1. Introduction
Hepatocellular carcinoma (HCC) is a common primary liver cancer, constituting about 75% of all liver cancer types and being one of the leading causes of cancer deaths internationally [
1]. While treatment options and mortality rates have improved for HCC, the current 2-year survivability rate is 50%, which decreases to 10% after 5 years [
1]. Males are 2-4 times more likely to develop HCC compared with females due to behavioral risk factors such as smoking and alcohol consumption, while Asian Americans, African Americans, and Hispanics have the highest rates based on ethnicity [
2]. Early detection of HCC can result in effective treatments such as liver transplantation or surgery, leading to an improved prognosis over time [
1]. Even so, most patients are diagnosed when HCC is in an advanced stage [
1]. Chemotherapy is a common cancer treatment used to inhibit cancer cell viability and proliferation, with the most common agent being sorafenib [
1]. While sorafenib’s mechanism of action is to block specific kinase pathways, it is often only able to increase survival by 7–10 months, indicating the need to develop other effective liver cancer treatment options [
1].
Non-alcoholic steatohepatitis (NASH), alcoholic cirrhosis, hepatitis B and C, and non-alcoholic fatty liver disease (NAFLD) are major contributors to the development of HCC in the United States population [
1]. Obesity, diabetes, metabolic syndrome, and a variety of other disease states increase the risk of the development and progression of HCC through exacerbation of NAFLD [
3]. McGlynn et al. reported that the risk of developing HCC was 2.6-fold higher in obese patients with NAFLD compared with non-obese patients with NAFLD [
3]. Obesity has been shown to be a strong underlying factor in the development of NASH and NAFLD, specifically through the upregulation of certain adipokines, subsequently leading to tumorigenesis [
3]. Clinical data show a correlation between NAFLD and obesity, with the high prevalence of steatohepatitis in those with class I–II obesity being 65% and 85% in those with class III obesity [
4]. According to the US Cancer Statistics database, 4.7% of cancer cases in men and 9.6% of cancer cases in women were due to excess body weight, with 51% of those cases being liver or gallbladder cancer [
5]. Data have also shown that obesity is associated with a 13% risk of recurrence and poor outcomes in survivors [
5]. Obesity is indicative of a high level of adipose tissue, which plays a role in the impairment of metabolic and hormonal control, thus leading to carcinogenesis [
6]. While the obesity-cancer connection is multifactorial, research has shown that there are three primary mechanisms responsible for the increased risk of cancer in obese individuals [
6]. For one, adipose tissue can increase the production of hormones and adipokines, which may be anti-apoptotic and pro-proliferative when excreted in excess concentrations by adipocytes [
6]. The overproduction of these hormones, such as leptin, leads to proinflammatory environments with excess and sustained levels of obesity-associated hormones. However, when overexpressed, they can contribute to tumorigenesis through the activation of pro-proliferative and anti-apoptotic mechanisms by binding to its receptor and stimulating pro-tumorigenic cellular changes [
6]. Furthermore, obesity is often associated with hyperinsulinemia, with high levels of insulin and insulin-like growth factor 1 (IGF-1), a pro-tumorigenic hormone, aiding in the development of cancer. In certain cases, this can lead to progression due to its pro-proliferative and anti-apoptotic characteristics [
6,
7]. While the hormone itself is not responsible for the appearance of cancer cells, it contributes to the uncontrolled growth and survival of tumor cells [
7]. Those properties exacerbated by obesity, such as resistance to cell death, proliferative signaling, and cell immortality, have been deemed to be just a few of the hallmarks of cancer that are adversely affected by obesity.
Obesity is the primary cause of NAFLD development, a precursor to NASH, in about 50% of HCC patients, both of which are characterized by lipid accumulation and inflammation in the liver [
8]. Prolonged lipid accumulation will subsequently lead to the progression of liver disease, evidenced by the presence of steatohepatitis and ballooning of the hepatocytes. NASH is the result of enlarged hepatocytes and an inflamed liver due to steatohepatitis, eventually leading to fibrosis, cirrhosis, and the development of liver cancer if untreated [
8]. Rajesh et al. suggested that insulin resistance and a high-caloric intake, often associated with obesity, can lead to a buildup of adipocytes, thus producing a pro-inflammatory microenvironment where hepatic cancer cells are able to survive [
9]. Chronic injury to the liver, such as cirrhosis exacerbated by NAFLD, can also result in an overexpression of nuclear factor-kB (NF-kB), a pro-tumorigenic transcription factor and inflammatory mediator associated with liver cancer [
10]. Obesity-induced leptin and IGF-1 can activate NF-kB transcription, leading to an increase in cytokines [
10,
11]. The release of these pro-inflammatory cytokines, such as interleukin (IL)-6, interleukin-1β (IL-1β), and tumor necrosis factor alpha (TNF-α), leads to the perpetuation of an inflammatory response, resulting in increased activation of signaling pathways that are associated with the hallmarks of cancer, including survivability, proliferation, reactive oxygen species (ROS) production, and invasion into other tissues [
7,
12].
