
The Potential Regulators of Amyloidogenic Pathway of APP Processing in Alzheimer’s Disease




Abstract
1. Alzheimer’s Disease
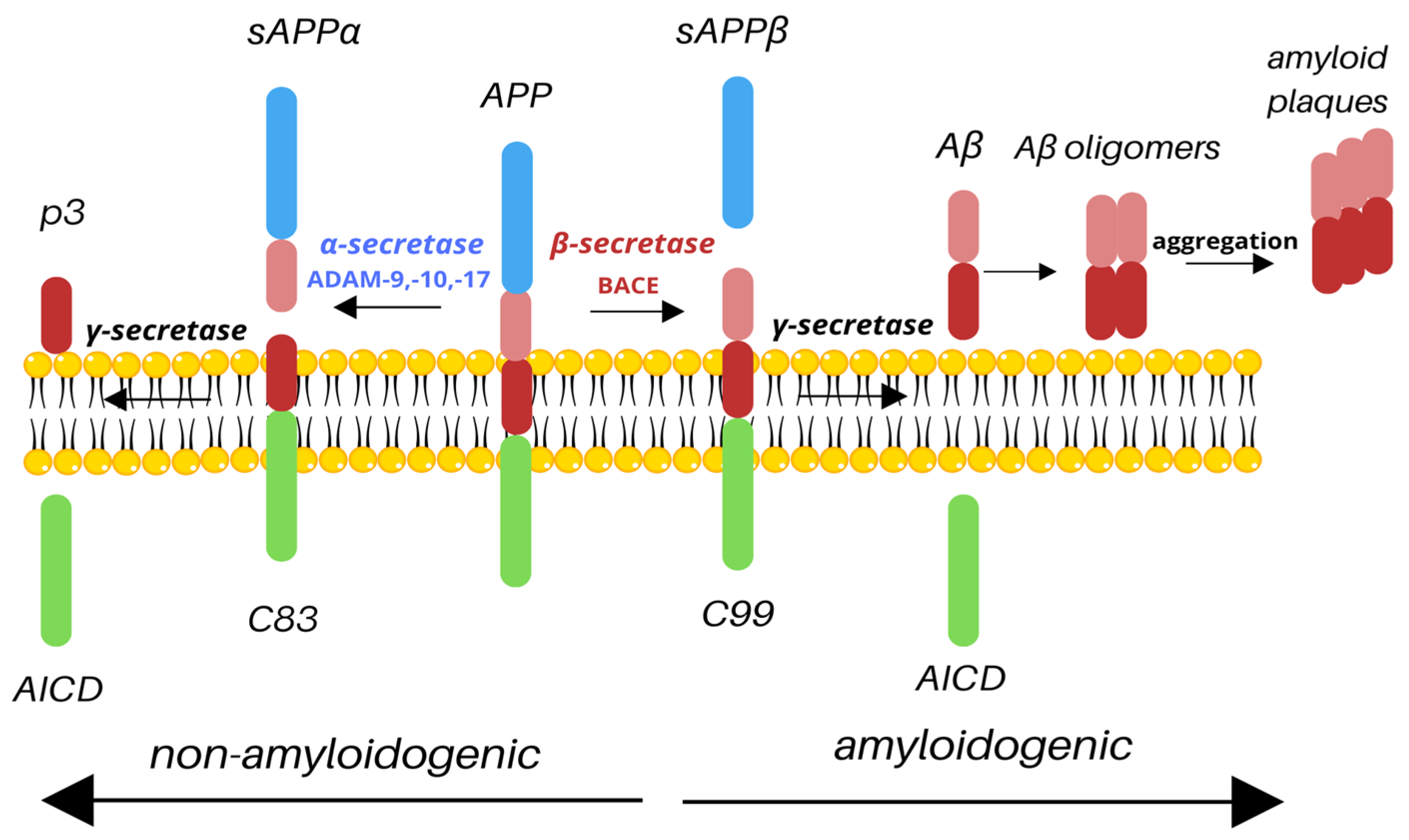
2. APP Processing
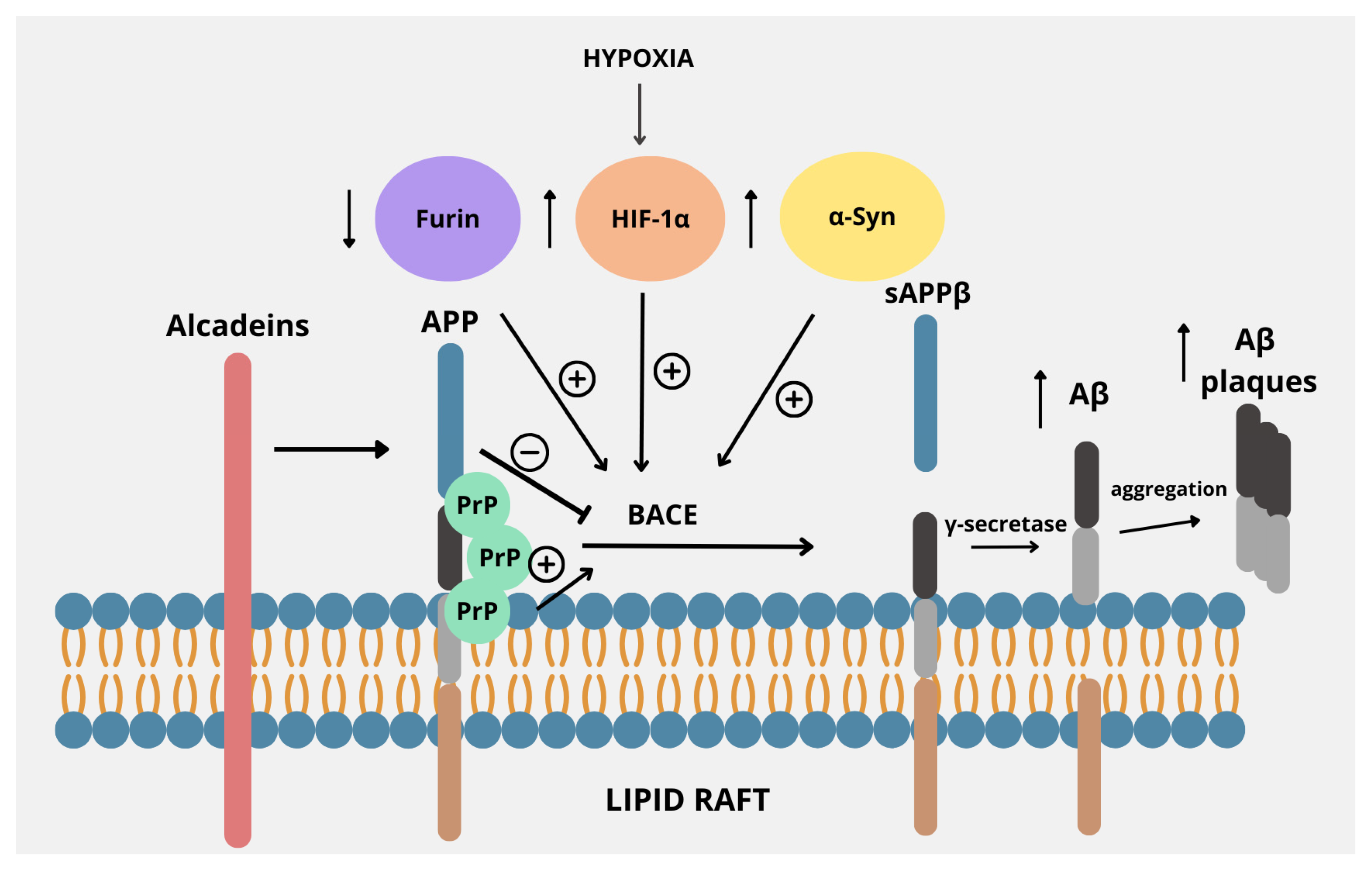
3. Regulators of Amyloidogenic Pathway
3.1. Lipid Rafts
3.2. Alcadeins
3.3. Furin
3.4. Hypoxia-Inducible Factor-1
3.5. Cellular Prion Protein
3.6. α-Synuclein
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wong, W. Economic burden of Alzheimer disease and managed care considerations. Am. J. Manag. Care 2020, 26 (Suppl. 8), S177–S183. [Google Scholar] [CrossRef]
- Rostagno, A.A. Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 107. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.S.; Cummings, J.; Jack, C.R., Jr.; Morris, J.C.; Sperling, R.; Frölich, L.; Jones, R.W.; Dowsett, S.A.; Matthews, B.R.; Raskin, J.; et al. On the path to 2025: Understanding the Alzheimer’s disease continuum. Alzheimer’s Res. Ther. 2017, 9, 60. [Google Scholar] [CrossRef]
- Zvěřová, M. Clinical aspects of Alzheimer’s disease. Clin. Biochem. 2019, 72, 3–6. [Google Scholar] [CrossRef]
- Nasb, M.; Tao, W.; Chen, N. Alzheimer’s Disease Puzzle: Delving into Pathogenesis Hypotheses. Aging Dis. 2024, 15, 43–73. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, H.K.; Jayaraman, V.; Krishna, K.; Wang, T.; Bei, K.; Changalath, C.; Rajasekaran, J.J. An overview of the genes and biomarkers in Alzheimer’s disease. Ageing Res. Rev. 2025, 104, 102599. [Google Scholar] [CrossRef] [PubMed]
- Orobets, K.S.; Karamyshev, A.L. Amyloid Precursor Protein and Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 14794. [Google Scholar] [CrossRef]
- Al-Kuraishy, H.M.; Jabir, M.S.; Al-Gareeb, A.I.; Albuhadily, A.K.; Albukhaty, S.; Sulaiman, G.M.; Batiha, G.E. Evaluation and targeting of amyloid precursor protein (APP)/amyloid beta (Aβ) axis in amyloidogenic and non-amyloidogenic pathways: A time outside the tunnel. Ageing Res. Rev. 2023, 92, 102119. [Google Scholar] [CrossRef]
- Young-Pearse, T.L.; Chen, A.C.; Chang, R.; Marquez, C.; Selkoe, D.J. Secreted APP regulates the function of full-length APP in neurite outgrowth through interaction with integrin β1. Neural Dev. 2008, 3, 15. [Google Scholar] [CrossRef]
- Hefter, D.; Draguhn, A. APP as a Protective Factor in Acute Neuronal Insults. Front. Mol. Neurosci. 2017, 10, 22. [Google Scholar] [CrossRef]
- Obregon, D.; Hou, H.; Deng, J.; Giunta, B.; Tian, J.; Darlington, D.; Shahaduzzaman, M.; Zhu, Y.; Mori, T.; Mattson, M.P.; et al. Soluble amyloid precursor protein-α modulates β-secretase activity and amyloid-β generation. Nat. Commun. 2012, 3, 777. [Google Scholar] [CrossRef] [PubMed]
- Taylor, H.A.; Przemylska, L.; Clavane, E.M.; Meakin, P.J. BACE1: More than just a β-secretase. Obes. Rev. 2022, 23, e13430. [Google Scholar] [CrossRef] [PubMed]
- Kamenetz, F.; Tomita, T.; Hsieh, H.; Seabrook, G.; Borchelt, D.; Iwatsubo, T.; Sisodia, S.; Malinow, R. APP processing and synaptic function. Neuron 2003, 37, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Song, M.; Liu, X.; Su Kang, S.; Duong, D.M.; Seyfried, N.T.; Cao, X.; Cheng, L.; Sun, Y.E.; Ping Yu, S.; et al. Delta-secretase cleaves amyloid precursor protein and regulates the pathogenesis in Alzheimer’s disease. Nat. Commun. 2015, 6, 8762. [Google Scholar] [CrossRef]
