
Regulation of Stemness by NR1D2 in Colorectal Cancer

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Analysis of p53 Mutations in CRC Samples
2.3. RNA Extraction and qPCR
2.4. Cell Culture and Reagents
2.5. Transfection Protocol
2.6. Immunoblotting
2.7. Cell Cycle Analysis
2.8. Apoptosis Assay
2.9. Aldefluor Assay and Cell Surface Markers Analysis
2.10. MTT Assay
2.11. Clonogenic Assay
2.12. Spheres Formation Assay
2.13. Statistical Analysis
3. Results
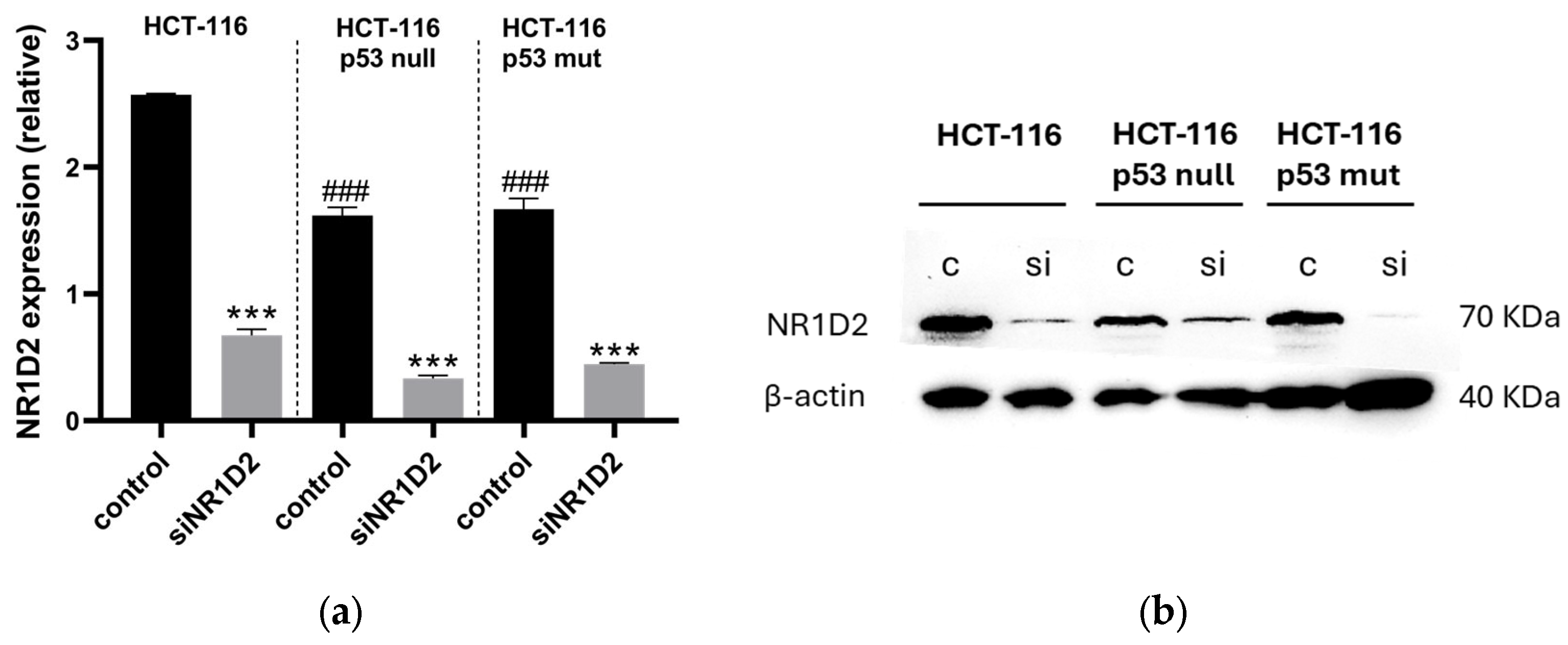
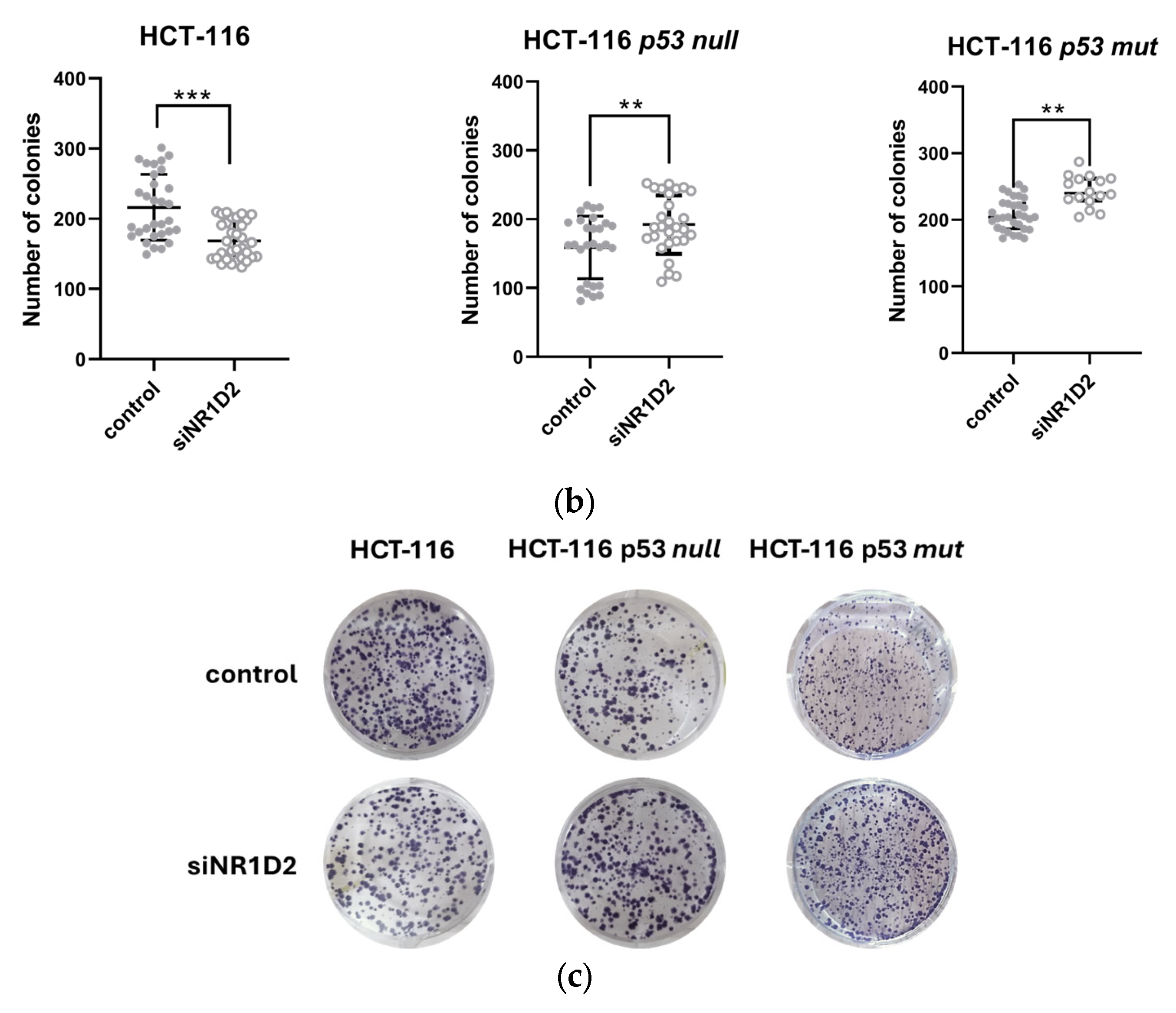
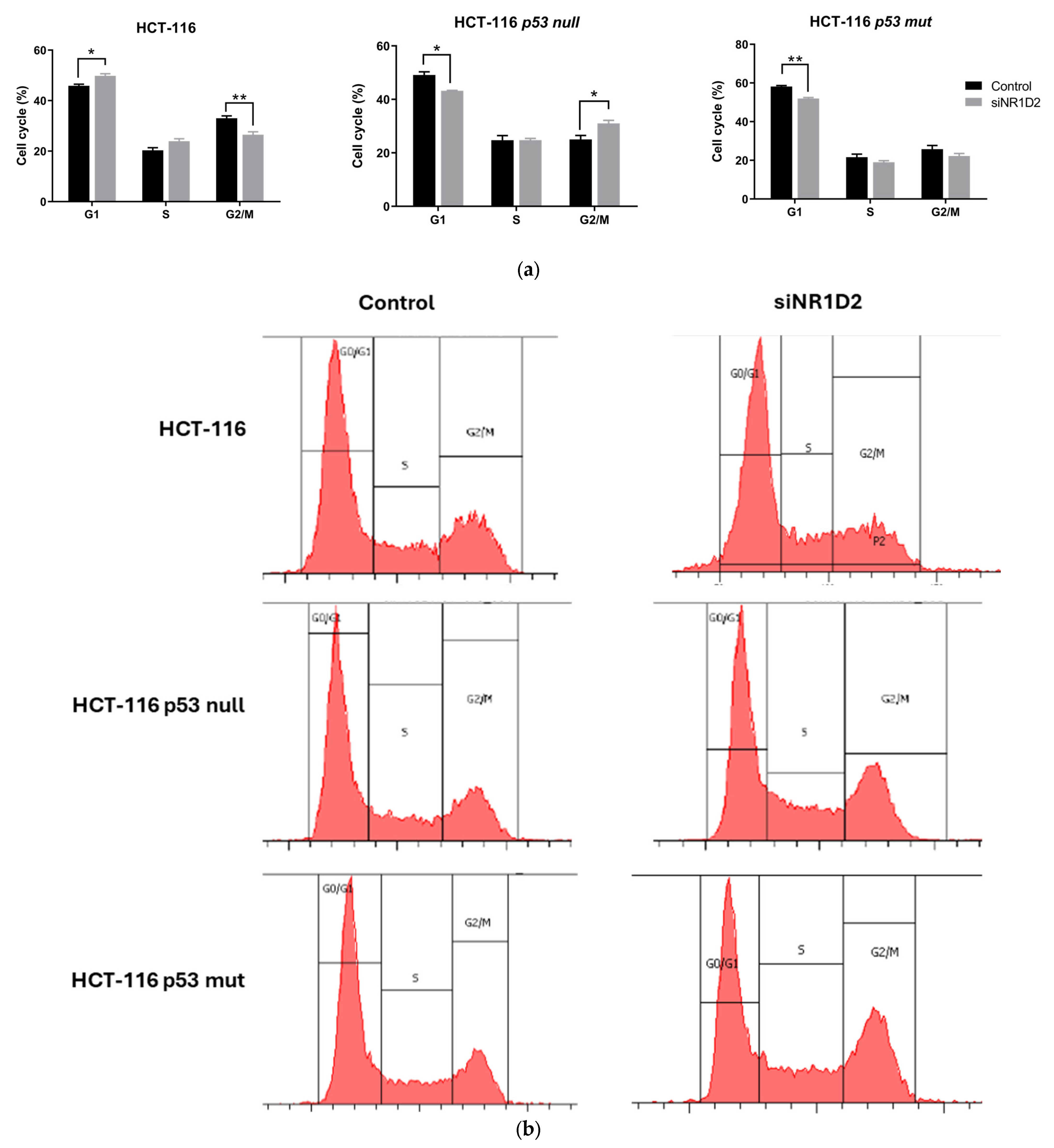
3.1. NR1D2 Regulates Growth and Apoptosis of CRC Cells
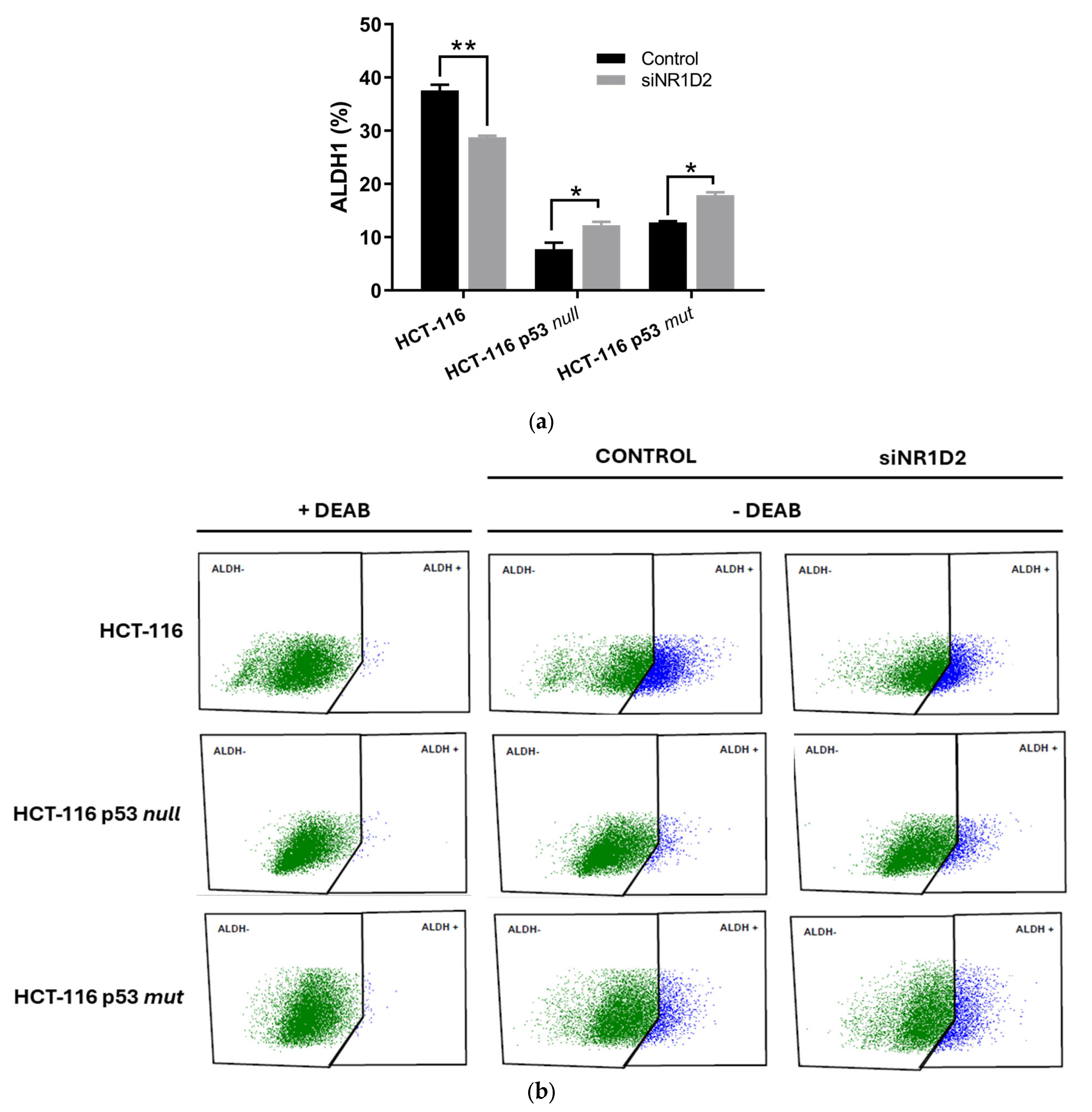
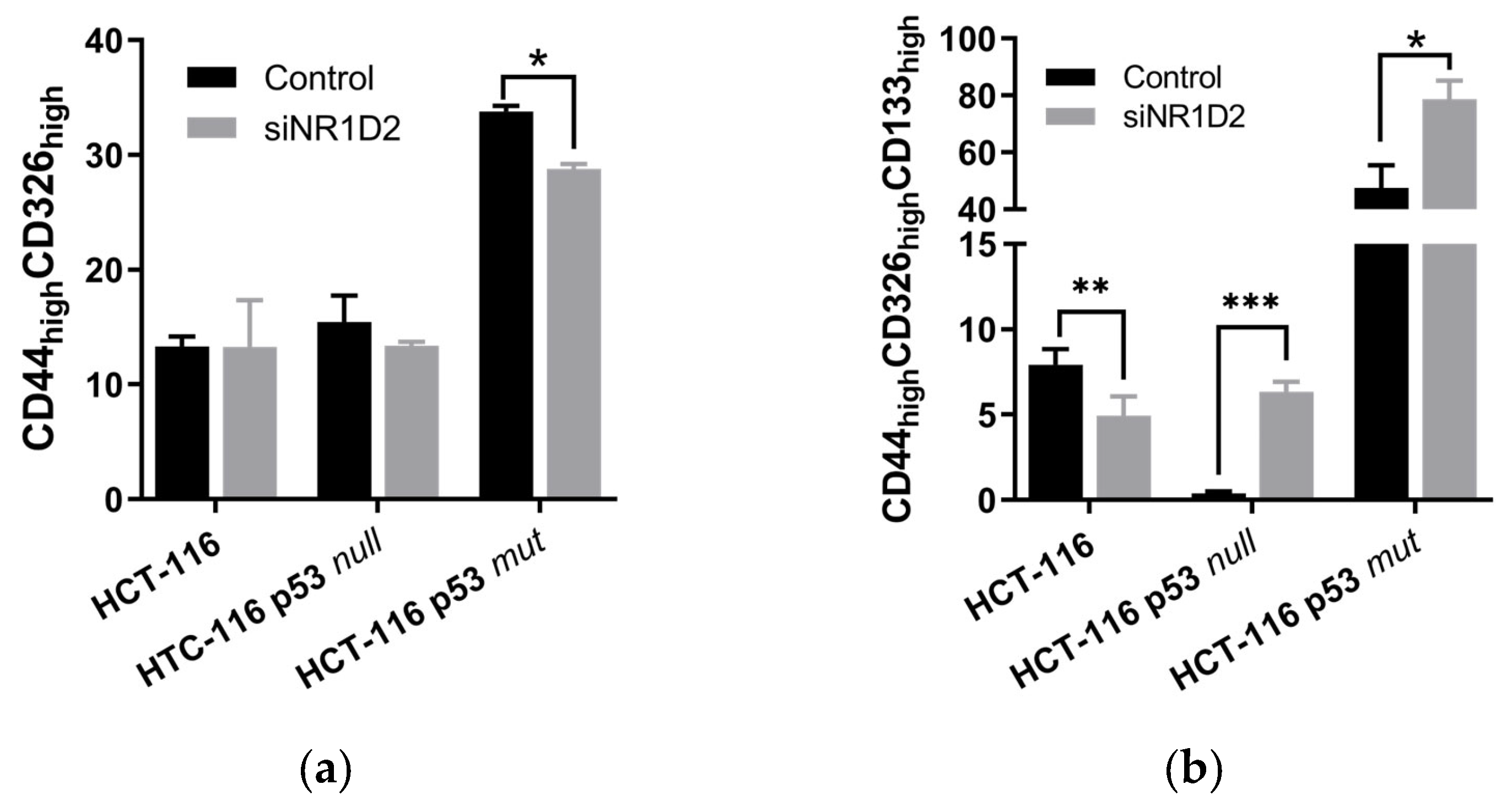
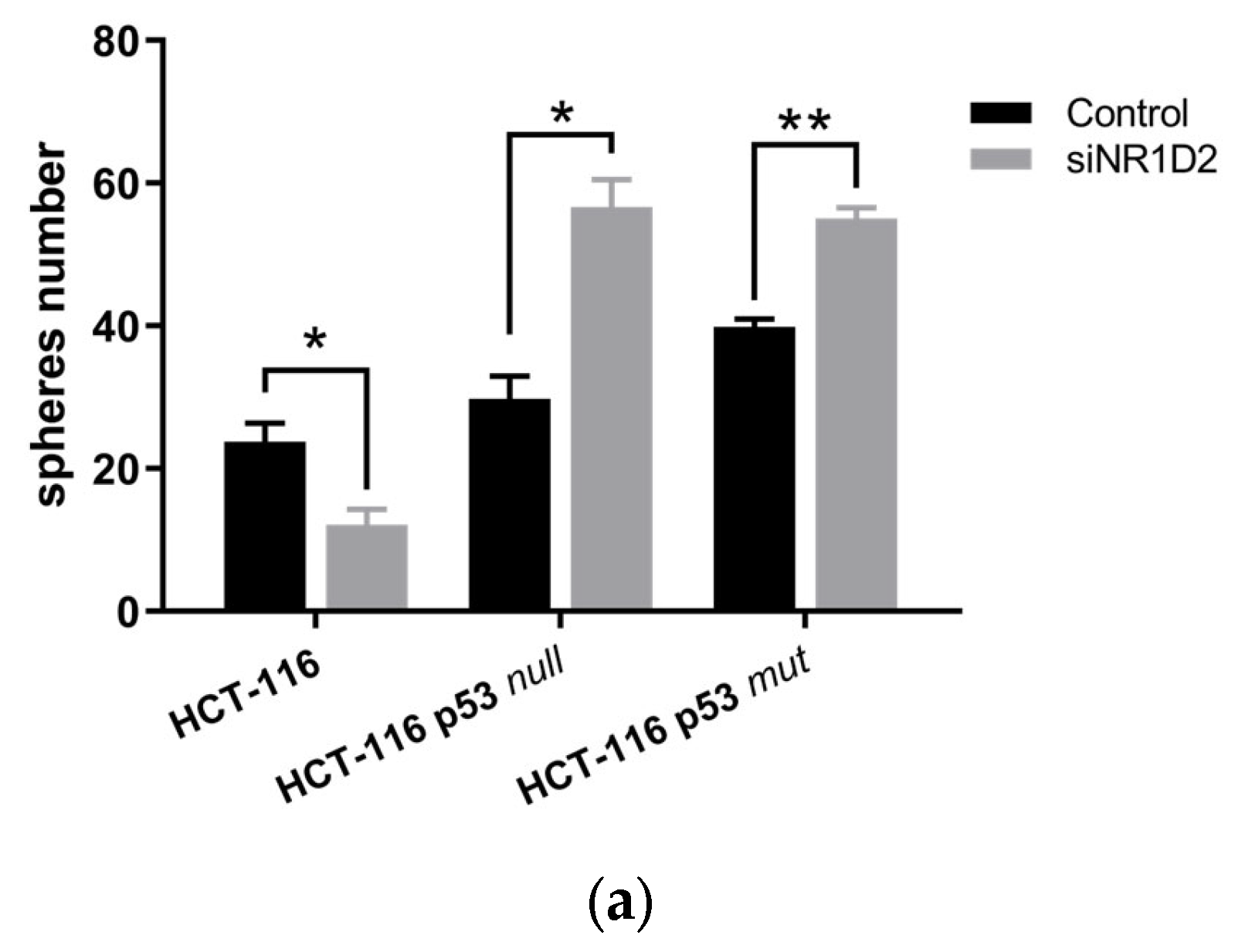
3.2. Stemness Regulation by NR1D2 in CRC Cells In Vitro
3.3. NR1D2 Expression Correlates with Low Differentiation Grade in Tumour Tissues of CRC Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CSCs | Cancer stem cells |
| CICs | Cancer-initiating cells |
| CRC | Colorectal cancer |
| NR1D2 | Nuclear Receptor Subfamily 1 Group D Member 2 |
| SCN | Suprachiasmatic nucleus |
| RHT | Retino-hypothalamic tract |
| TTFL | Transcription/translation feedback loops |
| CCG | Clock-controlled genes |
| TME | Tumour microenvironment |
| UBC | Ubiquitin C |
| TBP | TATA binding protein |
| RSP13 | Ribosomal protein S13 |
| PI | Propidium iodide |
| ALDH1 | Aldehyde dehydrogenase 1 |
| PE | Phycoerythrin |
| APC | Allophycocyanin |
| FITC | Fluorescein IsoTioCyanate |
References
- Siegel, R.L.; Kratzer, T.B.; Giaquinto, A.N.; Sung, H.; Jemal, A. Cancer statistics, 2025. CA A Cancer J. Clin. 2025, 75, 10–45. [Google Scholar] [CrossRef]
- Shin, A.E.; Giancotti, F.G.; Rustgi, A.K. Metastatic colorectal cancer: Mechanisms and emerging therapeutics. Trends Pharmacol. Sci. 2023, 44, 222–236. [Google Scholar] [CrossRef]
- Swartjes, H.; van Lankveld, D.W.P.; van Erning, F.N.; Verheul, H.M.W.; de Wilt, J.H.W.; Koëter, T.; Vissers, P.A.J. Locoregionally Recurrent Colon Cancer: How Far Have We Come? A Population-Based, Retrospective Cohort Study. Ann. Surg. Oncol. 2022, 30, 1726–1734. [Google Scholar] [CrossRef]