There is a plethora of mechanistic pathways involved in the relationship between obesity and HCC. Nicotinamide ribosyl transferase (NAMPT) is an enzyme that is highly expressed in the liver and is involved in the progression of tumors in obesogenic environments, having been shown to have increased concentrations in the context of obesity [
13]. NAMPT has two derivatives with varying functions: extracellular NAMPT (eNAMPT) and intracellular NAMPT (iNAMPT). eNAMPT, also known as visfatin, is an adipokine with an important role, though its involvement in carcinogenesis is not fully understood [
13]. Recent research has shown that eNAMPT overexpression has been associated with apoptotic resistance, cancer cell proliferation, and activation of the pro-inflammatory pathways which are associated with NAD biosynthesis [
13]. iNAMPT is known to be the rate-limiting enzyme in the synthesis of NAD, an essential coenzyme involved in numerous redox reactions that progress into the production of the cell’s means of energy, ATP, which promotes a pro-growth environment in the context of obesity [
13]. While the role of iNAMPT in HCC is a novel concept, overexpressed iNAMPT levels have been detected in many other cancer types, including prostate, colorectal, and gastric tumors [
14]. iNAMPT has been shown to have anti-apoptotic and proliferative properties on tumor cells and is associated with increased metastasis and tumor growth [
13,
14]. Because iNAMPT and cellular energetics, a hallmark of cancer, play an important role in liver cancer progression, it has become a target for therapies focused on inhibiting its function in cancer cells [
15,
16]. FK866, an FDA-approved iNAMPT inhibitor, has been shown to have antitumor properties through repression of cancer cell growth, dephosphorylation of the Erk and Akt pathways, and apoptotic induction, which could play an important role in cancers that are dependent on these pathways.
The purpose of this study was to determine the contribution of NAD biosynthesis in obesity-induced liver cancer progression. We hypothesized that liver cancer cells exposed to an obese person’s serum will show suppressed viability and increased apoptotic ability when treated with the iNAMPT inhibitor FK866. We also hypothesized through using a syngeneic mouse model of liver cancer that obese mice injected with an iNAMPT inhibitor will show a decrease in tumor incidence and growth. For both, we demonstrate that inhibition of iNAMPT resulted in suppression of tumor growth in the context of obesity. Identifying pre-clinical strategies to reverse the impact of obesity on liver cancer progression is important due to the strong increased risk of liver cancer and its poor prognosis. Future translational research studies could build upon pre-clinical foundational research.
2. Materials and Methods
2.1. Cell Culture
Both human and mouse liver carcinoma cell lines (SNU-449 and Hepa 1-6, respectively) were purchased from the American Type Tissue Culture Collection (ATCC). SNU-449 cells were cultured in RPMI plus 10% fetal bovine serum (FBS), and both HepG2 and Hepa 1-6 cells were cultured in EMEM plus 10% FBS. All cells were maintained at 37 °C in a 5% (v/v) CO2 humidified incubator. Cells were exposed to one of five experimental conditions: 5% obese (OB) mouse serum, 5% OB serum + FK866 (10 nM), 5% control (CR) mouse serum, and 5% CR + FK866 (10 nM). FK866 (cat # HY-50876) was obtained from Medchemexpress USA (Monmouth Junction, NJ, USA). The times of exposure will be explained further in each assay described below.
2.2. Mouse Serum
Mouse serum from C57BL/6J mice was purchased from Jackson Laboratories (Bar Harbor, ME, USA). Post weaning, the mice were randomized to a high-fat diet (60% kcal from fat) (OB) or control diet (CR) and continued their respective diets for the remainder of the study. For the OB group, 12 mice sera were pooled at 14 weeks of age. For the CR group, 12 mice sera were pooled at 14 weeks of age. OB and CR sera were used for in vitro experiments at a concentration of 5%, according to previous research [
17].
2.3. MTT Assay
To measure cell viability, an MTT assay was performed. HepG2, SNU-449, and Hepa 1-6 cells were seeded in a 96-well plate with 10,000 cells per well. The cells were incubated overnight in FBS. After 24 h, the cells were treated with the treatments stated above for 72 h. After 72 h, 20 µL of MTT reagent (5 mg/mL) was added to the well plate for 1.5 h. Then, the media along with the reagent was aspirated, and 100 µM of dimethyl sulfoxide (DMSO) was added to the well plate and shaken for 10 min at room temperature. Absorbance was measured at 540 nm using a Cytation 5 microplate reader (BioTek Instruments, Inc., Winooski, VT, USA).
2.4. Reactive Oxygen Species
The intracellular levels of reactive oxygen species (ROS) were assessed using a cell-permeable dichlorofluorescin diacetate (DCFH-DA) probe (ab113851) from Abcam (Cambridge, UK) according to the manufacturer’s procedure. SNU-449, HepG2, and Hepa 1-6 cells were seeded in a dark, clear-bottomed 96-well microplate. On day 2, the cells were exposed to the same experimental conditions described above. On day 3, the media were removed, and the cells were washed and incubated for 45 min with 25 μM DCFDA at 37 °C. The fluorescent intensity was measured at an excitation wavelength of 485 nm and an emission wavelength of 529 nm with a Cytation 5 microplate reader from BioTek Instruments, Inc. (Winooski, VT, USA).
2.5. Cytotoxicity
To assess cytotoxicity and early indicators of cell death, a lactate dehydrogenase (LDH) assay was used in the SNU-449, HepG2, and Hepa 1-6 cell lines according to the manufacturer’s instructions with Sigma’s Lactate Dehydrogenase Activity Assay Kit (MAK066-1KT) (MilliporeSigma, Burlington, MA, USA).
2.6. Invasion Assay
A Corning (Corning, NY, USA) BioCoat Matrigel Invasion Chamber (#354480) assay was utilized to assess the addition of FK866 and its ability to decrease the invasive capacity of the HepG2, SNU-449, and Hepa 1-6 cell lines when exposed to the treatments described above. The Matrigel chamber was hydrated in serum-free media for 2 h prior to seeding. After the chambers were rehydrated, cells were seeded on the upper side of the chamber at a concentration of 80,000 cells per well in a 24-well plate and treated with the conditions described above. The chemoattractant, 10% FBS, was added to the well below the chamber. After 48 h, the cells were stained with crystal violet, and images were captured using a Cytation 5 microplate reader from BioTek Instruments, Inc. (VT, USA). After imaging, the crystal violet stain was dissolved using a distain solution (methanol, acetic acid, and water), and absorbance was measured with the Cytation5 microplate reader.