- Wang, S.S.; Liu, Z.K.; Liu, J.J.; Cheng, Q.; Wang, Y.X.; Liu, Y.; Ni, W.W.; Chen, H.Z.; Song, M. Imaging asparaginyl endopeptidase (AEP) in the live brain as a biomarker for Alzheimer’s disease. J. Nanobiotechnol. 2021, 19, 249. [Google Scholar] [CrossRef]
- Afram, E.; Lauritzen, I.; Bourgeois, A.; El Manaa, W.; Duplan, E.; Chami, M.; Valverde, A.; Charlotte, B.; Pardossi-Piquard, R.; Checler, F. The η-secretase-derived APP fragment ηCTF is localized in Golgi, endosomes and extracellular vesicles and contributes to Aβ production. Cell. Mol. Life Sci. 2023, 80, 97. [Google Scholar] [CrossRef]
- Willem, M.; Tahirovic, S.; Busche, M.A.; Ovsepian, S.V.; Chafai, M.; Kootar, S.; Hornburg, D.; Evans, L.D.; Moore, S.; Daria, A.; et al. η-Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 2015, 526, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Pilat, D.; Paumier, J.M.; Louis, L.; Manrique, C.; García-González, L.; Stephan, D.; Bernard, A.; Pardossi-Piquard, R.; Checler, F.; Khrestchatisky, M.; et al. Suppression of MT5-MMP Reveals Early Modulation of Alzheimer’s Pathogenic Events in Primary Neuronal Cultures of 5xFAD Mice. Biomolecules 2024, 14, 1645. [Google Scholar] [CrossRef]
- Fu, J.; Lai, X.; Huang, Y.; Bao, T.; Yang, J.; Chen, S.; Chen, X.; Shang, H. Meta-analysis and systematic review of peripheral platelet-associated biomarkers to explore the pathophysiology of Alzheimer’s disease. BMC Neurol. 2023, 23, 66. [Google Scholar] [CrossRef]
- Li, T.R.; Liu, F.Q. β-Amyloid promotes platelet activation and activated platelets act as bridge between risk factors and Alzheimer’s disease. Mech. Ageing Dev. 2022, 207, 111725. [Google Scholar] [CrossRef]
- Al-Kuraishy, H.M.; Sulaiman, G.M.; Mohammed, H.A.; Dawood, R.A.; Albuhadily, A.K.; Al-Gareeb, A.I.; Abomughaid, M.M.; Klionsky, D.J. Alterations in the Processing of Platelet APP (Amyloid Beta Precursor Protein) in Alzheimer Disease: The Possible Nexus. Neuropsychopharmacol. Rep. 2025, 45, e12525. [Google Scholar] [CrossRef]
- Ramos-Cejudo, J.; Johnson, A.D.; Beiser, A.; Seshadri, S.; Salinas, J.; Berger, J.S.; Fillmore, N.R.; Do, N.; Zheng, C.; Kovbasyuk, Z.; et al. Platelet Function Is Associated with Dementia Risk in the Framingham Heart Study. J. Am. Heart Assoc. 2022, 11, e023918. [Google Scholar] [CrossRef] [PubMed]
- Burrinha, T.; Guimas Almeida, C. Aging impact on amyloid precursor protein neuronal trafficking. Curr. Opin. Neurobiol. 2022, 73, 102524. [Google Scholar] [CrossRef]
- Sasmita, A.O.; Depp, C.; Nazarenko, T.; Sun, T.; Siems, S.B.; Ong, E.C.; Nkeh, Y.B.; Böhler, C.; Yu, X.; Bues, B.; et al. Oligodendrocytes produce amyloid-β and contribute to plaque formation alongside neurons in Alzheimer’s disease model mice. Nat. Neurosci. 2024, 27, 1668–1674. [Google Scholar] [CrossRef]
- Wessels, A.M.; Lines, C.; Stern, R.A.; Kost, J.; Voss, T.; Mozley, L.H.; Furtek, C.; Mukai, Y.; Aisen, P.S.; Cummings, J.L.; et al. Cognitive outcomes in trials of two BACE inhibitors in Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Dunot, J.; Ribera, A.; Pousinha, P.A.; Marie, H. Spatiotemporal insights of APP function. Curr. Opin. Neurobiol. 2023, 82, 102754. [Google Scholar] [CrossRef]