- Xie, Y.-H.; Chen, Y.-X.; Fang, J.-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Ebrahimi, N.; Afshinpour, M.; Fakhr, S.S.; Kalkhoran, P.G.; Shadman-Manesh, V.; Adelian, S.; Beiranvand, S.; Rezaei-Tazangi, F.; Khorram, R.; Hamblin, M.R.; et al. Cancer stem cells in colorectal cancer: Signaling pathways involved in stemness and therapy resistance. Crit. Rev. Oncol. 2023, 182, 103920. [Google Scholar] [CrossRef]
- Ghatak, D.; Das Ghosh, D.; Roychoudhury, S. Cancer Stemness: p53 at the Wheel. Front. Oncol. 2021, 10, 604124. [Google Scholar] [CrossRef]
- Lee, Y.; Field, J.M.; Sehgal, A. Circadian Rhythms, Disease and Chronotherapy. J. Biol. Rhythm. 2021, 36, 503–531. [Google Scholar] [CrossRef]
- Hastings, M.H.; Maywood, E.S.; Brancaccio, M. Generation of circadian rhythms in the suprachiasmatic nucleus. Nat. Rev. Neurosci. 2018, 19, 453–469. [Google Scholar] [CrossRef]
- Takahashi, J.S. Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 2016, 18, 164–179. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhang, R.; Zhang, Y.; Xu, Y.; Wang, R.; Chen, S.; Lv, Y.; Chen, Y.; Ren, Y.; Luo, P.; et al. Circadian disruption in cancer hallmarks: Novel insight into the molecular mechanisms of tumorigenesis and cancer treatment. Cancer Lett. 2024, 604, 217273. [Google Scholar] [CrossRef]
- Zhu, X.; Maier, G.; Panda, S. Learning from circadian rhythm to transform cancer prevention, prognosis, and survivorship care. Trends Cancer 2023, 10, 196–207. [Google Scholar] [CrossRef]
- Zhu, H.; Chen, J.; Wen, Z.; Li, J.; Yu, Q.; Liao, W.; Luo, X. The role of circadian clock genes in colorectal carcinoma: Novel insights into regulatory mechanism and implications in clinical therapy. Life Sci. 2023, 333, 122145. [Google Scholar] [CrossRef]
- Aroca-Siendones, M.I.; Moreno-SanJuan, S.; Puentes-Pardo, J.D.; Verbeni, M.; Arnedo, J.; Escudero-Feliu, J.; García-Costela, M.; García-Robles, A.; Carazo, Á.; León, J. Core Circadian Clock Proteins as Biomarkers of Progression in Colorectal Cancer. Biomedicines 2021, 9, 967. [Google Scholar] [CrossRef]
- Wang, X.; Guo, Y.; Lin, P.; Yu, M.; Song, S.; Xu, W.; Kong, D.; Wang, Y.; Zhang, Y.; Lu, F.; et al. Nuclear receptor E75/NR1D2 promotes tumor malignant transformation by integrating Hippo and Notch pathways. EMBO J. 2024, 43, 6336–6363. [Google Scholar] [CrossRef]
- Lian, J.-W.; Li, S.-Y.; Clarke, R.B.; Howell, S.J.; Meng, Q.-J. Can we utilise the circadian clock to target cancer stem cells? Cancer Lett. 2024, 611, 217360. [Google Scholar] [CrossRef]