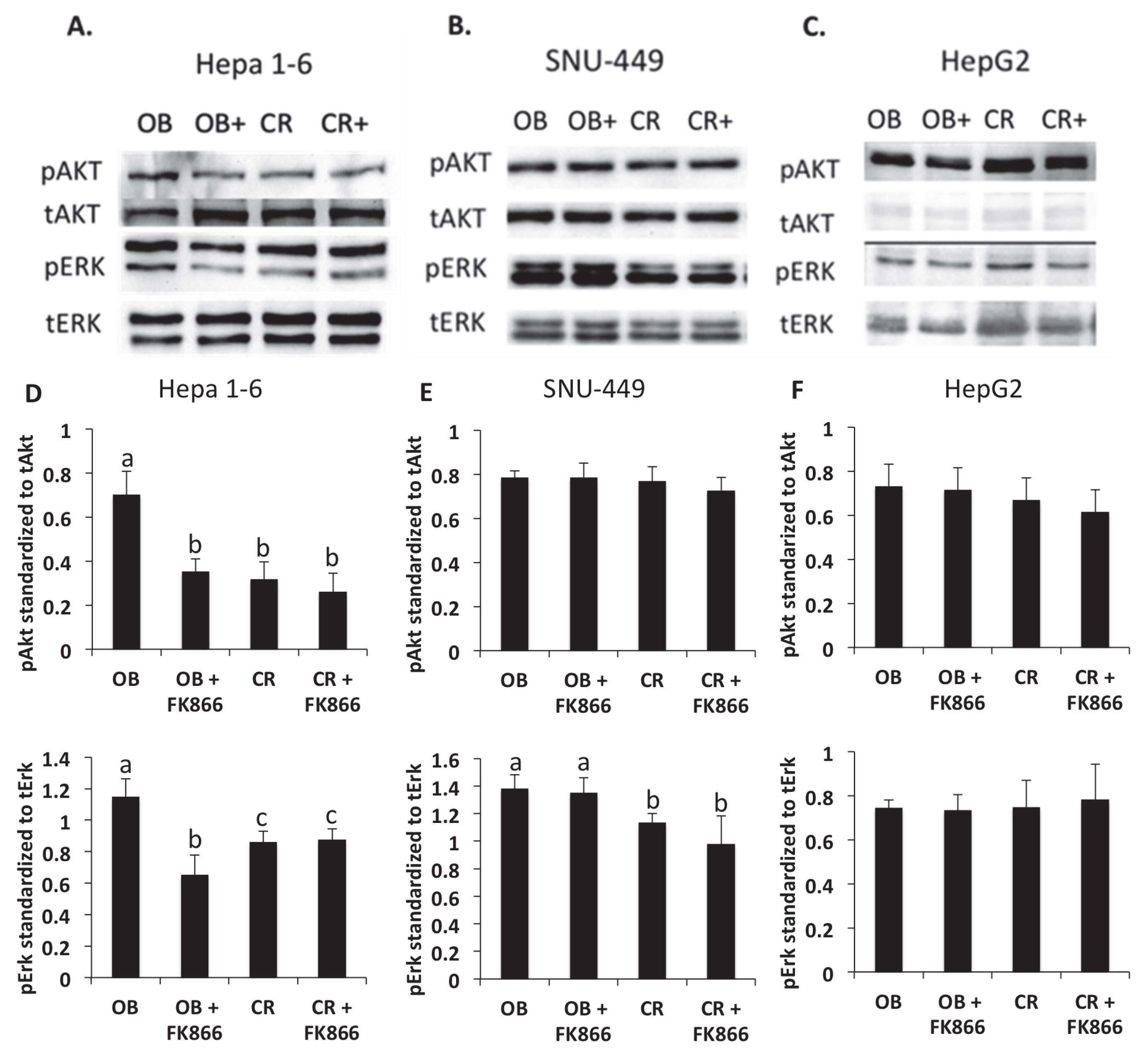
2.7. Immunoblot
To measure protein expression differences, SNU-449, HepG2, and Hepa 1-6 cells were plated at a density of 4 × 105 cells per well in a 6-well plate. After 24 h, the cells were starved of serum for 6 h. The cells were then exposed to their respective experimental conditions for 15 min. After treatment, the cells were harvested using lysis buffer (5 mL glycerol, 3.14 mL TRIS 1 M (pH: 6.8), 5 mL 10% SDS, and 36.86 mL ddH2O) and quantified using a Pierce BCA protein assay kit (Thermo Fisher Scientific, St. Louis, MO, USA). Then, 50 μg of protein lysate was electrophoresed through a 10% SDS PAGE gel. The proteins were then transferred to a nitrocellulose membrane. The membrane was blocked with 5% bovine serum albumin for 45 min. Protein lysates were subjected to immunodetection with rabbit anti-phospho-Erk, rabbit anti-total Erk, rabbit anti-phospho-Akt, and rabbit anti-total Akt. Images were acquired using a Fotodyne gel documentation system (Hartland, WI, USA). After measuring the phosphorylated proteins, blots were stripped with Restore Plus western blot stripping buffer before the total protein levels were measured. ImageJ version 1.54p was used to quantify the protein levels.