- Kao, Y.C.; Ho, P.C.; Tu, Y.K.; Jou, I.M.; Tsai, K.J. Lipids and Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1505. [Google Scholar] [CrossRef] [PubMed]
- Arbor, S.C.; LaFontaine, M.; Cumbay, M. Amyloid-beta Alzheimer targets—Protein processing, lipid rafts, and amyloid-beta pores. Yale J. Biol. Med. 2016, 89, 5–21. [Google Scholar]
- Cheng, H.; Vetrivel, K.S.; Gong, P.; Meckler, X.; Parent, A.; Thinakaran, G. Mechanisms of disease: New therapeutic strategies for Alzheimer’s disease—Targeting APP processing in lipid rafts. Nat. Clin. Pract. Neurol. 2007, 3, 374–382. [Google Scholar] [CrossRef]
- Cordy, J.M.; Hussain, I.; Dingwall, C.; Hooper, N.M.; Turner, A.J. Exclusively targeting β-secretase to lipid rafts by GPI-anchor addition up-regulates β-site processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2003, 100, 11735–11740. [Google Scholar] [CrossRef]
- Ehehalt, R.; Keller, P.; Haass, C.; Thiele, C.; Simons, K. Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 2003, 160, 113–123. [Google Scholar] [CrossRef]
- Díaz, M.; Fabelo, N.; Martín, V.; Ferrer, I.; Gómez, T.; Marín, R. Biophysical alterations in lipid rafts from human cerebral cortex associate with increased BACE1/AβPP interaction in early stages of Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 43, 1185–1198. [Google Scholar] [CrossRef] [PubMed]
- Fabelo, N.; Martín, V.; Marín, R.; Moreno, D.; Ferrer, I.; Díaz, M. Altered lipid composition in cortical lipid rafts occurs at early stages of sporadic Alzheimer’s disease and facilitates APP/BACE1 interactions. Neurobiol. Aging 2014, 35, 1801–1812. [Google Scholar] [CrossRef]
- Blaskovic, S.; Blanc, M.; van der Goot, F.G. What does S-palmitoylation do to membrane proteins? FEBS J. 2013, 280, 2766–2774. [Google Scholar] [CrossRef] [PubMed]
- Smotrys, J.E.; Linder, M.E. Palmitoylation of intracellular signaling proteins: Regulation and function. Annu. Rev. Biochem. 2004, 73, 559–587. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Vetrivel, K.S.; Drisdel, R.C.; Meckler, X.; Gong, P.; Leem, J.Y.; Li, T.; Carter, M.; Chen, Y.; Nguyen, P.; et al. S-palmitoylation of γ-secretase subunits nicastrin and APH-1. J. Biol. Chem. 2009, 284, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Um, J.W.; Pramanik, G.; Ko, J.S.; Song, M.Y.; Lee, D.; Kim, H.; Park, K.S.; Südhof, T.C.; Tabuchi, K.; Ko, J. Calsyntenins function as synaptogenic adhesion molecules in concert with neurexins. Cell Rep. 2014, 6, 1096–1109. [Google Scholar] [CrossRef]
- Piao, Y.; Kimura, A.; Urano, S.; Saito, Y.; Taru, H.; Yamamoto, T.; Hata, S.; Suzuki, T. Mechanism of intramembrane cleavage of alcadeins by γ-secretase. PLoS ONE 2013, 8, e62431. [Google Scholar] [CrossRef]
- Motodate, R.; Saito, Y.; Hata, S.; Suzuki, T. Expression and localization of X11 family proteins in neurons. Brain Res. 2016, 1646, 227–234. [Google Scholar] [CrossRef]
- Honda, K.; Takahashi, H.; Hata, S.; Abe, R.; Saito, T.; Saido, T.C.; Taru, H.; Sobu, Y.; Ando, K.; Yamamoto, T.; et al. Suppression of the amyloidogenic metabolism of APP and the accumulation of Aβ by alcadein α in the brain during aging. Sci. Rep. 2024, 14, 18471. [Google Scholar] [CrossRef]
