
Single-Cell Transcriptomic Analysis Unveils Key Regulators and Signaling Pathways in Lung Adenocarcinoma Progression



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. LUAD Datasets
2.2. scRNA-seq Data Preprocessing
2.3. Cell Clustering and Cell Lineage Analysis
2.4. Copy Number Variation from Single Cell RNAseq Data
2.5. LUAD Patient Subgroups
2.6. Gene Expression Regulatory Networks
2.7. Intercelluar Communication Networks
3. Results
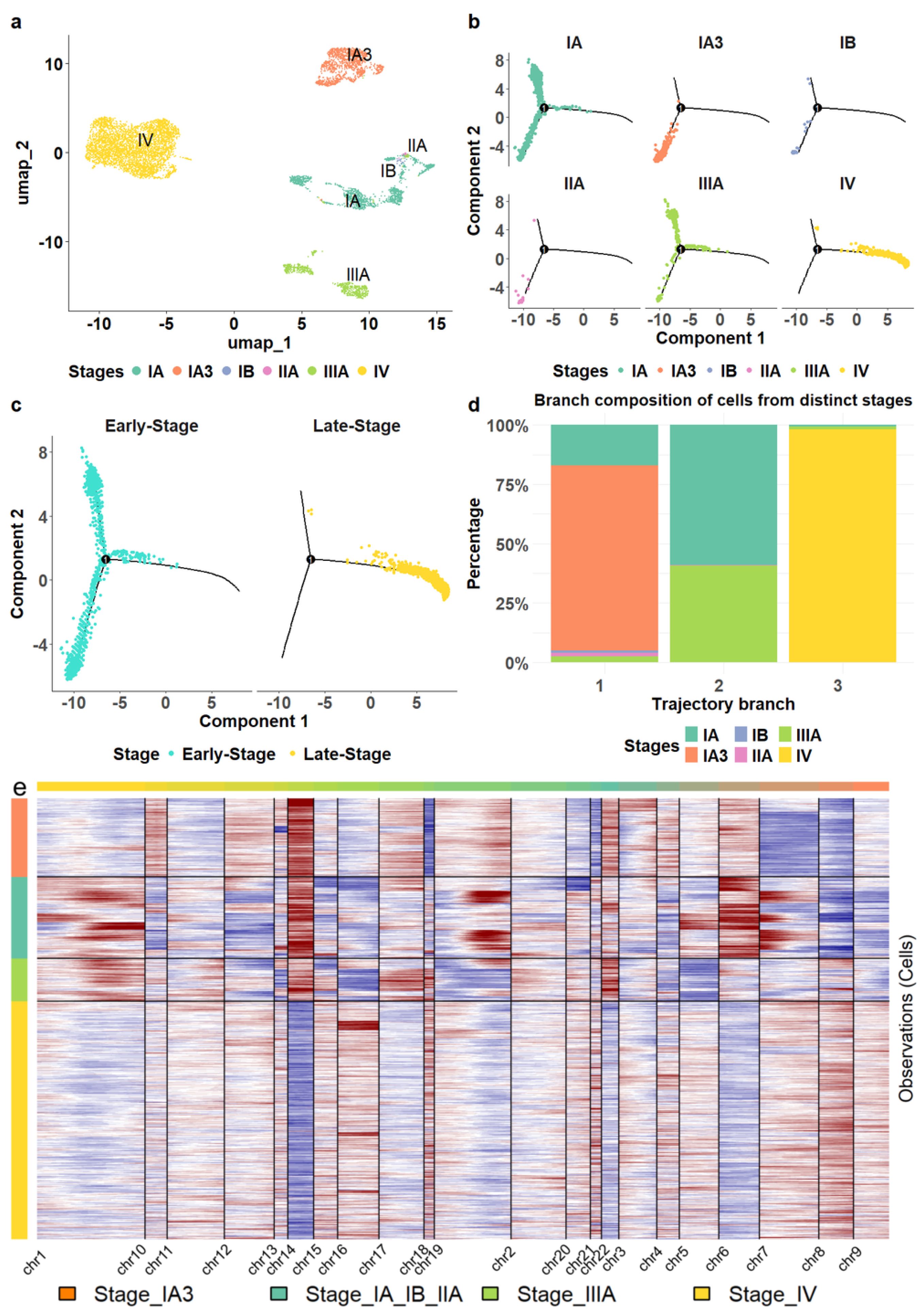
3.1. The Cellular Landscape of LUAD
3.2. Cellular Gene Markers Associated with LUAD Progression
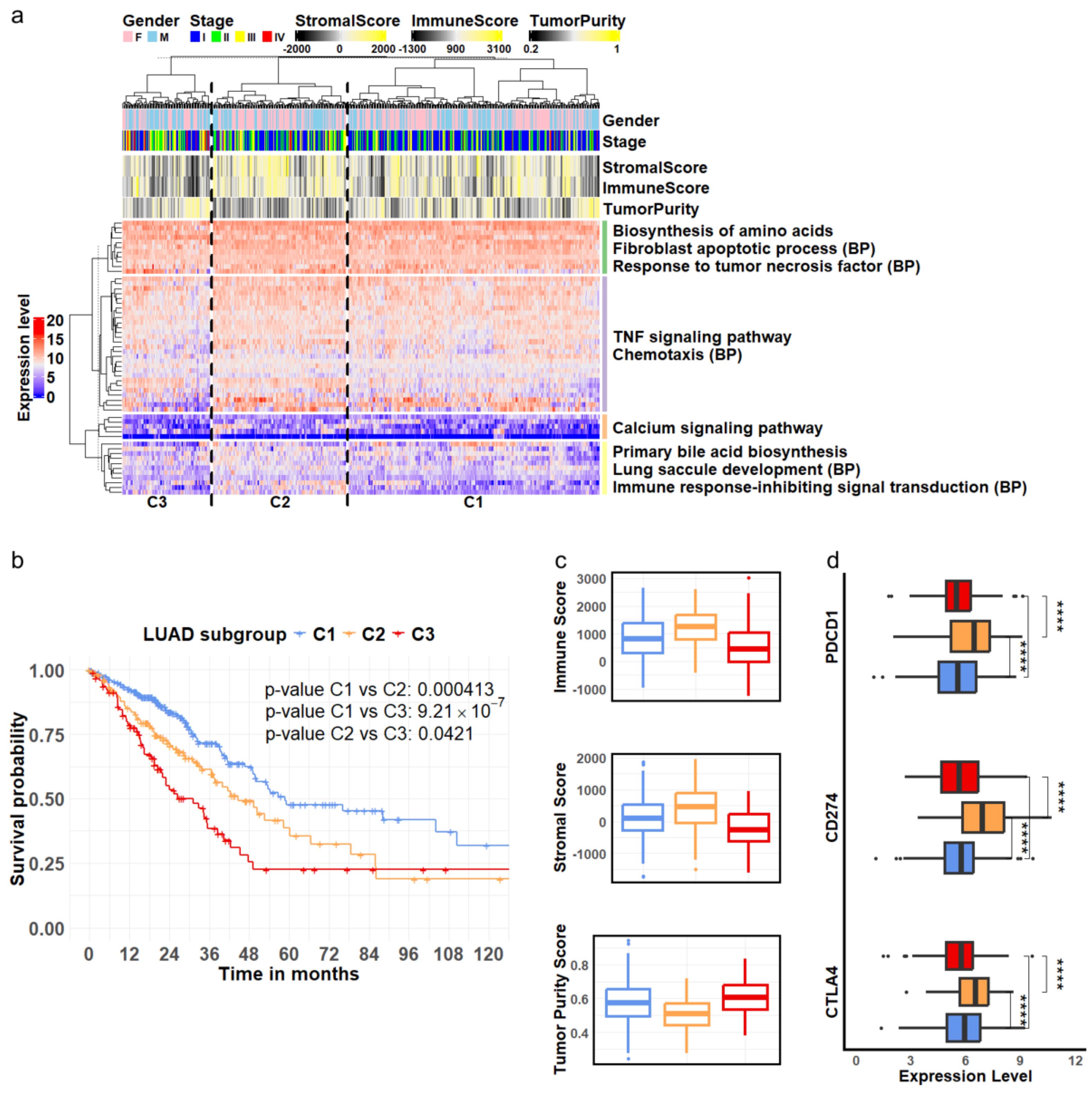
3.3. Clinical Implications of Cellular Marker Genes
3.4. Key Transcription Regulators of Progression-Related Gene Markers
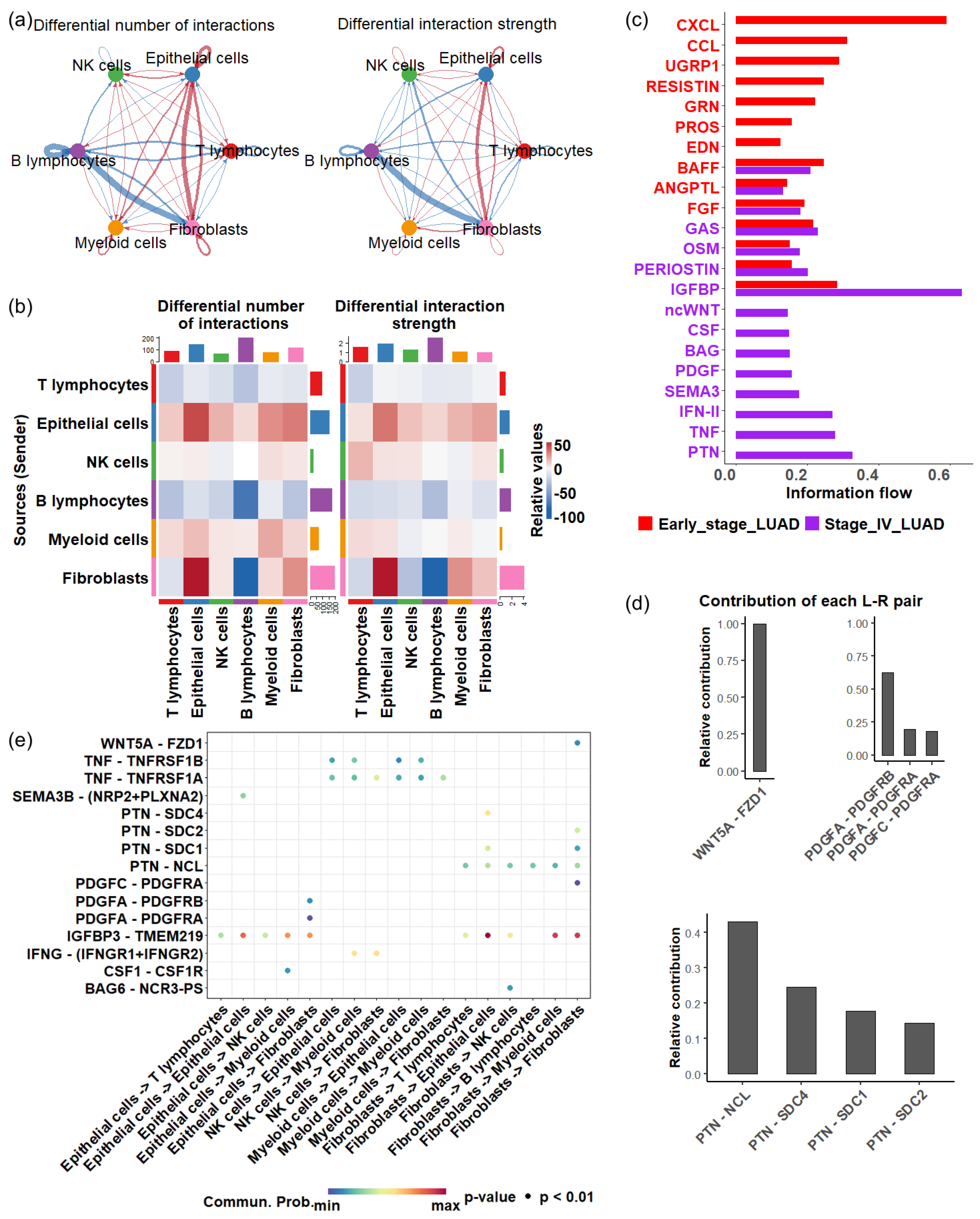
3.5. Cellular Communication Alterations in Response to Tumor Progression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Thandra, K.C.; Barsouk, A.; Saginala, K.; Aluru, J.S.; Barsouk, A. Epidemiology of lung cancer. Contemp. Oncol. Współczesna Onkol. 2021, 25, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zeng, T.; Zhang, H.; Li, Y.; Zhu, X.; Liu, H.; Sun, B.; Ji, C.; Li, T.; Huang, L.; et al. Nano-immunotherapy for lung cancer. Nano TransMed 2023, 2, e9130018. [Google Scholar] [CrossRef]
- Wu, Y.L.; Lu, S.; Zhou, Q.; Zhang, L.; Cheng, Y.; Wang, J.; Wang, B.; Hu, C.; Lin, L.; Zhong, W.; et al. Expert consensus on treatment for stage III non-small cell lung cancer. Med. Adv. 2023, 1, 3–13. [Google Scholar] [CrossRef]
- Cornelius, M.E.; Loretan, C.G.; Jamal, A.; Davis Lynn, B.C.; Mayer, M.; Alcantara, I.C.; Neff, L. Tobacco product use among adults–United States, 2021. MMWR. Morb. Mortal. Wkly. Rep. 2023, 72, 475–483. [Google Scholar] [CrossRef]
- Pelosof, L.; Ahn, C.; Gao, A.; Horn, L.; Madrigales, A.; Cox, J.; McGavic, D.; Minna, J.D.; Gazdar, A.F.; Schiller, J. Proportion of never-smoker non–small cell lung cancer patients at three diverse institutions. JNCI J. Natl. Cancer Inst. 2017, 109, djw295. [Google Scholar] [CrossRef]
- Gitlitz, B.J.; Novello, S.; Vavalà, T.; Bittoni, M.; Sable-Hunt, A.; Pavlick, D.; Hsu, R.; Park, S.L.; Chen, R.; Cooke, M.; et al. The genomics of young lung cancer: Comprehensive tissue genomic analysis in patients under 40 with lung cancer. JTO Clin. Res. Rep. 2021, 2, 100194. [Google Scholar] [CrossRef]
- Dubin, S.; Griffin, D. Lung cancer in non-smokers. Mo. Med. 2020, 117, 375–379. [Google Scholar]
- Ucvet, A.; Yazgan, S.; Gursoy, S.; Samancilar, O. Prognosis of resected non-small cell lung cancer with ipsilateral pulmonary metastasis. Thorac. Cardiovasc. Surg. 2020, 68, 176–182. [Google Scholar] [CrossRef]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Calles, A.; Riess, J.W.; Brahmer, J.R. Checkpoint blockade in lung cancer with driver mutation: Choose the road wisely. In Proceedings of the American Society of Clinical Oncology Educational Book; American Society of Clinical Oncology, Annual Meeting: Chicago, IL, USA, 2020; Volume 40, pp. 372–384. [Google Scholar]