2.8. Animal Model and Study Design
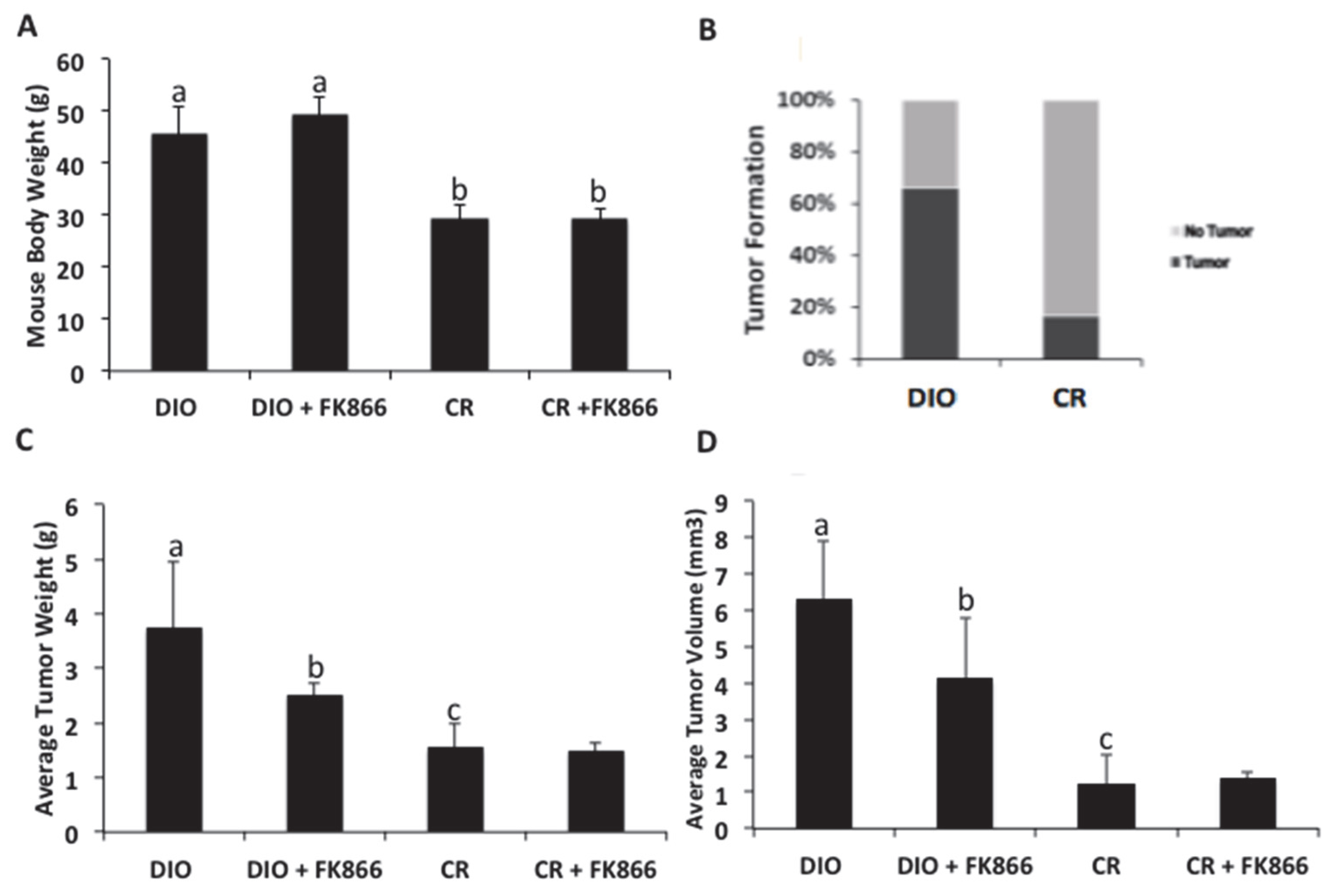
Twenty-one 8-week-old C57BL/6J male mice were ordered from Jackson Laboratories (Bar Harbor, ME, USA). Post weaning, 10 mice were randomized to a high-fat diet (ad libitum) that induces obesity (60% kcal from fat; D12492, referred to as DIO), and 11 mice were randomized to a control diet (10% kcal from fat; D12450B, referred to as CR). Both remained on these diets for the duration of the study. The mice received new food 2 times per week. The mice were weighed weekly to monitor weight gain. At 21 weeks of age, all mice were injected with 500,000 Hepa 1-6 liver cancer cells suspended in PBS for a total volume of 100 μL. At 23 weeks of age, FK866 was administered at a dose of 30 mg/kg dissolved in DMSO and phosphate buffered saline (PBS) for 3 days over 2 weeks. The DIO and CR mice were injected with a vehicle control consisting of DMSO and PBS at the same volume as the inhibitor. The concentration of FK866 was based on previous cancer mouse models [
18,
19]. The study was designed with a statistical power of 90% and a type 1 error of 5%. The sample size was determined by two primary outcomes of tumor and weight change, with a 50% reduction in tumor size and 30% increase in body weight when compared with their controls. The calculated minimum sample sizes were 2 mice per group for tumors and 3 mice for weight.
2.9. Tumor Injection, Incidence, and Growth
For each cancer cell injection, a 500,000 Hepa 1-6 tumor liver cancer cell suspension was added to PBS and 25% matrigel BME Type 3 (Cat # 3632-005-02) R&D Systems (Minneapolis, MN, USA). The ratio of the cell suspension to PBS was 1:1 with a maximum volume of 250 µL. The cell suspension was injected subcutaneously on the flank of the C56BL/6 mouse with a 25 g needle. The mice were monitored daily until a palpable tumor was detected. As a baseline, the mice were weighed on the day of implantation. Once a palpable tumor was detected, the mice were monitored daily. Twenty-four hours post cancer cell injection, the mice were monitored for palpable tumors. Tumor incidence was determined by counting the number of palpable tumors in each experimental group.