- Hata, S.; Saito, H.; Kakiuchi, T.; Fukumoto, D.; Yamamoto, S.; Kasuga, K.; Kimura, A.; Moteki, K.; Abe, R.; Adachi, S.; et al. Brain p3-Alcβ peptide restores neuronal viability impaired by Alzheimer’s amyloid β-peptide. EMBO Mol. Med. 2023, 15, e17052. [Google Scholar] [CrossRef] [PubMed]
- Hata, S.; Omori, C.; Kimura, A.; Saito, H.; Kimura, N.; Gupta, V.; Pedrini, S.; Hone, E.; Chatterjee, P.; Taddei, K.; et al. Decrease in p3-Alcβ37 and p3-Alcβ40, products of Alcadein β generated by γ-secretase cleavages, in aged monkeys and patients with Alzheimer’s disease. Alzheimer’s Dement. 2019, 5, 740–750. [Google Scholar] [CrossRef] [PubMed]
- Hata, S.; Fujishige, S.; Araki, Y.; Kato, N.; Araseki, M.; Nishimura, M.; Hartmann, D.; Saftig, P.; Fahrenholz, F.; Taniguchi, M.; et al. Alcadein cleavages by amyloid β-precursor protein (APP) α- and γ-secretases generate small peptides, p3-Alcs, indicating Alzheimer disease-related γ-secretase dysfunction. J. Biol. Chem. 2009, 284, 36024–36033. [Google Scholar] [CrossRef]
- Kamogawa, K.; Kohara, K.; Tabara, Y.; Takita, R.; Miki, T.; Konno, T.; Hata, S.; Suzuki, T. Potential utility of soluble p3-alcadein α plasma levels as a biomarker for sporadic Alzheimer’s disease. J. Alzheimer’s Dis. 2012, 31, 421–428. [Google Scholar] [CrossRef]
- Omori, C.; Kaneko, M.; Nakajima, E.; Akatsu, H.; Waragai, M.; Maeda, M.; Morishima-Kawashima, M.; Saito, Y.; Nakaya, T.; Taru, H.; et al. Japanese Alzheimer’s Disease Neuroimaging Initiative. Increased levels of plasma p3-alcα35, a major fragment of Alcadeinα by γ-secretase cleavage, in Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 39, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Konno, T.; Hata, S.; Hamada, Y.; Horikoshi-Sakuraba, Y.; Nakaya, T.; Saito, Y.; Yamamoto, T.; Yamamoto, T.; Maeda, M.; Ikeuchi, T.; et al. Japanese Alzheimer’s Disease Neuroimaging Initiative. Coordinated increase of γ-secretase reaction products in the plasma of some female Japanese sporadic Alzheimer’s disease patients: Quantitative analysis of p3-Alcα with a new ELISA system. Mol. Neurodegener. 2011, 6, 76. [Google Scholar] [CrossRef]
- Thomas, G. Furin at the cutting edge: From protein traffic to embryogenesis and disease. Nat. Rev. Mol. Cell Biol. 2002, 3, 753–766. [Google Scholar] [CrossRef]
- Bennett, B.D.; Denis, P.; Haniu, M.; Teplow, D.B.; Kahn, S.; Louis, J.C.; Citron, M.; Vassar, R. A furin-like convertase mediates propeptide cleavage of BACE, the Alzheimer’s β-secretase. J. Biol. Chem. 2000, 275, 37712–37717. [Google Scholar] [CrossRef]
- Hwang, E.M.; Kim, S.K.; Sohn, J.H.; Lee, J.Y.; Kim, Y.; Kim, Y.S.; Mook-Jung, I. Furin is an endogenous regulator of α-secretase associated APP processing. Biochem. Biophys. Res. Commun. 2006, 349, 654–659. [Google Scholar] [CrossRef]
- Yang, Y.; He, M.; Tian, X.; Guo, Y.; Liu, F.; Li, Y.; Zhang, H.; Lu, X.; Xu, D.; Zhou, R.; et al. Transgenic overexpression of furin increases epileptic susceptibility. Cell Death Dis. 2018, 9, 1058. [Google Scholar] [CrossRef]
