhat, G.R.; Mushtaq, I.; Parveen, S.; Hassan, R.; Hameed Zargar, M.; Afroze, D. Transforming Growth Factor-Beta (TGF-β) Signaling in Cancer-A Betrayal Within. Front. Pharmacol. 2022, 13, 1272. [Google Scholar] [CrossRef]
- Belnekar, M.; Virulkar, S.; Tulpule, S.; Kar, B. Jumping translocation of 3q21 in a patient with acute myeloid leukemia and poor clinical outcome. J. Cancer Res. Ther. 2024, 20, 1643–1646. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Tasaka, T.; Shimizu, R.; Hayashi, K.; Yamada, S.; Fukuda, H.; Hirose, T.; Takeuchi, A.; Sano, F.; Tokunaga, H.; et al. Jumping translocations of 1q in donor cell-derived myelodysplastic syndrome after cord blood transplantation: Case report and review of the literature. Mol. Clin. Oncol. 2020, 12, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-F.; Zhou, S.-W.; Zhang, X.; Ye, Z.-Q.; Zhang, J.-H.; Ma, X.; Zheng, T.; Li, H.-Z. Prostate androgen-regulated gene: A novel potential target for androgen-independent prostate cancer therapy. Asian J. Androl. 2006, 8, 455–462. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Authors | Year | Relevance | References |
|---|---|---|---|
| Hatakeyama et al. | 1999 | JTB gene was identified at 1q21 locus | [21] |
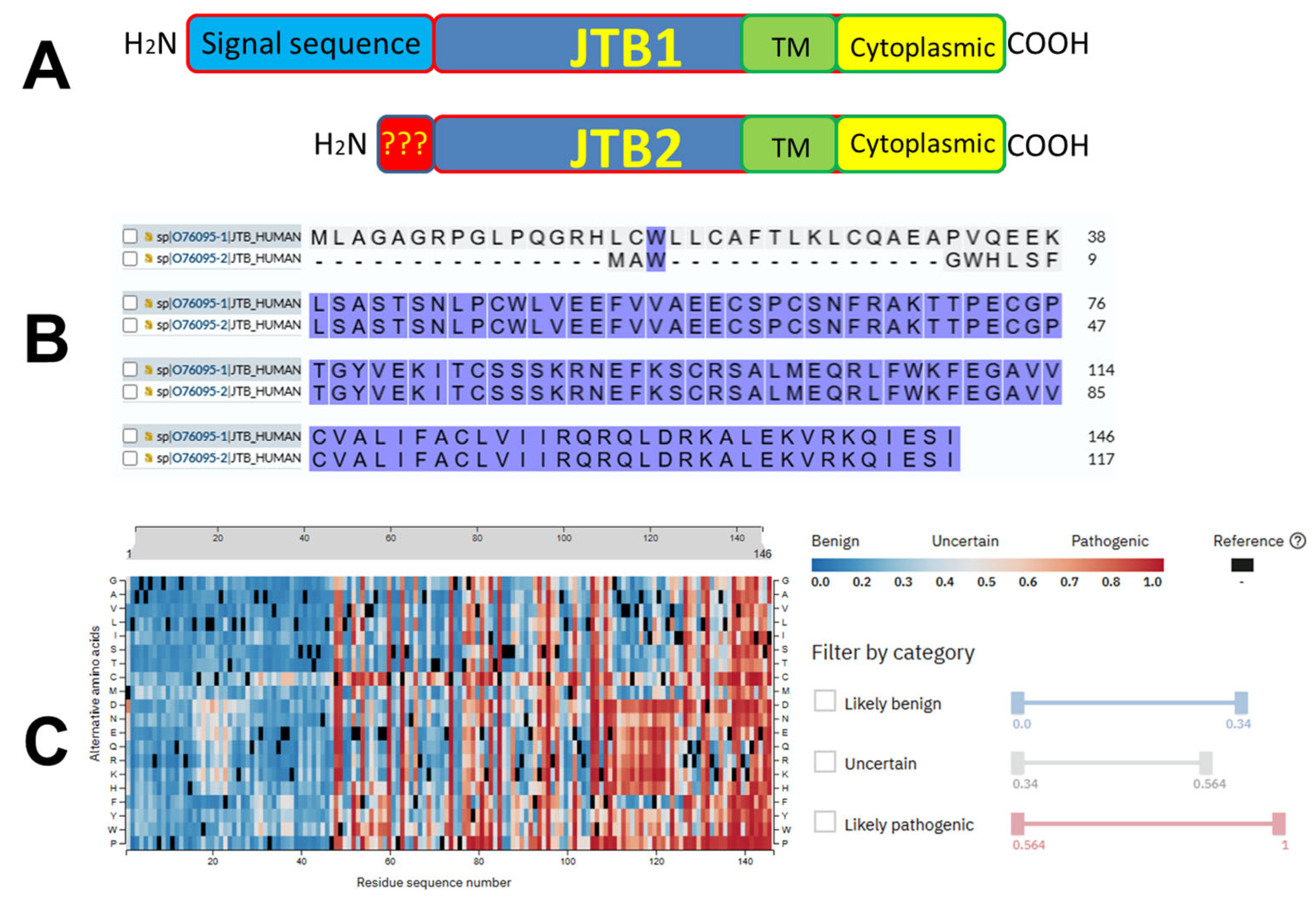
| JTB is a transmembrane protein of 16.4 kDa evolutionary conserved across diverse eukaryotic species | |||
| The N-terminal hydrophobic region likely serves as a signal sequence for polypeptide secretion or membrane compartment recruitment, as it is processed and removed during this process | |||