- Wang, Y.; Narasimamurthy, R.; Qu, M.; Shi, N.; Guo, H.; Xue, Y.; Barker, N. Circadian regulation of cancer stem cells and the tumor microenvironment during metastasis. Nat. Cancer 2024, 5, 546–556. [Google Scholar] [CrossRef]
- Puentes-Pardo, J.D.; Moreno-SanJuan, S.; Casado, J.; Escudero Feliu, J.; López-Pérez, D.; Sánchez-Uceta, P.; González-Novoa, P.; Galvez, J.; Carazo, Á.; León, J. PARP-1 Expression Influences Cancer Stem Cell Phenotype in Colorectal Cancer Depending on p53. Int. J. Mol. Sci. 2023, 24, 4787. [Google Scholar] [CrossRef]
- Casado, J.; Iñigo-Chaves, A.; Jiménez-Ruiz, S.M.; Ríos-Arrabal, S.; Carazo-Gallego, Á.; González-Puga, C.; Núñez, M.I.; Ruíz-Extremera, Á.; Salmerón, J.; León, J. AA-NAT, MT1 and MT2 Correlates with Cancer Stem-Like Cell Markers in Colorectal Cancer: Study of the Influence of Stage and p53 Status of Tumors. Int. J. Mol. Sci. 2017, 18, 1251. [Google Scholar] [CrossRef]
- Pietrantonio, F.; Biondani, P.; Perrone, F.; Di Bartolomeo, M.; Pacifici, M.; Milione, M.; Melotti, F.; Maggi, C.; Montemurro, G.; Bossi, I.; et al. TP53 Mutations in Advanced Colorectal Cancer: The Dark Side of the Moon. Oncology 2014, 86, 289–294. [Google Scholar] [CrossRef]
- Kandioler, D.; Mittlböck, M.; Kappel, S.; Puhalla, H.; Herbst, F.; Langner, C.; Wolf, B.; Tschmelitsch, J.; Schippinger, W.; Steger, G.; et al. TP53 Mutational Status and Prediction of Benefit from Adjuvant 5-Fluorouracil in Stage III Colon Cancer Patients. EBioMedicine 2015, 2, 825–830. [Google Scholar] [CrossRef]
- Rao, X.; Lin, L. Circadian clock as a possible control point in colorectal cancer progression (Review). Int. J. Oncol. 2022, 61, 149. [Google Scholar] [CrossRef]
- Heddes, M.; Altaha, B.; Niu, Y.; Reitmeier, S.; Kleigrewe, K.; Haller, D.; Kiessling, S. The intestinal clock drives the microbiome to maintain gastrointestinal homeostasis. Nat. Commun. 2022, 13, 6068. [Google Scholar] [CrossRef]
- Chun, S.K.; Fortin, B.M.; Fellows, R.C.; Habowski, A.N.; Verlande, A.; Song, W.A.; Mahieu, A.L.; Lefebvre, A.E.Y.T.; Sterrenberg, J.N.; Velez, L.M.; et al. Disruption of the circadian clock drives Apc loss of heterozygosity to accelerate colorectal cancer. Sci. Adv. 2022, 8, eabo2389. [Google Scholar] [CrossRef]