- Li, J.; Ding, Y.; Zhang, J.; Zhang, Y.; Cui, Y.; Zhang, Y.; Chang, S.; Chang, Y.Z.; Gao, G. Iron overload suppresses hippocampal neurogenesis in adult mice: Implication for iron dysregulation-linked neurological diseases. CNS Neurosci. Ther. 2024, 30, e14394. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.H.; Kwon, K.C.; Hwang, D.J.; Koo, J.H.; Um, H.S.; Song, H.S.; Kim, J.S.; Jang, Y.; Cho, J.Y. Treadmill Exercise Alleviates Brain Iron Dyshomeostasis Accelerating Neuronal Amyloid-β Production, Neuronal Cell Death, and Cognitive Impairment in Transgenic Mice Model of Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 3208–3223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bai, X.; Zhang, Y.; Yao, S.; Cui, Y.; You, L.H.; Yu, P.; Chang, Y.Z.; Gao, G. Hippocampal Iron Accumulation Impairs Synapses and Memory via Suppressing Furin Expression and Downregulating BDNF Maturation. Mol. Neurobiol. 2022, 59, 5574–5590. [Google Scholar] [CrossRef] [PubMed]
- Adeyemi, O.S.; Awakan, O.J.; Afolabi, L.B.; Rotimi, D.E.; Oluwayemi, E.; Otuechere, C.A.; Ibraheem, O.; Elebiyo, T.C.; Alejolowo, O.; Arowolo, A.T. Hypoxia and the Kynurenine Pathway: Implications and Therapeutic Prospects in Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2021, 2021, 5522981. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Huang, Z.T.; Yuan, M.H.; Jing, F.; Cai, R.L.; Zou, Q.; Pu, Y.S.; Wang, S.Y.; Chen, F.; Yi, W.M.; et al. Role of Hypoxia Inducible Factor-1α in Alzheimer’s Disease. J. Alzheimers Dis. 2021, 80, 949–961. [Google Scholar] [CrossRef]
- Liu, G.; Yang, C.; Wang, X.; Chen, X.; Wang, Y.; Le, W. Oxygen metabolism abnormality and Alzheimer’s disease: An update. Redox Biol. 2023, 68, 102955. [Google Scholar] [CrossRef]
- Tao, B.; Gong, W.; Xu, C.; Ma, Z.; Mei, J.; Chen, M. The relationship between hypoxia and Alzheimer’s disease: An updated review. Front. Aging Neurosci. 2024, 16, 1402774. [Google Scholar] [CrossRef]
- Elman-Shina, K.; Efrati, S. Ischemia as a common trigger for Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 1012779. [Google Scholar] [CrossRef]
- Desmond, D.W.; Moroney, J.T.; Sano, M.; Stern, Y. Incidence of dementia after ischemic stroke: Results of a longitudinal study. Stroke 2002, 33, 2254–2260. [Google Scholar] [CrossRef]
- Tarkowska, A. Hypoxic-Ischemic Brain Injury after Perinatal Asphyxia as a Possible Factor in the Pathology of Alzheimer’s Disease. In Cerebral Ischemia; Exon Publications: Brisbane, QLD, Australia, 2021; pp. 45–60. [Google Scholar]
- Sun, M.; Zhou, T.; Zhou, L.; Chen, Q.; Yu, Y.; Yang, H.; Zhong, K.; Zhang, X.; Xu, F.; Cai, S.; et al. Formononetin protects neurons against hypoxia-induced cytotoxicity through upregulation of ADAM10 and sAβPPα. J. Alzheimer’s Dis. 2012, 28, 795–808. [Google Scholar] [CrossRef]
- Auerbach, I.D.; Vinters, H.V. Effects of anoxia and hypoxia on amyloid precursor protein processing in cerebral microvascular smooth muscle cells. J. Neuropathol. Exp. Neurol. 2006, 65, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Lall, R.; Mohammed, R.; Ojha, U. What are the links between hypoxia and Alzheimer’s disease? Neuropsychiatr. Dis. Treat. 2019, 15, 1343–1354. [Google Scholar] [CrossRef]