- Mamdani, H.; Matosevic, S.; Khalid, A.B.; Durm, G.; Jalal, S.I. Immunotherapy in lung cancer: Current landscape and future directions. Front. Immunol. 2022, 13, 823618. [Google Scholar] [CrossRef] [PubMed]
- Kamata, T.; Sunami, K.; Yoshida, A.; Shiraishi, K.; Furuta, K.; Shimada, Y.; Katai, H.; Watanabe, S.i.; Asamura, H.; Kohno, T.; et al. Frequent BRAF or EGFR mutations in ciliated muconodular papillary tumors of the lung. J. Thorac. Oncol. 2016, 11, 261–265. [Google Scholar] [CrossRef]
- Cardarella, S.; Ogino, A.; Nishino, M.; Butaney, M.; Shen, J.; Lydon, C.; Yeap, B.Y.; Sholl, L.M.; Johnson, B.E.; Jänne, P.A. Clinical, pathologic, and biologic features associated with BRAF mutations in non–small cell lung cancer. Clin. Cancer Res. 2013, 19, 4532–4540. [Google Scholar] [CrossRef]
- Takeuchi, K.; Soda, M.; Togashi, Y.; Suzuki, R.; Sakata, S.; Hatano, S.; Asaka, R.; Hamanaka, W.; Ninomiya, H.; Uehara, H.; et al. RET, ROS1 and ALK fusions in lung cancer. Nat. Med. 2012, 18, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Nakaoku, T.; Tsuta, K.; Tsuchihara, K.; Matsumoto, S.; Yoh, K.; Goto, K. Beyond ALK-RET, ROS1 and other oncogene fusions in lung cancer. Transl. Lung Cancer Res. 2015, 4, 156–164. [Google Scholar] [PubMed]
- Conde, E.; Rojo, F.; Gómez, J.; Enguita, A.B.; Abdulkader, I.; González, A.; Lozano, D.; Mancheño, N.; Salas, C.; Salido, M.; et al. Molecular diagnosis in non-small-cell lung cancer: Expert opinion on ALK and ROS1 testing. J. Clin. Pathol. 2022, 75, 145–153. [Google Scholar] [CrossRef]
- Takuma, S.; Inoue, Y.; Karayama, M.; Tsuchiya, K.; Tsukui, H.; Hozumi, H.; Suzuki, Y.; Furuhashi, K.; Enomoto, N.; Fujisawa, T.; et al. EGFR-Mutated lung adenocarcinoma successfully treated with Osimertinib after spontaneous Transformation to SCLC and Adenocarcinoma with neuroendocrine differentiation: Case Report. JTO Clin. Res. Rep. 2022, 3, 100264. [Google Scholar] [CrossRef]
- Rivera-Concepcion, J.; Uprety, D.; Adjei, A.A. Challenges in the use of targeted therapies in non-small cell lung cancer. Cancer Res. Treat. 2022, 54, 315–329. [Google Scholar] [CrossRef]
- Gu, A.; Li, J.; Li, M.Y.; Liu, Y. Patient-derived xenograft model in cancer: Establishment and applications. MedComm 2025, 6, e70059. [Google Scholar] [CrossRef]
- Liu, W.; Cui, Y.; Zheng, X.; Yu, K.; Sun, G. Application status and future prospects of the PDX model in lung cancer. Front. Oncol. 2023, 13, 1098581. [Google Scholar] [CrossRef]
- Yang, M.; Shen, H.; Flodby, P.; Koss, M.D.; Bassiouni, R.; Liu, Y.; Jashashvili, T.; Neely, A.; Ogbolu, E.; Castillo, J.; et al. Alveolar type I cells can give rise to KRAS-induced lung adenocarcinoma. Cell Rep. 2023, 42, 113286. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Westcott, P.M.; Halliwill, K.D.; To, M.D.; Rashid, M.; Rust, A.G.; Keane, T.M.; Delrosario, R.; Jen, K.Y.; Gurley, K.E.; Kemp, C.J.; et al. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature 2015, 517, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Kotton, D.N.; Morrisey, E.E. Lung regeneration: Mechanisms, applications and emerging stem cell populations. Nat. Med. 2014, 20, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef]