| The C-terminal region is predicted to form the transmembrane domain, supporting the classification of JTB as a transmembrane protein | |||
| JT results in JTB truncation and a shortened protein variant that lacks the transmembrane and intracellular domains and is possibly secreted from cells | |||
| JTB is located in EDC | |||
| Platica et al. | 2000 | PAR gene was isolated from LNCaP-OM androgen-resistant subline of PCa | [26] |
| The complete sequence of the gene cDNA has 1029 nucleotides, with a continuous reading frame of 438 bases encoding for 146 amino acids | |||
| Amino acid sequence has motifs for myristoylation and phosphorylation by PKC | |||
| PAR gene was overexpressed in all PCa cell lines studied (LNCaP, DU145, PC3, and LNCaP-OM) compared to the normal prostatic tissue | |||
| PAR expression was higher in androgen-resistant prostate cancer lines (DU145, PC3, and LNCaP-OM) in comparison to androgen-sensitive cells (LNCaP) | |||
| PAR expression was downregulated by androgens in androgen-sensitive prostate cells, but not in the hormone-resistant cell lines | |||
| PAR gene is ubiquitously expressed in 29 normal studied tissues and overexpressed in most (67%) of their malignant counterparts | |||
| PAR expression was higher in the MCF7 and T47D BC cell lines, as well as in all primary breast tumors studied, compared to their normal tissue counterparts | |||