- Guglielmotto, M.; Aragno, M.; Autelli, R.; Giliberto, L.; Novo, E.; Colombatto, S.; Danni, O.; Parola, M.; Smith, M.A.; Perry, G.; et al. The up regulation of BACE1 mediated by hypoxia and ischemic injury: Role of oxidative stress and HIF1α. J. Neurochem. 2009, 108, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, K.; Wang, R.; Cui, J.; Lipton, S.A.; Liao, F.F.; Xu, H.; Zhang, Y.W. Hypoxia-inducible factor 1α (HIF-1α)-mediated hypoxia increases BACE1 expression and β-amyloid generation. J. Biol. Chem. 2007, 282, 10873–10880. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C.; Li, T.; Hattori, Y.; Chiu, D.; Frost, G.R.; Jonas, L.; Liu, C.; Anderson, C.J.; Wong, E.; Park, L.; et al. Hypoxia Inducible Factor-1α binds and activates γ-secretase for Aβ production under hypoxia and cerebral hypoperfusion. Mol. Psychiatry 2022, 27, 4264–4273. [Google Scholar] [CrossRef]
- Chrobak, A.A.; Adamek, D. New light on prions: Putative role of co-operation of PrPc and Aβ proteins in cognition. Folia Neuropathol. 2014, 52, 1–9. [Google Scholar] [CrossRef]
- Fułek, M.; Hachiya, N.; Gachowska, M.; Beszłej, J.A.; Bartoszewska, E.; Kurpas, D.; Kurpiński, T.; Adamska, H.; Poręba, R.; Urban, S.; et al. Cellular Prion Protein and Amyloid-β Oligomers in Alzheimer’s Disease—Are There Connections? Int. J. Mol. Sci. 2025, 26, 2097. [Google Scholar] [CrossRef]
- Del Bo, R.; Scarlato, M.; Ghezzi, S.; Martinelli-Boneschi, F.; Fenoglio, C.; Galimberti, G.; Galbiati, S.; Virgilio, R.; Galimberti, D.; Ferrarese, C.; et al. Is M129V of PRNP gene associated with Alzheimer’s disease? A case-control study and a meta-analysis. Neurobiol. Aging 2006, 27, 770.e1–770.e5. [Google Scholar] [CrossRef]
- Whitehouse, I.J.; Miners, J.S.; Glennon, E.B.; Kehoe, P.G.; Love, S.; Kellett, K.A.; Hooper, N.M. Prion protein is decreased in Alzheimer’s brain and inversely correlates with BACE1 activity, amyloid-β levels and Braak stage. PLoS ONE 2013, 8, e59554. [Google Scholar] [CrossRef]
- Parkin, E.T.; Watt, N.T.; Hussain, I.; Eckman, E.A.; Eckman, C.B.; Manson, J.C.; Baybutt, H.N.; Turner, A.J.; Hooper, N.M. Cellular prion protein regulates β-secretase cleavage of the Alzheimer’s amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2007, 104, 11062–11067. [Google Scholar] [CrossRef]
- Chung, E.; Ji, Y.; Sun, Y.; Kascsak, R.J.; Kascsak, R.B.; Mehta, P.D.; Strittmatter, S.M.; Wisniewski, T. Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer’s disease model mouse. BMC Neurosci. 2010, 11, 130. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Südhof, T.C. Cell Biology and Pathophysiology of α-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.F.; George, J.M. The synucleins: A family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998, 21, 249–254. [Google Scholar] [CrossRef]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Sagara, Y.; Mallory, M.; Hashimoto, M.; Mucke, L. β-Amyloid peptides enhance α-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 12245–12250. [Google Scholar] [CrossRef] [PubMed]
- Roberts, H.L.; Schneider, B.L.; Brown, D.R. α-Synuclein increases β-amyloid secretion by promoting β-/γ-secretase processing of APP. PLoS ONE 2017, 12, e0171925. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, X.; Xia, W.; Zhang, Y.; Wang, C. Targeting Amyloidogenic Processing of APP in Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 137. [Google Scholar] [CrossRef] [PubMed]