- Dohadwala, M.; Yang, S.C.; Luo, J.; Sharma, S.; Batra, R.K.; Huang, M.; Lin, Y.; Goodglick, L.; Krysan, K.; Fishbein, M.C.; et al. Cyclooxygenase-2–dependent regulation of E-cadherin: Prostaglandin E2 induces transcriptional repressors ZEB1 and Snail in non–small cell lung cancer. Cancer Res. 2006, 66, 5338–5345. [Google Scholar] [CrossRef]
- Kim, N.; Kim, H.K.; Lee, K.; Hong, Y.; Cho, J.H.; Choi, J.W.; Lee, J.I.; Suh, Y.L.; Ku, B.M.; Eum, H.H.; et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. 2020, 11, 2285. [Google Scholar] [CrossRef]
- Zilionis, R.; Engblom, C.; Pfirschke, C.; Savova, V.; Zemmour, D.; Saatcioglu, H.D.; Krishnan, I.; Maroni, G.; Meyerovitz, C.V.; Kerwin, C.M.; et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity 2019, 50, 1317–1334. [Google Scholar] [CrossRef]
- Chen, J.; Yang, H.; Teo, A.S.M.; Amer, L.B.; Sherbaf, F.G.; Tan, C.Q.; Alvarez, J.J.S.; Lu, B.; Lim, J.Q.; Takano, A.; et al. Genomic landscape of lung adenocarcinoma in East Asians. Nat. Genet. 2020, 52, 177–186. [Google Scholar] [CrossRef]
- Gene expression–based survival prediction in lung adenocarcinoma: A multi-site, blinded validation study. Nat. Med. 2008, 14, 822–827. [CrossRef]
- Schabath, M.B.; Welsh, E.A.; Fulp, W.J.; Chen, L.; Teer, J.K.; Thompson, Z.J.; Engel, B.E.; Xie, M.; Berglund, A.E.; Creelan, B.C.; et al. Differential association of STK11 and TP53 with KRAS mutation-associated gene expression, proliferation and immune surveillance in lung adenocarcinoma. Oncogene 2016, 35, 3209–3216. [Google Scholar] [CrossRef] [PubMed]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- de Bruijn, I.; Kundra, R.; Mastrogiacomo, B.; Tran, T.N.; Sikina, L.; Mazor, T.; Li, X.; Ochoa, A.; Zhao, G.; Lai, B.; et al. Analysis and visualization of longitudinal genomic and clinical data from the AACR project GENIE biopharma collaborative in cBioPortal. Cancer Res. 2023, 83, 3861–3867. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Revkov, E.; Kulshrestha, T.; Sung, K.W.K.; Skanderup, A.J. PUREE: Accurate pan-cancer tumor purity estimation from gene expression data. Commun. Biol. 2023, 6, 394. [Google Scholar] [CrossRef]
- Van de Sande, B.; Flerin, C.; Davie, K.; De Waegeneer, M.; Hulselmans, G.; Aibar, S.; Seurinck, R.; Saelens, W.; Cannoodt, R.; Rouchon, Q.; et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 2020, 15, 2247–2276. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-mesenchymal transition in cancer: A historical overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef] [PubMed]
- Tickle, T.; Tirosh, I.; Georgescu, C.; Brown, M.; Haas, B. inferCNV of the Trinity CTAT Project. 2019. Available online: https://github.com/broadinstitute/inferCNV (accessed on 21 October 2024).