2.10. Statistical Analysis
All in vitro experiments were replicated at least 3 times to ensure validity. GraphPad Prism version 10.5.0 (Boston, MA, USA) was used to analyze the results via one-way ANOVA with a Tukey’s post hoc test. Results containing p < 0.05 were considered statistically significant.
4. Discussion
Excess adiposity, as a result of obesity, produces pro-inflammatory adipokines and cytokines that support tumorigenesis by increasing proliferation and enhancing the biomolecules necessary for growth. One adipokine, NAMPT, and its different derivatives (eNAMPT and iNAMPT) are key targets for novel therapeutics. iNAMPT has been previously established as being overexpressed in other types of cancers and positively correlated to high levels of adiposity. As obesity increases, iNAMPT levels also increase as a result, leading to the upregulation of harmful and pro-tumorigenic changes in the liver, such as increased proliferation, ROS production, invasion, and survivability [
14]. The connection between iNAMPT in the presence of obesity and its role in tumorigenesis makes it an important target of inhibition in order to possibly halt cancer progression.
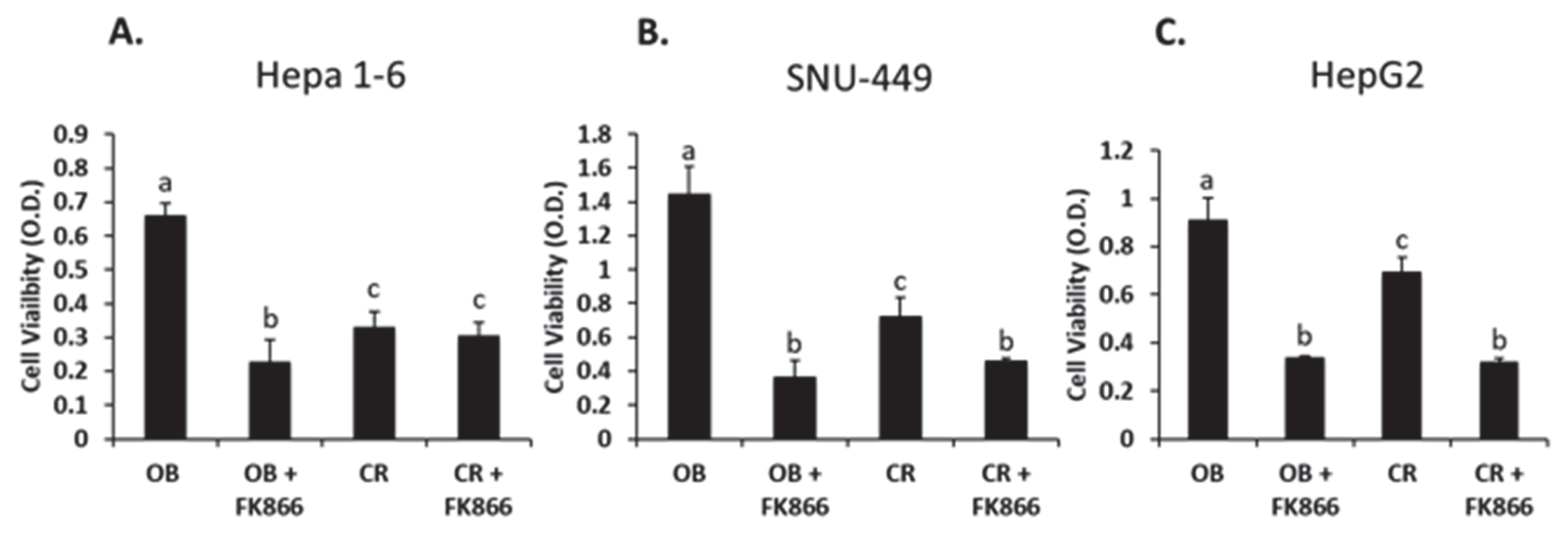
Studies have shown that obese phenotype serum increases cell proliferation compared with serum from a lean phenotype [
23]. The data in this study follow a similar trend, where the liver cancer cell lines treated with obese phenotype serum resulted in increased proliferation compared with the control group, thereby demonstrating that obesity promotes cell growth. On the other hand, other studies involving the use of FK866 as an NAMPT inhibitor have shown that FK866 reduces the cell viability of other hepatic cancer cell lines in high doses (5–40 nM) [
24,
25]. According to the data from this study, FK866 treatment in all three cell lines resulted in a similar trend, with cell proliferation being decreased with the addition of the NAMPT inhibitor. This suggests that blocking NAD synthesis with FK866 has a significant effect on reducing liver cancer cell viability in the context of obesity.