- Kalvodova, L.; Kahya, N.; Schwille, P.; Ehehalt, R.; Verkade, P.; Drechsel, D.; Simons, K. Lipids as modulators of proteolytic activity of BACE: Involvement of cholesterol, glycosphingolipids, and anionic phospholipids In Vitro. J. Biol. Chem. 2005, 280, 36815–36823. [Google Scholar] [CrossRef]
- Gotoh, N.; Saito, Y.; Hata, S.; Saito, H.; Ojima, D.; Murayama, C.; Shigeta, M.; Abe, T.; Konno, D.; Matsuzaki, F.; et al. Amyloidogenic processing of amyloid β protein precursor (APP) is enhanced in the brains of alcadein α-deficient mice. J. Biol. Chem. 2020, 295, 9650–9662. [Google Scholar] [CrossRef]
- Lin, T.-K.; Huang, C.-R.; Lin, K.-J.; Hsieh, Y.-H.; Chen, S.-D.; Lin, Y.-C.; Chao, A.-C.; Yang, D.-I. Potential Roles of Hypoxia-Inducible Factor-1 in Alzheimer’s Disease: Beneficial or Detrimental? Antioxidants 2024, 13, 1378. [Google Scholar] [CrossRef]
- Kostylev, M.A.; Kaufman, A.C.; Nygaard, H.B.; Patel, P.; Haas, L.T.; Gunther, E.C.; Vortmeyer, A.; Strittmatter, S.M. Prion-Protein-interacting Amyloid-β Oligomers of High Molecular Weight Are Tightly Correlated with Memory Impairment in Multiple Alzheimer Mouse Models. J. Biol. Chem. 2015, 290, 17415–17438. [Google Scholar] [CrossRef]
- Barry, A.E.; Klyubin, I.; Mc Donald, J.M.; Mably, A.J.; Farrell, M.A.; Scott, M.; Walsh, D.M.; Rowan, M.J. Alzheimer’s disease brain-derived amyloid-β-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J. Neurosci. 2011, 31, 7259–7263. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Leitão, A.D.G.; Fang, S.; Gu, Y.; Barber, S.; Gilliard-Telefoni, R.; Castro, A.; Sung, K.; Shen, R.; Florio, J.B.; et al. Overexpression of alpha synuclein disrupts APP and Endolysosomal axonal trafficking in a mouse model of synucleinopathy. Neurobiol. Dis. 2023, 178, 106010. [Google Scholar] [CrossRef]
- Cao, Y.; Zhao, L.W.; Chen, Z.X.; Li, S.H. New insights in lipid metabolism: Potential therapeutic targets for the treatment of Alz-heimer’s disease. Front. Neurosci. 2024, 18, 1430465. [Google Scholar] [CrossRef]
- Marin, R.; Rojo, J.A.; Fabelo, N.; Fernandez, C.E.; Diaz, M. Lipid raft disarrangement as a result of neuro-pathological progresses: A novel strategy for early diagnosis? Neuroscience 2013, 245, 26–39. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, X.; Bai, X.; Yao, S.; Chang, Y.Z.; Gao, G. The emerging role of furin in neurodegenerative and neuropsychiatric diseases. Transl. Neurodegener. 2022, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Porel, P.; Bala, K.; Aran, K.R. Exploring the role of HIF-1α on pathogenesis in Alzheimer’s disease and potential therapeutic approaches. Inflammopharmacology 2025, 33, 669–678. [Google Scholar] [CrossRef]
- Laurén, J. Cellular prion protein as a therapeutic target in Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 38, 227–244. [Google Scholar] [CrossRef] [PubMed]
- Jácome, D.; Cotrufo, T.; Andrés-Benito, P.; Lidón, L.; Martí, E.; Ferrer, I.; Del Río, J.A.; Gavín, R. miR-519a-3p, found to regulate cellular prion protein during Alzheimer’s disease pathogenesis, as a biomarker of asymptomatic stages. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2024, 1870, 167187. [Google Scholar] [CrossRef]