- Wang, X.; Lin, Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef]
- Roussos, E.T.; Condeelis, J.S.; Patsialou, A. Chemotaxis in cancer. Nat. Rev. Cancer 2011, 11, 573–587. [Google Scholar] [CrossRef]
- Sadras, F.; Monteith, G.R.; Roberts-Thomson, S.J. An emerging role for calcium signaling in cancer-associated fibroblasts. Int. J. Mol. Sci. 2021, 22, 11366. [Google Scholar] [CrossRef] [PubMed]
- Monteith, G.R.; Prevarskaya, N.; Roberts-Thomson, S.J. The calcium–cancer signalling nexus. Nat. Rev. Cancer 2017, 17, 373–380. [Google Scholar] [CrossRef]
- Fu, J.; Yu, M.; Xu, W.; Yu, S. Research progress of bile acids in cancer. Front. Oncol. 2022, 11, 778258. [Google Scholar] [CrossRef] [PubMed]
- Vettore, L.; Westbrook, R.L.; Tennant, D.A. New aspects of amino acid metabolism in cancer. Br. J. Cancer 2020, 122, 150–156. [Google Scholar] [CrossRef]
- Boussiotis, V.A.; Chatterjee, P.; Li, L. Biochemical signaling of PD-1 on T cells and its functional implications. Cancer J. 2014, 20, 265–271. [Google Scholar] [CrossRef]
- Parvez, A.; Choudhary, F.; Mudgal, P.; Khan, R.; Qureshi, K.A.; Farooqi, H.; Aspatwar, A. PD-1 and PD-L1: Architects of immune symphony and immunotherapy breakthroughs in cancer treatment. Front. Immunol. 2023, 14, 1296341. [Google Scholar] [CrossRef]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Liu, D.; Lobie, P. Transcriptional activation of p53 by Pitx1. Cell Death Differ. 2007, 14, 1893–1907. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Liu, Y.; Yang, Y.; Qiu, Y.; Wang, Z.; Li, X.; Zhang, W. Emerging roles of activating transcription factor (ATF) family members in tumourigenesis and immunity: Implications in cancer immunotherapy. Genes Dis. 2022, 9, 981–999. [Google Scholar] [CrossRef] [PubMed]
- Wortel, I.M.; van der Meer, L.T.; Kilberg, M.S.; van Leeuwen, F.N. Surviving stress: Modulation of ATF4-mediated stress responses in normal and malignant cells. Trends Endocrinol. Metab. 2017, 28, 794–806. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef]
- Gumilar, K.E.; Chin, Y.; Ibrahim, I.H.; Tjokroprawiro, B.A.; Yang, J.Y.; Zhou, M.; Gassman, N.R.; Tan, M. Heat Shock Factor 1 Inhibition: A Novel Anti-Cancer Strategy with Promise for Precision Oncology. Cancers 2023, 15, 5167. [Google Scholar] [CrossRef] [PubMed]
- Ghaleb, A.M.; Yang, V.W. Krüppel-like factor 4 (KLF4): What we currently know. Gene 2017, 611, 27–37. [Google Scholar] [CrossRef]
- Li, S.; Huang, L.; Gu, J.; Wu, J.; Ou, W.; Feng, J.; Liu, B.; Cui, X.; Zhou, Y. Restoration of KLF4 inhibits invasion and metastases of lung adenocarcinoma through suppressing MMP2. J. Cancer 2017, 8, 3480–3489. [Google Scholar] [CrossRef]
- Chen, K.S.; Lim, J.W.; Richards, L.J.; Bunt, J. The convergent roles of the nuclear factor I transcription factors in development and cancer. Cancer Lett. 2017, 410, 124–138. [Google Scholar] [CrossRef]