| PAR biological function is still unknown | |||
| Putative PAR involvement in basic cellular processes and malignant transformation | |||
| Platica et al. | 2001 | PAR tends to be overexpressed in tumor cells | [40] |
| PAR biological function is still unknown | |||
| Putative PAR implication in malignant transformation | |||
| Transfection of DU145 PCa cells with antisense PAR cDNA for PAR silencing led to decreased cell proliferation (arrest in G2/M phase) in tissue culture, low efficiency of colony formation in soft agar, and decreased tumor growth in SCID mice | |||
| Platica et al. | 2004 | PAR is a 1038 bp gene located in chromosome 1 within EDC | [9] |
| PAR is overexpressed in malignant tissues (proto-oncogene) | |||
| Transfection of NIH3T3 fibroblasts with PAR cDNA led to enhanced growth in culture, colony formation in soft agar, accelerated cell growth (shortened G1 and S phases), tumor formation in SCID mice | |||
| Transfection of NIH3T3 with 22-mer oligonucleotide in antisense orientation with PAR mRNA suppressed tumorigenic behaviors and abrogated colony formation in soft agar | |||
| Xu et al. | 2006 | PAR expression was higher in PC3 PCa cells (more aggressive) than that in LNCaP cells | [45] |
| DHT modulated PAR mRNA expression in LNCaP cells, and this effect was blocked by the AR antagonists | |||
| DHT did not affect PAR expression in PC3 cells | |||
| Reintroduction of AR into PC3 cells via stable transfection restored the androgen effect on PAR upregulation | |||