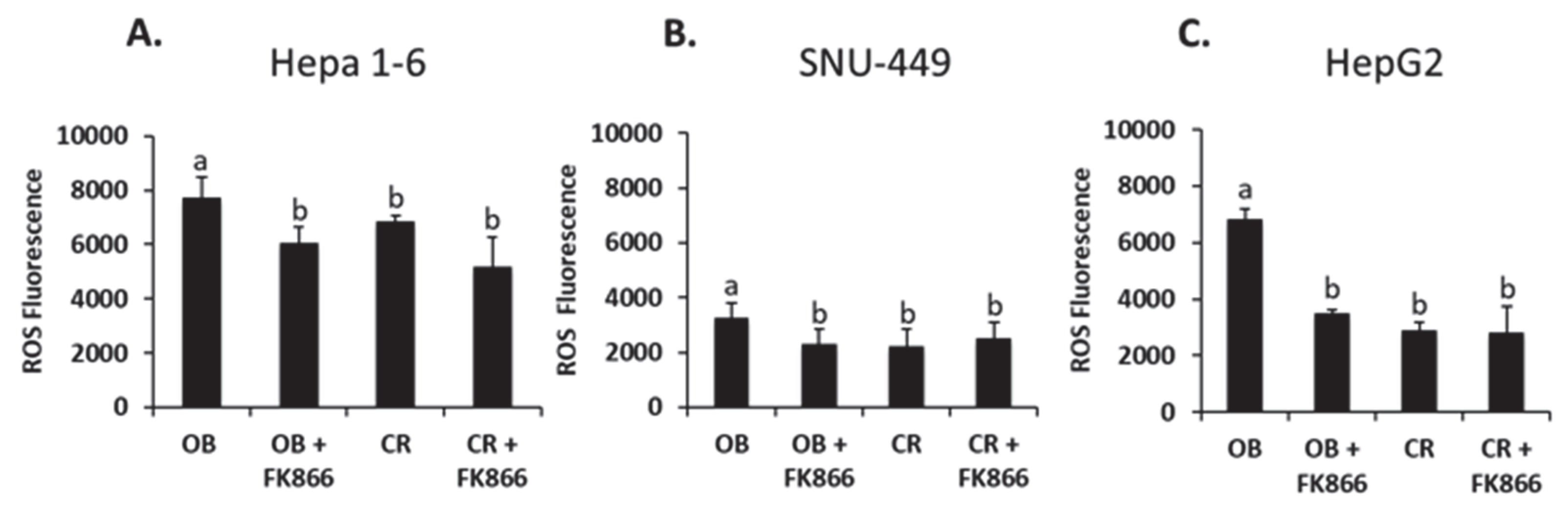
Oxidative stress because of ROS production from inflammation and metabolic processes is common in many disease states, including cancer and increased adiposity [
26]. The intracellular destruction that follows the production of ROS creates an ideal environment for cancer to progress. The presence of obesity has been shown to be a pathological process that may induce oxidative stress due to the excretion of pro-inflammatory cytokines from adipocytes such as IL-6 and TNF-α. When inflammation is exacerbated by obesity due to secretion of adipocytokines, mitochondrial function can be altered, which leads to increased production of ROS [
27]. One study demonstrated a reversal in ROS production with the introduction of FK866 to cells treated with hydrogen peroxide [
28]. The data from the current study show that the cells treated with obese phenotype serum had a significant increase in ROS production compared with the control, whereas the addition of FK866 to the obese group decreased ROS in all three cell lines. The high levels of adiposity associated with obesity create a pro-inflammatory environment that degrades metabolic processes, possibly leading to the accumulation of ROS and thus perpetuating inflammation. Previous research on ROS levels in tumorigenesis have shown that ROS, as well as other molecules, are responsible for the activation of signaling cascades that lead to proliferation, invasion, suppression of apoptosis, and thus the formation of larger tumors [
29]. A larger tumor size can have an effect on prognosis and treatment outcomes in those with HCC. Collectively, the combined in vitro and in vivo data from the current study also suggest that the inhibition of NAD biosynthesis via FK866 suppresses ROS production and may decrease tumor growth. However, this study did not test the mechanism by which ROS suppression inhibits tumor growth. In addition, while the murine Hepa 1-6 and human HepG2 liver cancer cell lines had significant decreases in ROS production with the addition of FK866, the same pattern was not as robust in SNU-449. This difference may be due to metabolic differences in the enzymatic pathways and genetic background between the liver cell lines used in this study. Because ROS is a byproduct of cellular metabolism, future directions may involve investigating the mechanisms by which NAD inhibition slows the release of ROS in order to decrease the overall production of ROS in an obesogenic environment.
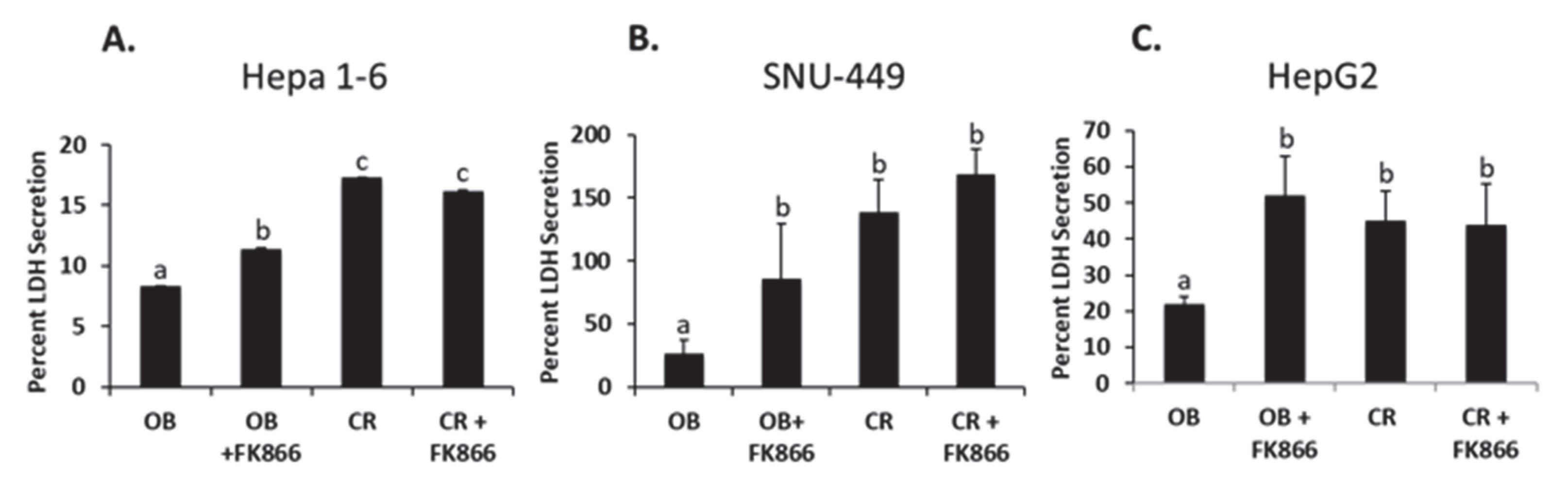
Cell survivability and the avoidance of cell death are two of the hallmarks of cancer and have become popular points of interest for treatment [
7]. LDH is commonly excreted by cells in the early stages of cell death, also known as apoptosis [