- Nilsson, J.; Helou, K.; Kovács, A.; Bendahl, P.O.; Bjursell, G.; Fernö, M.; Carlsson, P.; Kannius-Janson, M. Nuclear Janus-activated kinase 2/nuclear factor 1-C2 suppresses tumorigenesis and epithelial-to-mesenchymal transition by repressing Forkhead box F1. Cancer Res. 2010, 70, 2020–2029. [Google Scholar] [CrossRef]
- Brun, M.; Jain, S.; Monckton, E.A.; Godbout, R. Nuclear factor I represses the notch effector HEY1 in glioblastoma. Neoplasia 2018, 20, 1023–1037. [Google Scholar] [CrossRef]
- Zhang, H.; Luo, Z.; Tang, J.; Tian, J.; Xiao, Y.; Sun, C.; Wang, T. Transcription factor NFIC functions as a tumor suppressor in lung squamous cell carcinoma progression by modulating lncRNA CASC2. Cell Cycle 2022, 21, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Affo, S.; Nair, A.; Brundu, F.; Ravichandra, A.; Bhattacharjee, S.; Matsuda, M.; Chin, L.; Filliol, A.; Wen, W.; Song, X.; et al. Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell 2021, 39, 866–882. [Google Scholar] [CrossRef] [PubMed]
- Unterleuthner, D.; Neuhold, P.; Schwarz, K.; Janker, L.; Neuditschko, B.; Nivarthi, H.; Crncec, I.; Kramer, N.; Unger, C.; Hengstschläger, M.; et al. Cancer-associated fibroblast-derived WNT2 increases tumor angiogenesis in colon cancer. Angiogenesis 2020, 23, 159–177. [Google Scholar] [CrossRef]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10+ GPR77+ cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell 2018, 172, 841–856. [Google Scholar] [CrossRef]
- Deng, J.; Fleming, J.B. Inflammation and myeloid cells in cancer progression and metastasis. Front. Cell Dev. Biol. 2022, 9, 759691. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Xue, W.; Yang, L.; Chen, C.; Ashrafizadeh, M.; Tian, Y.; Sun, R. Wnt/β-catenin-driven EMT regulation in human cancers. Cell. Mol. Life Sci. 2024, 81, 79. [Google Scholar] [CrossRef]
- Jechlinger, M.; Grunert, S.; Tamir, I.H.; Janda, E.; Lüdemann, S.; Waerner, T.; Seither, P.; Weith, A.; Beug, H.; Kraut, N. Expression profiling of epithelial plasticity in tumor progression. Oncogene 2003, 22, 7155–7169. [Google Scholar] [CrossRef]
- Jechlinger, M.; Sommer, A.; Moriggl, R.; Seither, P.; Kraut, N.; Capodiecci, P.; Donovan, M.; Cordon-Cardo, C.; Beug, H.; Grünert, S.; et al. Autocrine PDGFR signaling promotes mammary cancer metastasis. J. Clin. Investig. 2006, 116, 1561–1570. [Google Scholar] [CrossRef]
- Elahouel, R.; Blanc, C.; Carpentier, G.; Frechault, S.; Cascone, I.; Destouches, D.; Delbé, J.; Courty, J.; Hamma-Kourbali, Y. Pleiotrophin exerts its migration and invasion effect through the neuropilin-1 pathway. Neoplasia 2015, 17, 613–624. [Google Scholar] [CrossRef]
- Masiuk, M.; Urasinska, E.; Domagala, W. Intranuclear nucleolin distribution during cell cycle progression in human invasive ductal breast carcinomas in relation to estrogen receptor status. Anticancer Res. 2007, 27, 3957–3962. [Google Scholar] [PubMed]