| siRNA transfection for PAR silencing in PC3 cells led to a reversal of the malignant phenotype | |||
| It is possible that PAR is downstream from the AR | |||
| PAR contributes to malignant proliferation in androgen-independent PCa cells | |||
| PAR could be a potential therapeutic target for androgen-independent PCa with AR signaling pathway alteration | |||
| Kanome et al. | 2007 | JTB expression is suppressed in many cancers from different organs | [7] |
| JTB plays a role in the neoplastic transformation of cells | |||
| JTB was isolated as a TGF-β1-inducible clone via differential screening | |||
| JTB may be processed at the N-terminus and is located mostly in mitochondria | |||
| JTB-induced clustering of mitochondria around the nuclear periphery and swelling of each mitochondrion | |||
| Mitochondria membrane potential was significantly reduced | |||
| JTB retarded the growth of the cells and conferred resistance to TGF-β1-induced apoptosis | |||
| These activities were dependent on the N-terminal processing and induced by wild-type JTB, but not by a mutant resistant to cleavage | |||
| Alterations in the structure or expression of JTB can lead to neoplastic changes in cells by disrupting mitochondrial function, resulting in uncontrolled cell growth and/or cell death | |||
| Platica et al. | 2011 | PAR possesses oncogenic activity | [10] |
| PAR has a dynamic expression throughout the cell cycle (lowest at G1/S, peaks in G2/M) | |||