- Ogunlusi, O.; Ghosh, A.; Sarkar, M.; Carter, K.; Davuluri, H.; Chakraborty, M.; Eckel-Mahan, K.; Keene, A.; Menet, J.S.; Bell-Pedersen, D.; et al. Rhythm is essential: Unraveling the relation between the circadian clock and cancer. Crit. Rev. Oncol. 2025, 208, 104632. [Google Scholar] [CrossRef]
- Brown, K.G.M.; Koh, C.E. Surgical management of recurrent colon cancer. J Gastrointest Oncol. 2020, 11, 513–525. [Google Scholar] [CrossRef]
- Roberson, J.; Maguire, L. Epidemiological trends in stage IV colorectal cancer. Semin. Colon Rectal Surg. 2023, 34, 100967. [Google Scholar] [CrossRef]
- Stephenson, E.M.; Usselmann, L.E.J.; Tergaonkar, V.; Virshup, D.M.; Dallmann, R. Cancer clocks in tumourigenesis: The p53 pathway and beyond. Endocr.-Relat. Cancer 2021, 28, R95–R110. [Google Scholar] [CrossRef]
- Gotoh, T.; Kim, J.K.; Liu, J.; Vila-Caballer, M.; Stauffer, P.E.; Tyson, J.J.; Finkielstein, C.V. Model-driven experimental approach reveals the complex regulatory distribution of p53 by the circadian factor Period 2. Proc. Natl. Acad. Sci. USA 2016, 113, 13516–13521. [Google Scholar] [CrossRef]
- Jiang, W.; Zhao, S.; Jiang, X.; Zhang, E.; Hu, G.; Hu, B.; Zheng, P.; Xiao, J.; Lu, Z.; Lu, Y.; et al. The circadian clock gene Bmal1 acts as a potential anti-oncogene in pancreatic cancer by activating the p53 tumor suppressor pathway. Cancer Lett. 2016, 371, 314–325. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, Q.; Hu, Y.; Wang, F. The Circadian Gene Per1 Plays an Important Role in Radiation-Induced Apoptosis and DNA Damage in Glioma. Asian Pac. J. Cancer Prev. 2019, 20, 2195–2201. [Google Scholar] [CrossRef]
- Chan, A.B.; Parico, G.C.G.; Fribourgh, J.L.; Ibrahim, L.H.; Bollong, M.J.; Partch, C.L.; Lamia, K.A. CRY2 missense mutations suppress P53 and enhance cell growth. Proc. Natl. Acad. Sci. USA 2021, 118, e2101416118. [Google Scholar] [CrossRef]
- Miki, T.; Matsumoto, T.; Zhao, Z.; Lee, C.C. p53 regulates Period2 expression and the circadian clock. Nat. Commun. 2013, 4, 2444. [Google Scholar] [CrossRef]
- Yu, M.; Li, W.; Wang, Q.; Wang, Y.; Lu, F. Circadian regulator NR1D2 regulates glioblastoma cell proliferation and motility. Oncogene 2018, 37, 4838–4853. [Google Scholar] [CrossRef]
- Friedmann-Morvinski, D.; Verma, I.M. Dedifferentiation and reprogramming: Origins of cancer stem cells. EMBO Rep. 2014, 15, 244–253. [Google Scholar] [CrossRef]