- Cicchillitti, L.; Di Michele, M.; Urbani, A.; Ferlini, C.; Donati, M.B.; Scambia, G.; Rotilio, D. Comparative proteomic analysis of paclitaxel sensitive A2780 epithelial ovarian cancer cell line and its resistant counterpart A2780TC1 by 2D-DIGE: The role of ERp57. J. Proteome Res. 2009, 8, 1902–1912. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Zhou, F.; Zhang, Q.; Sun, X.; Shi, X.; Liang, Y.; Wang, X.; Yue, L. Overexpression of nucleolin and different expression sites both related to the prognosis of gastric cancer. Apmis 2013, 121, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Bambury, R.M.; Van Allen, E.M.; Drabkin, H.A.; Lara, P.N.; Harzstark, A.L.; Wagle, N.; Figlin, R.A.; Smith, G.W.; Garraway, L.A.; et al. A phase II trial of AS1411 (a novel nucleolin-targeted DNA aptamer) in metastatic renal cell carcinoma. Investig. New Drugs 2014, 32, 178–187. [Google Scholar] [CrossRef]
- Xu, J.y.; Lu, S.; Xu, X.y.; Hu, S.l.; Li, B.; Li, W.x.; Chang, J.Y. Prognostic significance of nuclear or cytoplasmic nucleolin expression in human non-small cell lung cancer and its relationship with DNA-PKcs. Tumor Biol. 2016, 37, 10349–10356. [Google Scholar] [CrossRef]
- Chen, Z.; Xu, X. Roles of nucleolin: Focus on cancer and anti-cancer therapy. Saudi Med. J. 2016, 37, 1312–1318. [Google Scholar] [CrossRef]
- Ren, Q.; Zhu, P.; Zhang, H.; Ye, T.; Liu, D.; Gong, Z.; Xia, X. Identification and validation of stromal-tumor microenvironment-based subtypes tightly associated with PD-1/PD-L1 immunotherapy and outcomes in patients with gastric cancer. Cancer Cell Int. 2020, 20, 92. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Dai, Y.; Zhang, Q.; Tang, P.; Fu, Q.; Xiong, G. Stromal score-based gene signature: A prognostic prediction model for colon cancer. Front. Genet. 2021, 12, 655855. [Google Scholar] [CrossRef]
- Ahrendt, S.A.; Hu, Y.; Buta, M.; McDermott, M.P.; Benoit, N.; Yang, S.C.; Wu, L.; Sidransky, D. p53 mutations and survival in stage I non-small-cell lung cancer: Results of a prospective study. J. Natl. Cancer Inst. 2003, 95, 961–970. [Google Scholar] [CrossRef]
- Robles, A.I.; Jen, J.; Harris, C.C. Clinical outcomes of TP53 mutations in cancers. Cold Spring Harb. Perspect. Med. 2016, 6, a026294. [Google Scholar] [CrossRef]
- Cui, Y.; Guo, G. Immunomodulatory function of the tumor suppressor p53 in host immune response and the tumor microenvironment. Int. J. Mol. Sci. 2016, 17, 1942. [Google Scholar] [CrossRef] [PubMed]
- Agupitan, A.D.; Neeson, P.; Williams, S.; Howitt, J.; Haupt, S.; Haupt, Y. P53: A guardian of immunity becomes its saboteur through mutation. Int. J. Mol. Sci. 2020, 21, 3452. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.A.; Vallejos, C.S.; Raez, L.E.; Mas, L.A.; Ruiz, R.; Torres-Roman, J.S.; Morante, Z.; Araujo, J.M.; Gómez, H.L.; Aguilar, A.; et al. Gender and outcomes in non-small cell lung cancer: An old prognostic variable comes back for targeted therapy and immunotherapy? ESMO Open 2018, 3, e000344. [Google Scholar] [CrossRef]