| PAR’s subcellular localization shifts dynamically throughout mitosis | |||
| PAR is functionally related to CPP (mainly AURKA) | |||
| PAR changes AURKB activity | |||
| PAR silencing leads to defects during mitosis | |||
| PAR is overexpressed in cancer (OC, BC, lung, uterus, and colon cancer) | |||
| PAR is overexpressed in MCF7, T47D (BC cell lines), DU145, and LNCaP (PCa cell lines) | |||
| PAR degradation can occur by the ubiquitin–proteasome pathway | |||
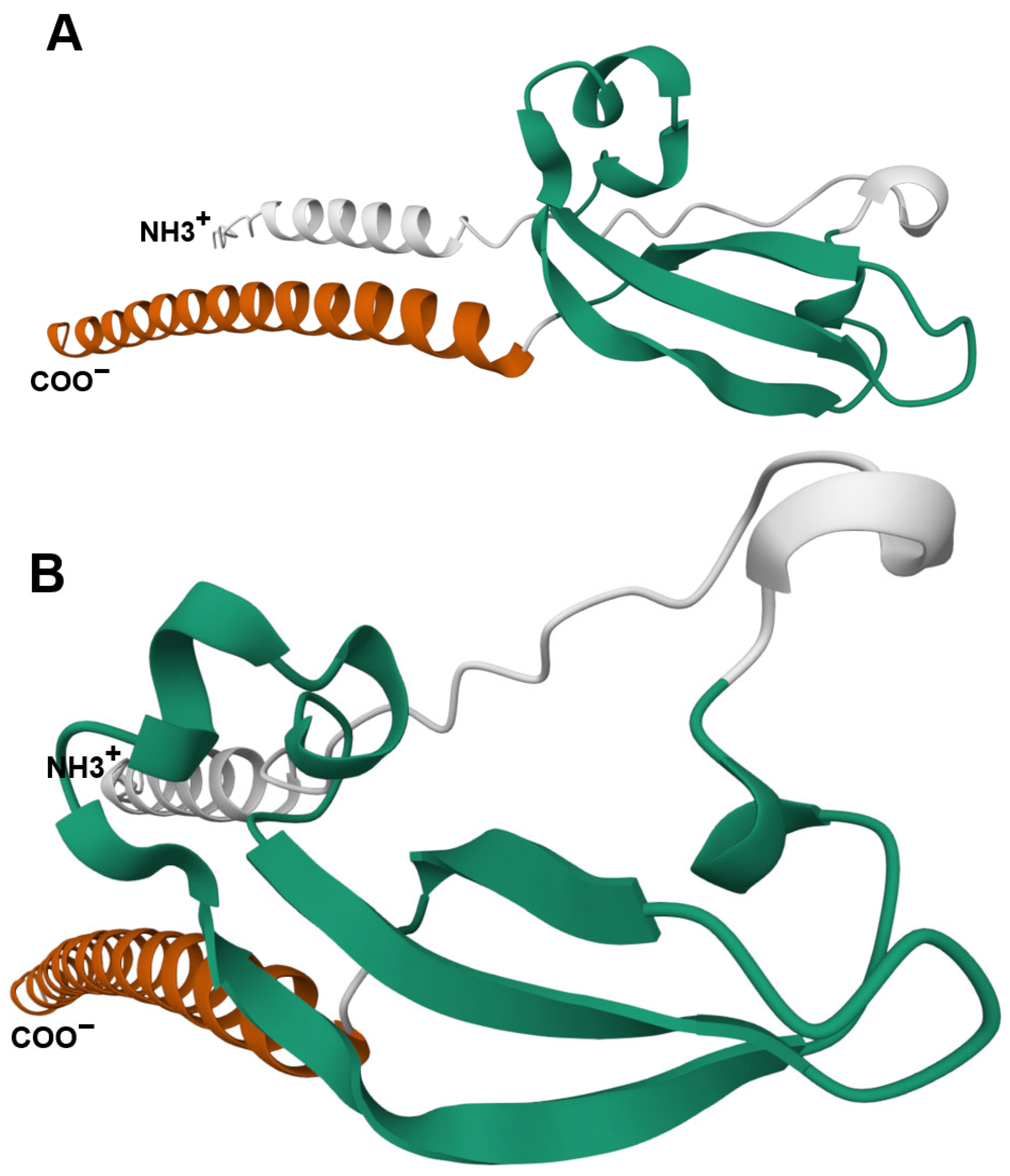
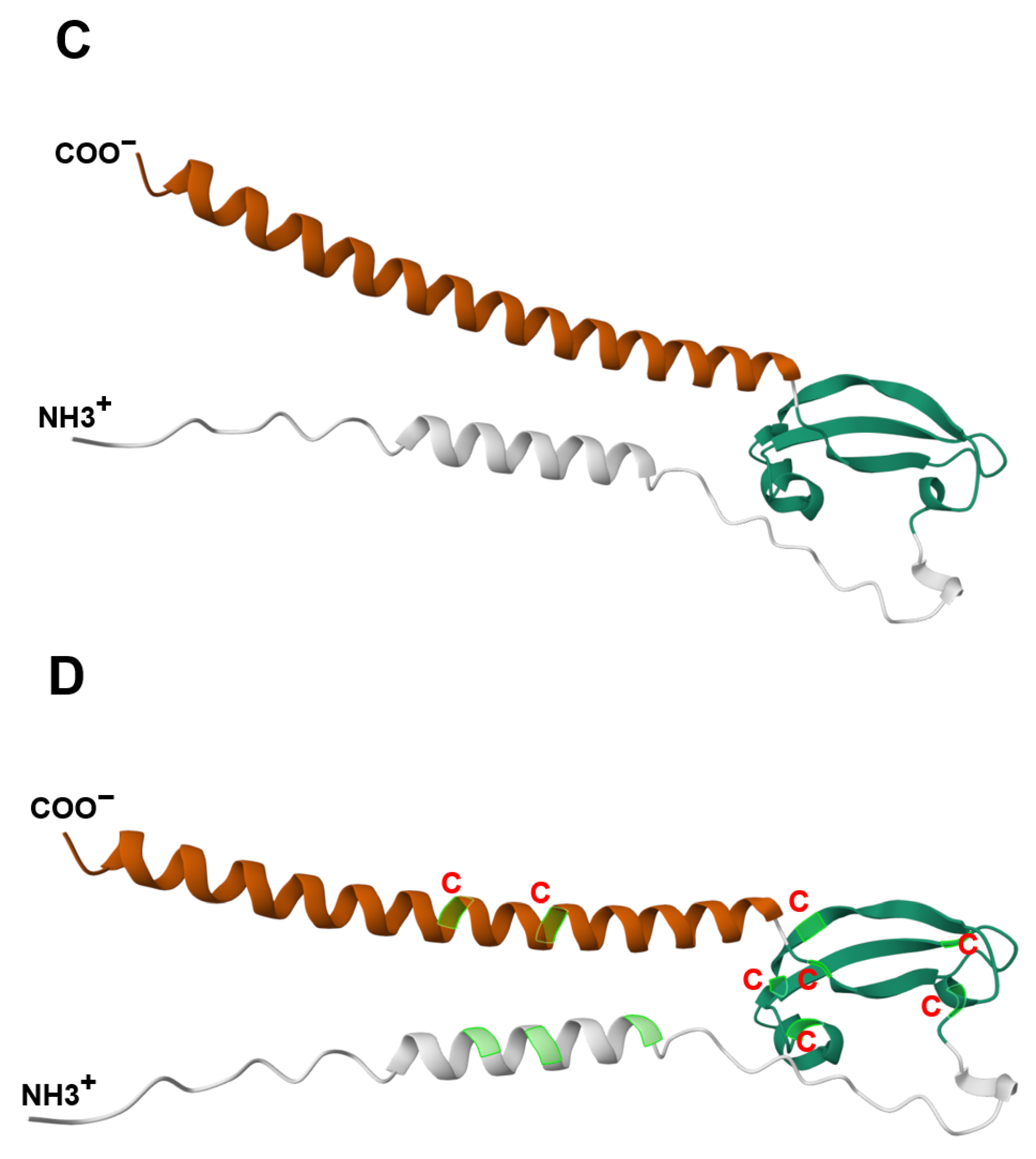
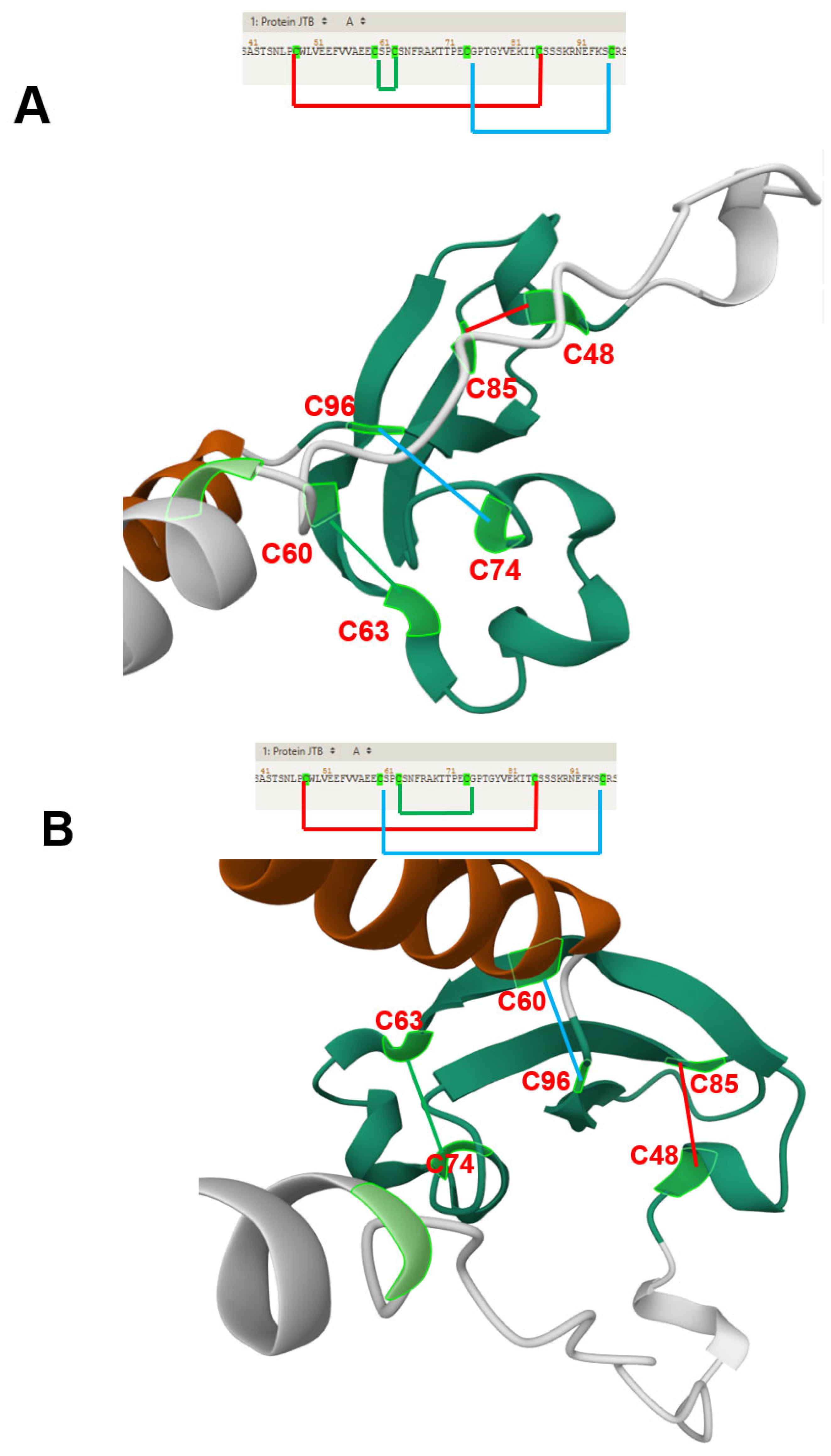
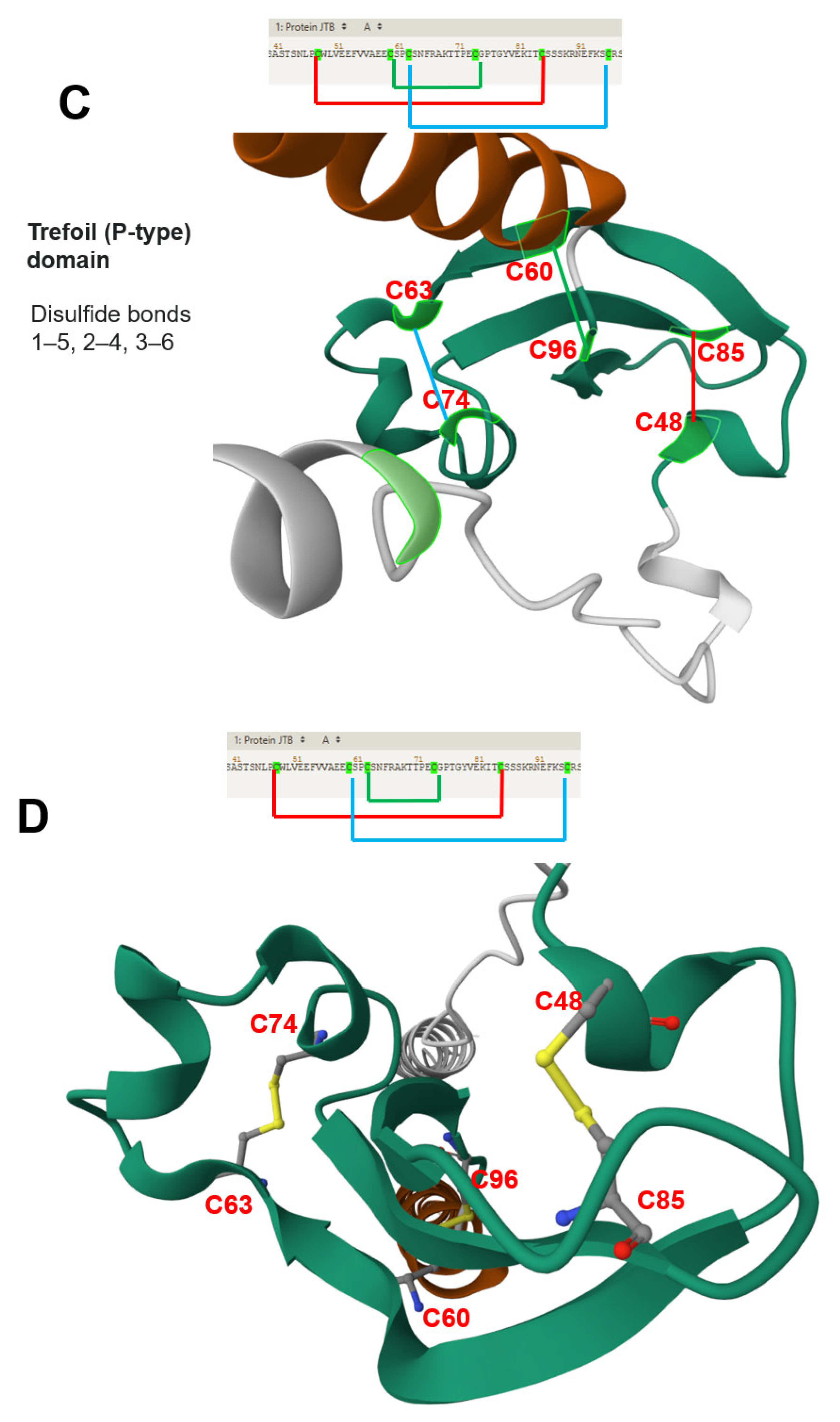
| Rousseau et al. | 2012 | JTB is an orphan receptor | [11] |
| NMR analysis reveals a novel three-stranded antiparallel β-meander in the N-terminal ectodomain of JTB | |||
| JTB shows distant structural relation to midkine/pleiotrophin, especially in conserved disulfide bonds | |||
| Extracellular domain of JTB may be secreted and interact with proteins or ECM, suggesting roles in yet-undefined biological processes | |||
| Jayathirtha et al. | 2021 | Study supports the hypothesis that JTB plays a role in tumorigenesis, particularly in BC, where it is frequently overexpressed | [17] |
| Proteomic analysis of MCF7 cells with both upregulated and downregulated JTB expression emphasized in dysregulated proteins potentially linked to cancer-related pathways | |||