- Welch, R.D.; Billon, C.; Valfort, A.-C.; Burris, T.P.; Flaveny, C.A. Pharmacological inhibition of REV-ERB stimulates differentiation, inhibits turnover and reduces fibrosis in dystrophic muscle. Sci. Rep. 2017, 7, 17142. [Google Scholar] [CrossRef]
- Anabtawi, N.; Cvammen, W.; Kemp, M.G. Pharmacological inhibition of cryptochrome and REV-ERB promotes DNA repair and cell cycle arrest in cisplatin-treated human cells. Sci. Rep. 2021, 11, 17997. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, T.; Zhao, Z.; Zhang, H.; Yuan, P.; Wang, G.; Zhao, Z.; An, J.; Lyu, Z.; Xing, J.; et al. Down-regulation of BMAL1 by MiR-494-3p Promotes Hepatocellular Carcinoma Growth and Metastasis by Increasing GPAM-mediated Lipid Biosynthesis. Int. J. Biol. Sci. 2022, 18, 6129–6144. [Google Scholar] [CrossRef]
- Sulli, G.; Rommel, A.; Wang, X.; Kolar, M.J.; Puca, F.; Saghatelian, A.; Plikus, M.V.; Verma, I.M.; Panda, S. Pharmacological activation of REV-ERBs is lethal in cancer and oncogene-induced senescence. Nature 2018, 553, 351–355. [Google Scholar] [CrossRef]
- Dierickx, P.; Emmett, M.J.; Jiang, C.; Uehara, K.; Liu, M.; Adlanmerini, M.; Lazar, M.A. SR9009 has REV-ERB–independent effects on cell proliferation and metabolism. Proc. Natl. Acad. Sci. USA 2019, 116, 12147–12152. [Google Scholar] [CrossRef]
- Liu, A.C.; Tran, H.G.; Zhang, E.E.; Priest, A.A.; Welsh, D.K.; Kay, S.A.; Takahashi, J.S. Redundant Function of REV-ERBα and β and Non-Essential Role for Bmal1 Cycling in Transcriptional Regulation of Intracellular Circadian Rhythms. PLoS Genet. 2008, 4, e1000023. [Google Scholar] [CrossRef]
- Amador, A.; Campbell, S.; Kazantzis, M.; Lan, G.; Burris, T.P.; Solt, L.A.; Foulkes, N.S. Distinct roles for REV-ERBα and REV-ERBβ in oxidative capacity and mitochondrial biogenesis in skeletal muscle. PLoS ONE 2018, 13, e0196787. [Google Scholar] [CrossRef]
- Bhoumik, S.; Lee, Y. Core Molecular Clock Factors Regulate Osteosarcoma Stem Cell Survival and Behavior via CSC/EMT Pathways and Lipid Droplet Biogenesis. Cells 2025, 14, 517. [Google Scholar] [CrossRef]