30]. In other words, LDH secretion is a precursor event prior to initiating the steps leading to cell death. One study found that leptin, an adipocytokine known to have increased levels during cancer development, impaired natural killer cell cytotoxicity. While the methods in this study assessed cytotoxicity through the secretion of LDH, the data remained consistent, as obesity decreased LDH secretion and thus cytotoxicity. In another study, it was shown that treating glioblastoma cells with FK866 increased LDH’s release, suggesting that more cells were undergoing apoptosis when NAD biosynthesis was inhibited [
30]. The findings of this study followed a similar pattern, with OB by itself decreasing LDH secretion and the addition of FK866 increasing secretion in all three cell lines. The decrease in LDH indicates less cytotoxicity, meaning the cells were not undergoing apoptosis. This may suggest that the presence of obesity protects cells from cell death. Though the exact mechanism is unknown, it may be associated with iNAMPT. Despite this, the addition of FK866 shows that there is a key pathway that can be blocked with the inhibition of NAD biosynthesis, thus sensitizing cells to apoptosis. These data are also supported by the cell viability assay used in this study, where viability was shown to have increased in the presence of obesity yet decreased with the addition of FK866. To extend these findings, future studies may investigate analyzing the specific mechanisms behind cytokines released by adipocytes and its protective ability against cell death. On the other hand, future studies may also investigate strategies to sensitize cells to apoptosis in order to reverse the protective ability of obesity-associated tumorigenic proteins.
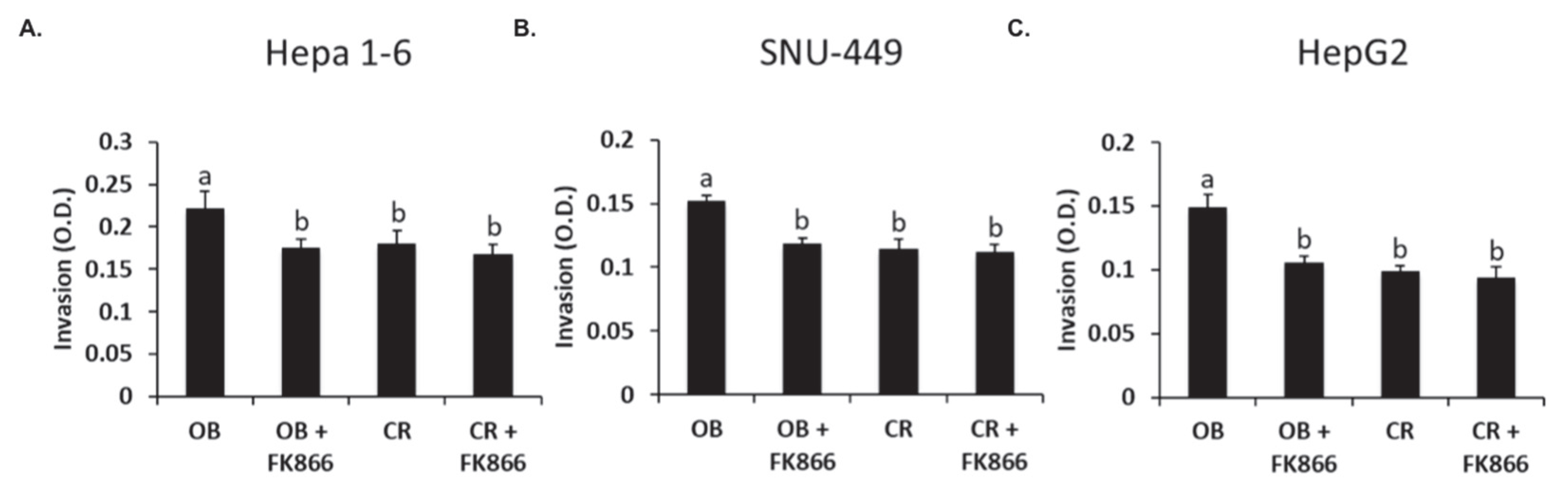
Cellular invasion is a critical step in metastasis, which is the process of cancer cells spreading to other tissues and causing destruction to other body systems. One study shows that the addition of adipocytes to prostate cancer cells increased cell invasion significantly compared with cells that were not treated with adipocytes [
31]. In another study, when MHCC97-H cells were treated with the NAMPT inhibitor FK866, the cells were shown to be significantly less invasive than those without the inhibitor [
24]. Thus, previous research aligns with the results observed in this study, which showed that all cell lines exposed to OB serum had an increase in invasion compared with the control, whereas the addition of FK866 to the OB groups significantly decreased invasion in all three lines. These results suggest that the presence of obesity-associated proteins such as hormones and cytokines may increase the invasive capacity of tumor cells but can be decreased via the inhibition of NAD biosynthesis. In addition to invasion, future studies could investigate the role of obesity-associated hormones in enhancing the metastatic cascade and the role of visfatin in promoting the epithelial to mesenchymal transition, which is a key process in the early stages of invasion.