| Jayathirtha et al. | 2022 | Cellular proteomics: MCF7 BC cells transfected with sense orientation of JTB cDNA for JTB overexpression; SDS-PAGE and nLC-MS/MS | [13] |
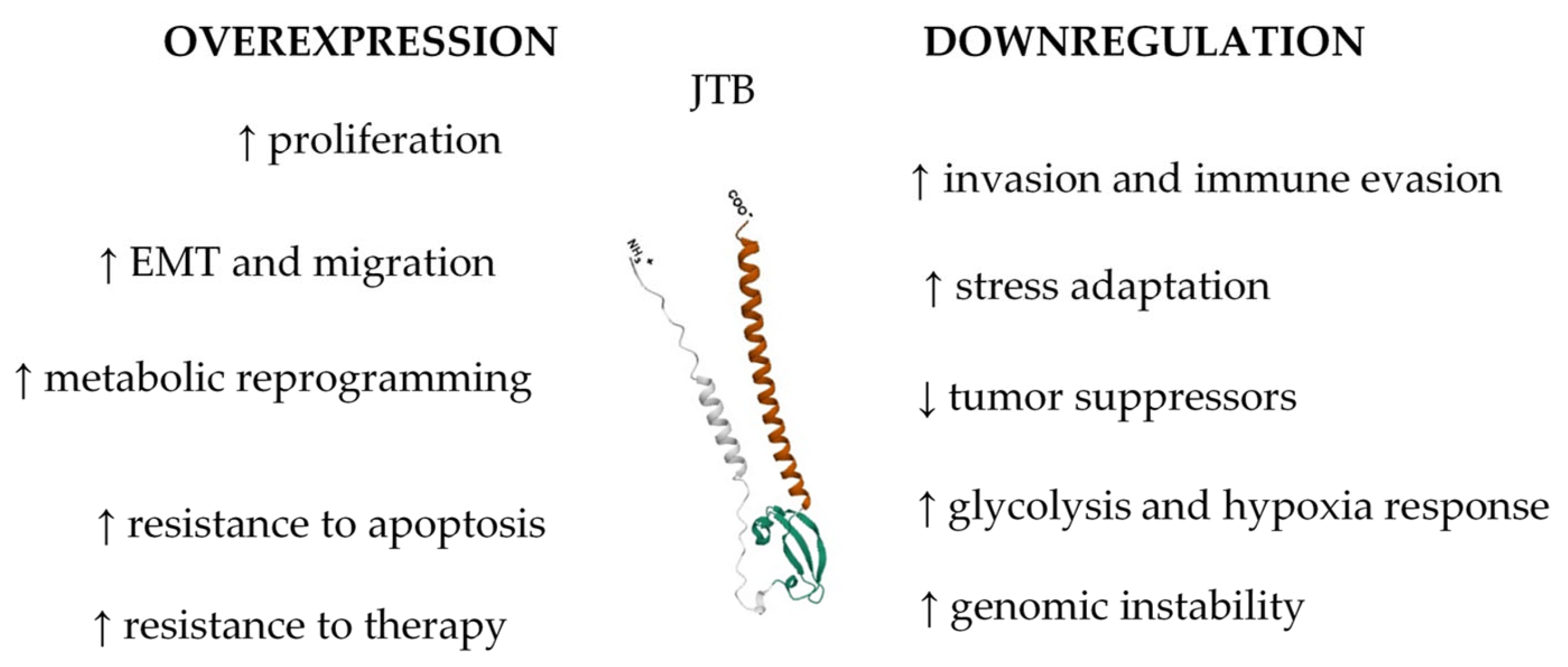
| Overexpression of JTB in MCF7 BC cells led to significant dysregulation in various cellular pathways (mitotic spindle assembly, estrogen response, and EMT) | |||
| Key proteins related to cell division, cytoskeletal organization, estrogen response, lipid biogenesis, migration, and metastasis were upregulated | |||
| Overexpressed JTB was associated with altered metabolic and stress response pathways, as well as resistance to cancer therapies | |||
| JTB contributes to tumorigenesis, regulating cell division, estrogen signaling, and cellular responses to environmental changes | |||
| Jayathirtha et al. | 2022 | Cellular proteomics: MCF7 BC cells transfected with shRNA plasmids for JTB silencing; SDS-PAGE and nLC-MS/MS | [14] |
| Upregulation of proteins that promote actin cytoskeleton reorganization, EMT, cell motility, invasion, metabolic reprogramming, and immune evasion | |||
| Key pathways affected include glycolysis, FA metabolism, cell cycle regulation, inflammatory signaling, response to OS, and hypoxia | |||
| JTB downregulation drives MCF7 cells toward a phenotype characterized by enhanced proliferation, migration, invasion, and resistance to hostile TME | |||
| Jayathirtha et al. | 2022 |