- Chakravarti, B.; Siddiqui, J.A.; Sinha, R.A.; Raza, S. Targeting autophagy and lipid metabolism in cancer stem cells. Biochem. Pharmacol. 2023, 212, 115550. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Debnath, J.; Gammoh, N.; Ryan, K.M. Autophagy and autophagy-related pathways in cancer. Nat. Rev. Mol. Cell Biol. 2023, 24, 560–575. [Google Scholar] [CrossRef]
- Saito, T.; Kuma, A.; Sugiura, Y.; Ichimura, Y.; Obata, M.; Kitamura, H.; Okuda, S.; Lee, H.-C.; Ikeda, K.; Kanegae, Y.; et al. Autophagy regulates lipid metabolism through selective turnover of NCoR1. Nat. Commun. 2019, 10, 1567. [Google Scholar] [CrossRef]
- Toledo, M.; Batista-Gonzalez, A.; Merheb, E.; Aoun, M.L.; Tarabra, E.; Feng, D.; Sarparanta, J.; Merlo, P.; Botrè, F.; Schwartz, G.J.; et al. Autophagy Regulates the Liver Clock and Glucose Metabolism by Degrading CRY1. Cell Metab. 2018, 28, 268–281.e4. [Google Scholar] [CrossRef]
- Marescal, O.; Cheeseman, I.M. Cellular Mechanisms and Regulation of Quiescence. Dev. Cell 2020, 55, 259–271. [Google Scholar] [CrossRef]
- Li, D.; Peng, X.; He, G.; Liu, J.; Li, X.; Lin, W.; Fang, J.; Li, X.; Yang, S.; Yang, L.; et al. Crosstalk between autophagy and CSCs: Molecular mechanisms and translational implications. Cell Death Dis. 2023, 14, 409. [Google Scholar] [CrossRef]
- Luo, W. Nasopharyngeal carcinoma ecology theory: Cancer as multidimensional spatiotemporal “unity of ecology and evolution” pathological ecosystem. Theranostics 2023, 13, 1607–1631. [Google Scholar] [CrossRef] [PubMed]
- Vo-Quang, E.; Gandini, A.; Taly, V.; Laurent-Puig, P.; Zaanan, A.; Taieb, J. Circulating tumor DNA: A new tool to predict recurrence and guide treatment of colorectal cancer. Clin. Res. Hepatol. Gastroenterol. 2025, 49, 102611. [Google Scholar] [CrossRef] [PubMed]
- Symonds, E.L.; Pedersen, S.K.; Yeo, B.; Al Naji, H.; Byrne, S.E.; Roy, A.; Young, G.P. Assessment of tumor burden and response to therapy in patients with colorectal cancer using a quantitative ctDNA test for methylated BCAT1/IKZF1. Mol. Oncol. 2022, 16, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, M.; Sharma, M.R.; Kuboki, Y.; Perets, R.; Cohen, J.; Raimbourg, J.; Nakajima, T.E.; Cruz-Correa, M.; Ghiringhelli, F.; Raghav, K.P.S.; et al. Genomic alterations in circulating tumor DNA (ctDNA) and response to telisotuzumab adizutecan (ABBV-400) treatment in patients (pts) with colorectal cancer (CRC). J. Clin. Oncol. 2025, 43, 258. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, C.; Ling, S.; Wei, R.; Wang, J.; Xu, X. The metabolic flexibility of quiescent CSC: Implications for chemotherapy resistance. Cell Death Dis. 2021, 12, 835. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Lahouel, K.; Lo, S.N.; Wang, Y.; Kosmider, S.; Wong, R.; Shapiro, J.; Lee, M.; Harris, S.; et al. Circulating Tumor DNA Analysis Guiding Adjuvant Therapy in Stage II Colon Cancer. N. Engl. J. Med. 2022, 386, 2261–2272. [Google Scholar] [CrossRef]
- Rebuzzi, S.E.; Pesola, G.; Martelli, V.; Sobrero, A.F. Adjuvant Chemotherapy for Stage II Colon Cancer. Cancers 2020, 12, 2584. [Google Scholar] [CrossRef]
- Haldavnekar, R.; Ganesh, S.; Venkatakrishnan, K.; Tan, B. Cancer Stem Cell DNA Enabled Real-Time Genotyping with Self-Functionalized Quantum Superstructures—Overcoming the Barriers of Noninvasive cfDNA Cancer Diagnostics. Small Methods 2022, 6, e2101467. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | N (%) | |
|---|---|---|
| Age | <72 | 92 (46.9) |
| ≥72 | 104 (53.1) | |
| Sex | Female | 74 (38.1) |
| Male | 122 (61.9) | |
| Location | Colon | 184 (93.4) |
| Rectum | 12 (6.6) | |
| DG 1 | Well differentiated | 43 (21.9) |
| Moderately differentiated | 131 (66.8) | |
| Poorly differentiated | 22 (11.3) | |
| T stage | T1 | 4 (2.0) |
| T2 | 26 (13.27) | |
| T3 | 126 (64.3) | |
| T4 | 40 (20.4) | |
| N stage | N0 | 103 (52.6) |
| N1 | 50 (25.5) | |
| N2 | 43 (21.9) | |
| M stage | M0 | 177 (90.3) |
| M1 | 19 (9.7) | |
| pTMN stage | Stage I | 22 (11.3) |
| Stage II | 74 (37.9) | |
| Stage III | 82 (41.5) | |
| Stage IV | 18 (9.2) | |
| All 1 | wtp53 2 | mtp53 3 | |||||
|---|---|---|---|---|---|---|---|
| Characteristic | Median ± IQR 5 | p | Median ± IQR 5 | p | Median ± IQR 5 | p | |
| Age (y) *4 | <72 | 0.90 (0.62–1.27) | 0.453 | 0.83 (0.68–1.11) | 0.509 | 0.93 (0.79–1.41) | 0.704 |
| ≥72 | 0.85 (0.54– 1.21) | 0.84 (0.51–1.17) | 0.99 (0.65–1.27) | ||||
| Sex * | Male | 0.92 (0.58–1.28) | 0.077 | 0.90 (0.52–1.24) | 0.442 | 1.01 (0.79–1.41) | 0.077 |
| Female | 0.75 (0.56–1.07) | 0.74 (0.66–1.04) | 0.90 (0.48–1.18) | ||||
| Location * | Colon | 0. 89 (0.58–1.22) | 0.508 | 0.84 (0.58–1.16) | 0.353 | 0.95 (0.65–1.31) | 0.352 |
| Rectum | 0.86 (0.42–1.52) | 0.62 (0.42–1.30) | 1.50 (0.86–1.27) | ||||