- Sakurai, H.; Asamura, H.; Goya, T.; Eguchi, K.; Nakanishi, Y.; Sawabata, N.; Okumura, M.; Miyaoka, E.; Fujii, Y.; Japanese Joint Committee for Lung Cancer Registration. Survival differences by gender for resected non-small cell lung cancer: A retrospective analysis of 12,509 cases in a Japanese Lung Cancer Registry study. J. Thorac. Oncol. 2010, 5, 1594–1601. [Google Scholar] [CrossRef]
- Kim, S.K.; Cho, S.W. The evasion mechanisms of cancer immunity and drug intervention in the tumor microenvironment. Front. Pharmacol. 2022, 13, 868695. [Google Scholar] [CrossRef]
- Schleimer, R.P.; Kato, A.; Kern, R.; Kuperman, D.; Avila, P.C. Epithelium: At the interface of innate and adaptive immune responses. J. Allergy Clin. Immunol. 2007, 120, 1279–1284. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Sarkar, M.; Nguyen, T.; Gundre, E.; Ogunlusi, O.; El-Sobky, M.; Giri, B.; Sarkar, T.R. Cancer-associated fibroblasts: The chief architect in the tumor microenvironment. Front. Cell Dev. Biol. 2023, 11, 1089068. [Google Scholar] [CrossRef]
- De Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Zhang, S.; Gong, Z.; Li, X.; Cao, K.; Deng, H.; He, Y.; et al. The role of microenvironment in tumor angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 204. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Zhu, Y.T.; Chen, S.Y.; Tseng, S.C. Wnt signaling induces epithelial–mesenchymal transition with proliferation in ARPE-19 cells upon loss of contact inhibition. Lab. Investig. 2012, 92, 676–687. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Martin-Orozco, E.; Sanchez-Fernandez, A.; Ortiz-Parra, I.; Ayala-San Nicolas, M. WNT signaling in tumors: The way to evade drugs and immunity. Front. Immunol. 2019, 10, 2854. [Google Scholar] [CrossRef]
- Dostert, C.; Grusdat, M.; Letellier, E.; Brenner, D. The TNF family of ligands and receptors: Communication modules in the immune system and beyond. Physiol. Rev. 2018, 99, 115–160. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Ping, Y.F.; Zhou, W.; He, Z.C.; Chen, C.; Bian, B.S.J.; Zhang, L.; Chen, L.; Lan, X.; Zhang, X.C.; et al. Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat. Commun. 2017, 8, 15080. [Google Scholar] [CrossRef] [PubMed]
- Mikelis, C.; Koutsioumpa, M.; Papadimitriou, E. Pleiotrophin as a possible new target for angiogenesis-related diseases and cancer. Recent Patents Anti-Cancer Drug Discov. 2007, 2, 175–186. [Google Scholar] [CrossRef]
- Pandey, P.; Khan, F.; Upadhyay, T.K.; Seungjoon, M.; Park, M.N.; Kim, B. New insights about the PDGF/PDGFR signaling pathway as a promising target to develop cancer therapeutic strategies. Biomed. Pharmacother. 2023, 161, 114491. [Google Scholar] [CrossRef]
- Zou, X.; Tang, X.Y.; Qu, Z.Y.; Sun, Z.W.; Ji, C.F.; Li, Y.J.; Guo, S.D. Targeting the PDGF/PDGFR signaling pathway for cancer therapy: A review. Int. J. Biol. Macromol. 2022, 202, 539–557. [Google Scholar] [CrossRef]
- Sorrelle, N.; Dominguez, A.T.; Brekken, R.A. From top to bottom: Midkine and pleiotrophin as emerging players in immune regulation. J. Leukoc. Biol. 2017, 102, 277–286. [Google Scholar] [CrossRef]