Cellular proteomics: MCF7 BC cells transfected with sense orientation of the JTB cDNA for JTB upregulation and shRNA plasmid targeting the JTB mRNA for silencing; in-solution digestion-based cellular proteomics, nLC-MS/MS | [15] |
| JTB dysregulation (both overexpression and downregulation) in the MCF7 BC cell line alters key biological processes (EMT, cytoskeleton organization, metabolic reprogramming, and cellular proteostasis) | |||
| JTB influences mitochondrial function, OS response, apoptosis, and interferon signaling pathways | |||
| JTB emerges as a potential biomarker and therapeutic target in BC, warranting further investigation into its molecular mechanisms and interactions | |||
| Jayathirtha et al. | 2023 | Cellular proteomics: MCF7 cells transfected for JTB upregulation; 2D-PAGE coupled with LC-MS/MS | [12] |
| JTB has a dual function as both a potential oncogene and a tumor suppressor, highlighting the context-dependent nature of JTB’s role | |||
| Data support the potential of JTB as a biomarker in BC and underscore the need for further mechanistic studies to elucidate its contribution to tumor initiation and progression | |||
| Jayathirtha et al. | 2023 | Cellular proteomics: MCF7 cells transfected with shRNA plasmids for JTB downregulation; 2D-PAGE coupled with LC-MS/MS | [16] |
| JTB interacting DEPs involved in key pro-tumorigenic pathways (EMT, ERK/MAPK, PI3K/AKT, Wnt/β-catenin, mTOR signaling) | |||