Erk and Akt are key signaling pathways that are known to be upregulated in both obesity and cancer. In one study, both kinases were shown to be increased in the presence of adipocytes, confirming the results found in this study, where pAkt and pErk protein levels were increased in the presence of obesity-associated hormones and cytokines [
29]. Studies have shown that the upregulation of these kinase pathways has been implicated in obesity-induced cancer progression through stimulating signal cascades that increase cell proliferation, growth, and metabolism [
29]. Another study demonstrated the role that IGF-1, a hormone elevated in obesity, plays in activating downstream Erk and Akt signaling pathways, showing that upregulation of the pathways via IGF-1 led to the promotion of cancer cell invasion and proliferation [
30]. One study demonstrated that the inhibition of IGF-1 signaling reduced Akt and Erk phosphorylation and thus slowed cancer proliferation, though this was correlative [
32]. The results of this study were shown to be consistent with other findings, where inhibition of NAD via FK866 reduced the Akt protein levels in Hepa 1-6. However, the results were not consistent among all three cell lines, indicating that each cell line may have altered pathways associated with tumorigenesis. Obesity-associated hormones activate kinases that can promote downstream transcription factors, such as NF-kB, which leads to the production of more cytokines and proteins that affect invasion and proliferation. The mechanism by which FK866 inhibits the signal mediating these physiological outcomes needs to be explored further. Furthermore, mechanistic studies, a limitation of this study, may also investigate the mechanisms by which NAD inhibition mediates Erk and Akt signaling differently in liver cancer cell lines through proof of principle studies, such as gene silencing or knockdown models.
A high-fat diet has been previously shown to alter the metabolic ability of liver cells, eventually leading to liver cancer progression through increased ROS production and DNA damage. Feeding mice a high-fat diet has been shown to accelerate liver cancer development. This aligns with the results found in this study, where approximately 70% of the mice fed a high-fat diet developed tumors, while only 1% of the mice on a control diet developed tumors. An obesogenic environment promotes tumor growth, evidenced by increased weights and volumes. Importantly, NAD biosynthesis and cellular energetics play a role in obesity-induced growth. This may be a beneficial target, as suppressing this enzyme decreased these effects. These data suggest that an obese environment plays a significant role in the development and sustainability of liver cancer growth.
Elevated expression of the NAMPT enzyme and extracellular NAMPT has been found in the tumor tissues of HCC patients [
33,
34]. Both the enzyme and ligand function of NAMPT play an important role in supplying energy needs within cells. This is relevant to cancer as deregulated cellular energetics is a hallmark of cancer. Upregulated NAMPT in the scavenger pathway is a central contributor to the NAD+ pool, which can supply cancer cells with the ability to meet increased energy needs. However, NAMPT has different roles in various inflammatory-associated diseases related to infection, diabetes, and anti-microbial responses [
34,
35,
36,
37,
38,
39,
40,
41,
42]. Therefore, when considering the suppression of NAMPT as an intervention, it is essential to consider the mechanistic context and tissue-specific effects.
While FK866 has demonstrated promising anti-cancer effects through iNAMPT inhibition and subsequent NAD+ depletion, several studies have reported potential side effects that warrant caution. In a Phase I clinical trial involving cancer patients with advanced solid tumors refractory to standard therapies, thrombocytopenia was identified as a dose-limiting toxicity at higher doses, along with fatigue, nausea, and decreased lymphocyte counts [
18]. Although the authors reported patient side effects to range between mild and moderate [
18], pre-clinical data have identified additional side effects. In healthy cardiomyocytes, FK866 treatment impaired mitochondrial metabolism and stress responses, suggesting potential cardiovascular risks in non-cancerous tissues [
43]. In HCC cells, higher concentrations of FK866 (20–40 nM) were noted to potentially harm normal cells, prompting the authors to utilize lower FK866 doses (0–10 nM) in their research and study design, which is in alignment with the dosage utilized in this study [
24]. Interestingly, FK866 treatment increased ROS activity in cholangiocarcinoma cells [
44], which contrasts with our findings, where FK866 reduced ROS, suggesting that the effects of FK866 on oxidative stress may be cell type-, dosage-, and context-specific. As with many chemotherapeutic or adjuvant interventions, the adverse effects on non-cancerous tissues should be considered when designing future studies. Collectively, these findings demonstrate that FK866 holds therapeutic promise.
The data found in this study contribute to understanding the impact that obesity has on tumorigenesis and possible interventions that may block obesity-induced progression. Still, further analyses need to be performed to understand the role of energy production and cancer development, such as looking at the NAD/NADH ratio to assess FK866’s efficacy in blocking NAD synthesis, MMP-9, and inflammatory markers in tissue and completing serum analysis of the current mouse model. One strength of this study is the novel strategy to break the obesity-liver cancer link through the exploitation of cellular energetics, specifically blocking NAD biosynthesis. Currently, cancer therapeutics primarily focus on inhibition angiogenesis, signal interruption to inhibit cell growth, and promoting cancer cell death through apoptotic induction or through the delivery of toxins, radiation, and other cell-killing substances. Another strength of this study is the use of pooled obese sera to look at the collective effects of combined hormones, though this may also be considered a limitation since it is difficult to assess the specificity of what hormones or enzymes in sera promote pro-tumorigenic effects. In future studies, it may be useful to determine the contribution of individual hormones through the addition of a neutralizing antibody to target specific proteins upregulated in obesity. Finally, while the primary aim of this study was to look at the efficacy of FK866 in an obesity-indued liver cancer model, the mechanistic actions behind FK866 should be elucidated to improve understanding of its role in NAD inhibition.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}