| DG †& | Well | 0.72 (0.44–1.17) | 0.380 | 0.61 (0.43–0.85) | 0.043 | 0.95 (0.46–1.58) | 0.780 |
| Moderately | 0.91 (0.59–1.23) | 0.86 (0.62–1.19) | 1.01 (0.74–1.40) | ||||
| Poor | 0.90 (0.69–1.25) | 0.97 (0.68–1.28) | 0.91 (0.89–1.40) | ||||
| T stage * | T1+T2 | 1.02 (0.73–1.34) | 0.191 | 0.80 (0.64–1.08) | 0.772 | 1.18 (0.97–1.74) | 0.257 |
| T3+T4 | 0.87 (0.55–1.19) | 0.83 (0.53–1.17) | 0.92 (0.58–1.30) | ||||
| N stage † | N0 | 0.81 (0.53–1.14) | 0.361 | 0.74 (0.49–1.15) | 0.875 | 0.92 (0.54–1.17) | 0.075 |
| N1 | 0.89 (0.52–1.29) | 0.70 (0.52–1.29) | 0.90 (0.67–1.77) | ||||
| N2 | 0.97 (0.78–1.23) | 0.92 (0.73–1.00) | 1.16 (0.92–1.56) | ||||
| M stage * | M0 | 0.89 (0.57–1.23) | 0.927 | 0.84 (0.53–1.18) | 0.685 | 0.96 (0.76–1.40) | 0.719 |
| M1 | 0.85 (0.63–1.20) | 0.81 (0.61–1.01) | 1.06 (0.56–1.56) | ||||
| pTNM Stage * | Stage I+II | 0.94 (0.71–1.14) | 0.080 | 0.79 (0.52–0.99) | 0.427 | 1.02 (0.91–1.32) | 0.097 |
| Stage III+IV | 0.91 (0.64–1.27) | 0.88 (0.63–1.17) | 1.11 (0.72–1.35) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alonso-García, S.; Sánchez-Uceta, P.; Moreno-SanJuan, S.; Casado, J.; Puentes-Pardo, J.D.; Khaldy, H.; Lopez-Pérez, D.; Zurita-Saavedra, M.S.; González-Puga, C.; Carazo, A.; et al. Regulation of Stemness by NR1D2 in Colorectal Cancer. Biomedicines 2025, 13, 1500. https://doi.org/10.3390/biomedicines13061500
Alonso-García S, Sánchez-Uceta P, Moreno-SanJuan S, Casado J, Puentes-Pardo JD, Khaldy H, Lopez-Pérez D, Zurita-Saavedra MS, González-Puga C, Carazo A, et al. Regulation of Stemness by NR1D2 in Colorectal Cancer. Biomedicines. 2025; 13(6):1500. https://doi.org/10.3390/biomedicines13061500
Chicago/Turabian StyleAlonso-García, Sandra, Paula Sánchez-Uceta, Sara Moreno-SanJuan, Jorge Casado, Jose D. Puentes-Pardo, Huda Khaldy, David Lopez-Pérez, María Sol Zurita-Saavedra, Cristina González-Puga, Angel Carazo, and et al. 2025. "Regulation of Stemness by NR1D2 in Colorectal Cancer" Biomedicines 13, no. 6: 1500. https://doi.org/10.3390/biomedicines13061500
APA StyleAlonso-García, S., Sánchez-Uceta, P., Moreno-SanJuan, S., Casado, J., Puentes-Pardo, J. D., Khaldy, H., Lopez-Pérez, D., Zurita-Saavedra, M. S., González-Puga, C., Carazo, A., & León, J. (2025). Regulation of Stemness by NR1D2 in Colorectal Cancer. Biomedicines, 13(6), 1500. https://doi.org/10.3390/biomedicines13061500