| DEPs are linked to enhanced proliferation, invasion, metabolic reprogramming, immune evasion, and maintenance of stemness, indicating that JTB silencing contributes to a more aggressive neoplastic phenotype | |||
| JTB protein may be a potential tumor suppressor in BC | |||
| Data emphasize the potential of JTB as both biomarker and therapeutic target in BC, warranting further functional and clinical investigation | |||
| Importance of a multi-platform proteomic approach for a comprehensive understanding of JTB-associated molecular mechanisms |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jayaweera, T.M.; Jayathirtha, M.; Weraduwage, K.; Kraus, P.; Darie, C.C.; Neagu, A.-N. From Jumping Gene to Cancer: Revisiting the Role of JTB Protein. Biomedicines 2025, 13, 1705. https://doi.org/10.3390/biomedicines13071705
Jayaweera TM, Jayathirtha M, Weraduwage K, Kraus P, Darie CC, Neagu A-N. From Jumping Gene to Cancer: Revisiting the Role of JTB Protein. Biomedicines. 2025; 13(7):1705. https://doi.org/10.3390/biomedicines13071705
Chicago/Turabian StyleJayaweera, Taniya M., Madhuri Jayathirtha, Krishan Weraduwage, Petra Kraus, Costel C. Darie, and Anca-Narcisa Neagu. 2025. "From Jumping Gene to Cancer: Revisiting the Role of JTB Protein" Biomedicines 13, no. 7: 1705. https://doi.org/10.3390/biomedicines13071705
APA StyleJayaweera, T. M., Jayathirtha, M., Weraduwage, K., Kraus, P., Darie, C. C., & Neagu, A.-N. (2025). From Jumping Gene to Cancer: Revisiting the Role of JTB Protein. Biomedicines, 13(7), 1705. https://doi.org/10.3390/biomedicines13071705