- Heldin, C.H. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun. Signal. 2013, 11, 97. [Google Scholar] [CrossRef] [PubMed]
- Pietras, K.; Rubin, K.; Sjoblom, T.; Buchdunger, E.; Sjoquist, M.; Heldin, C.H.; Ostman, A. Inhibition of PDGF receptor signaling in tumor stroma enhances antitumor effect of chemotherapy. Cancer Res. 2002, 62, 5476–5484. [Google Scholar]
- Liu, J.; Liao, S.; Huang, Y.; Samuel, R.; Shi, T.; Naxerova, K.; Huang, P.; Kamoun, W.; Jain, R.K.; Fukumura, D.; et al. PDGF-D improves drug delivery and efficacy via vascular normalization, but promotes lymphatic metastasis by activating CXCR4 in breast cancer. Clin. Cancer Res. 2011, 17, 3638–3648. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Li, G.Y.; Wang, B.; Han, S.X.; Sun, X.; Jiang, Y.N.; Shen, Y.W.; Zhou, C.; Feng, J.; Lu, S.Y.; et al. Metformin inhibits metastatic breast cancer progression and improves chemosensitivity by inducing vessel normalization via PDGF-B downregulation. J. Exp. Clin. Cancer Res. 2019, 38, 235. [Google Scholar] [CrossRef]
- Akiyama, T.; Yasuda, T.; Uchihara, T.; Yasuda-Yoshihara, N.; Tan, B.J.; Yonemura, A.; Semba, T.; Yamasaki, J.; Komohara, Y.; Ohnishi, K.; et al. Stromal reprogramming through dual PDGFRα/β blockade boosts the efficacy of anti–PD-1 immunotherapy in fibrotic tumors. Cancer Res. 2023, 83, 753–770. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Guo, Z.; Song, M.; Pan, Q.; Zhao, J.; Huang, Y.; Han, Y.; Ouyang, D.; Yang, C.; Chen, H.; et al. Lenvatinib improves anti-PD-1 therapeutic efficacy by promoting vascular normalization via the NRP-1-PDGFRβ complex in hepatocellular carcinoma. Front. Immunol. 2023, 14, 1212577. [Google Scholar] [CrossRef]
- Luo, W. Nasopharyngeal carcinoma ecology theory: Cancer as multidimensional spatiotemporal “unity of ecology and evolution” pathological ecosystem. Theranostics 2023, 13, 1607–1631. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, J.; McQuay, C.; Talburt, J.; Tiwari, A.K.; Yang, M.Q. Single-Cell Transcriptomic Analysis Unveils Key Regulators and Signaling Pathways in Lung Adenocarcinoma Progression. Biomedicines 2025, 13, 1606. https://doi.org/10.3390/biomedicines13071606
Ma J, McQuay C, Talburt J, Tiwari AK, Yang MQ. Single-Cell Transcriptomic Analysis Unveils Key Regulators and Signaling Pathways in Lung Adenocarcinoma Progression. Biomedicines. 2025; 13(7):1606. https://doi.org/10.3390/biomedicines13071606
Chicago/Turabian StyleMa, Jialu, Caleb McQuay, John Talburt, Amit K. Tiwari, and Mary Qu Yang. 2025. "Single-Cell Transcriptomic Analysis Unveils Key Regulators and Signaling Pathways in Lung Adenocarcinoma Progression" Biomedicines 13, no. 7: 1606. https://doi.org/10.3390/biomedicines13071606
APA StyleMa, J., McQuay, C., Talburt, J., Tiwari, A. K., & Yang, M. Q. (2025). Single-Cell Transcriptomic Analysis Unveils Key Regulators and Signaling Pathways in Lung Adenocarcinoma Progression. Biomedicines, 13(7), 1606. https://doi.org/10.3390/biomedicines13071606