- Das, B.; Yan, R. A Close Look at BACE1 Inhibitors for Alzheimer’s Disease Treatment. CNS Drugs 2019, 33, 251–263. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Protein | Group | Mean Value | Method | Source |
|---|---|---|---|---|
| CSF | ||||
| p3-Alcβ37 | Patients along the AD continuum | ELISA | [41] | |
| A− | 3928 pg/mL | |||
| A+T−N− | 1889 pg/mL | |||
| A+T+N− | 2899 pg/mL | |||
| A+T+N+ | 3808 pg/mL | |||
| p3-Alcβ37 | I cohort: | ELISA | [42] | |
| controls vs. AD | 9394 pg/mL vs. 6614 pg/mL | |||
| controls vs. MCI | 9394 pg/mL vs. 9596 pg/mL | |||
| MCI vs. AD | 9596 pg/mL vs. 6614 pg/mL | |||
| II cohort: | ||||
| controls vs. AD | 6421 pg/mL vs. 4780 pg/mL | |||
| controls vs. MCI | 6421 pg/mL vs. 6296 pg/mL | |||
| MCI vs. AD | 6296 pg/mL vs. 4780 pg/mL | |||
| p3-Alcβ40 | I cohort: | ELISA | [42] | |
| controls vs. AD | 1212 pg/mL vs. 884.1 pg/mL | |||
| controls vs. MCI | 1212 pg/mL vs. 1177 pg/mL | |||
| MCI vs. AD | 1177 pg/mL vs. 884.1 pg/mL | |||
| II cohort: | ||||
| controls vs. AD | 1032 pg/mL vs. 931.5 pg/mL | |||
| controls vs. MCI | 1032 pg/mL vs. 1065 pg/mL | |||
| MCI vs. AD | 1065 pg/mL vs. 931.5 pg/mL | |||
| III cohort: | ||||
| controls vs. AD | 651.4 pg/mL vs. 509.8 pg/mL | |||
| controls vs. MCI | 509.8 pg/mL vs. 544.1 pg/mL | |||
| MCI vs. AD | 544.1 pg/mL vs. 509.8 pg/mL | |||
| Plasma | ||||
| p3-Alcα35 | controls vs. AD | 189.1 pg/mL vs. 224.7 pg/mL | ELISA | [44] |
| controls vs. MCI | 189.1 pg/mL vs. 223.3 pg/mL | |||
| MCI vs. AD | 223.3 pg/mL vs. 224.7 pg/mL | |||
| p3-Alcα35 | I cohort: | ELISA | [45] | |
| controls vs. AD | 140.1 pg/mL vs. 178.7 pg/mL | |||
| controls vs. MCI | 140.1 pg/mL vs. 166.3 pg/mL | |||
| MCI vs. AD | 166.3 pg/mL vs. 178.7 pg/mL | |||
| II cohort: | ||||
| controls vs. AD | 148 pg/mL vs. 196.5 pg/mL | |||
| controls vs. MCI | 148 pg/mL vs. 166.4 pg/mL | |||
| MCI vs. AD | 166.4 pg/mL vs. 196.5 pg/mL | |||
| III cohort: | ||||
| controls vs. AD | 128.6 pg/mL vs. 130.8 pg/mL | |||
| controls vs. MCI | 128.6 pg/mL vs. 146.0 pg/mL | |||
| MCI vs. AD | 146.0 pg/mL vs. 130.8 pg/mL | |||
| total p3-Alcα | controls vs. AD | 163 pg/mL vs. 232 pg/mL | ELISA | [46] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krawczuk, D.; Kulczyńska-Przybik, A.; Mroczko, B. The Potential Regulators of Amyloidogenic Pathway of APP Processing in Alzheimer’s Disease. Biomedicines 2025, 13, 1513. https://doi.org/10.3390/biomedicines13071513
Krawczuk D, Kulczyńska-Przybik A, Mroczko B. The Potential Regulators of Amyloidogenic Pathway of APP Processing in Alzheimer’s Disease. Biomedicines. 2025; 13(7):1513. https://doi.org/10.3390/biomedicines13071513
Chicago/Turabian StyleKrawczuk, Daria, Agnieszka Kulczyńska-Przybik, and Barbara Mroczko. 2025. "The Potential Regulators of Amyloidogenic Pathway of APP Processing in Alzheimer’s Disease" Biomedicines 13, no. 7: 1513. https://doi.org/10.3390/biomedicines13071513
APA StyleKrawczuk, D., Kulczyńska-Przybik, A., & Mroczko, B. (2025). The Potential Regulators of Amyloidogenic Pathway of APP Processing in Alzheimer’s Disease. Biomedicines, 13(7), 1513. https://doi.org/10.3390/biomedicines13071513