
Vitamin D Attenuates Hepatic Sinusoidal Capillarization in Type 2 Diabetes Mellitus– Metabolic Dysfunction-Associated Fatty Liver Disease via Dual Autophagy Activation and Pyroptosis Suppression in Liver Sinusoidal Endothelial Cells
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability Assay
2.3. Cell Navigator Fluorescence Method for Lipid Droplet Detection
2.4. Enzyme-Linked Immunosorbent Assay (ELISA)
2.5. Western Blotting
2.6. Electron Microscopy
2.7. Adenoviral Transduction
2.8. Short Hairpin RNA (shRNA) Treatment
2.9. Animals
2.10. Histological Analysis
2.11. Serum Biochemical Analysis
2.12. Statistical Analysis
3. Results
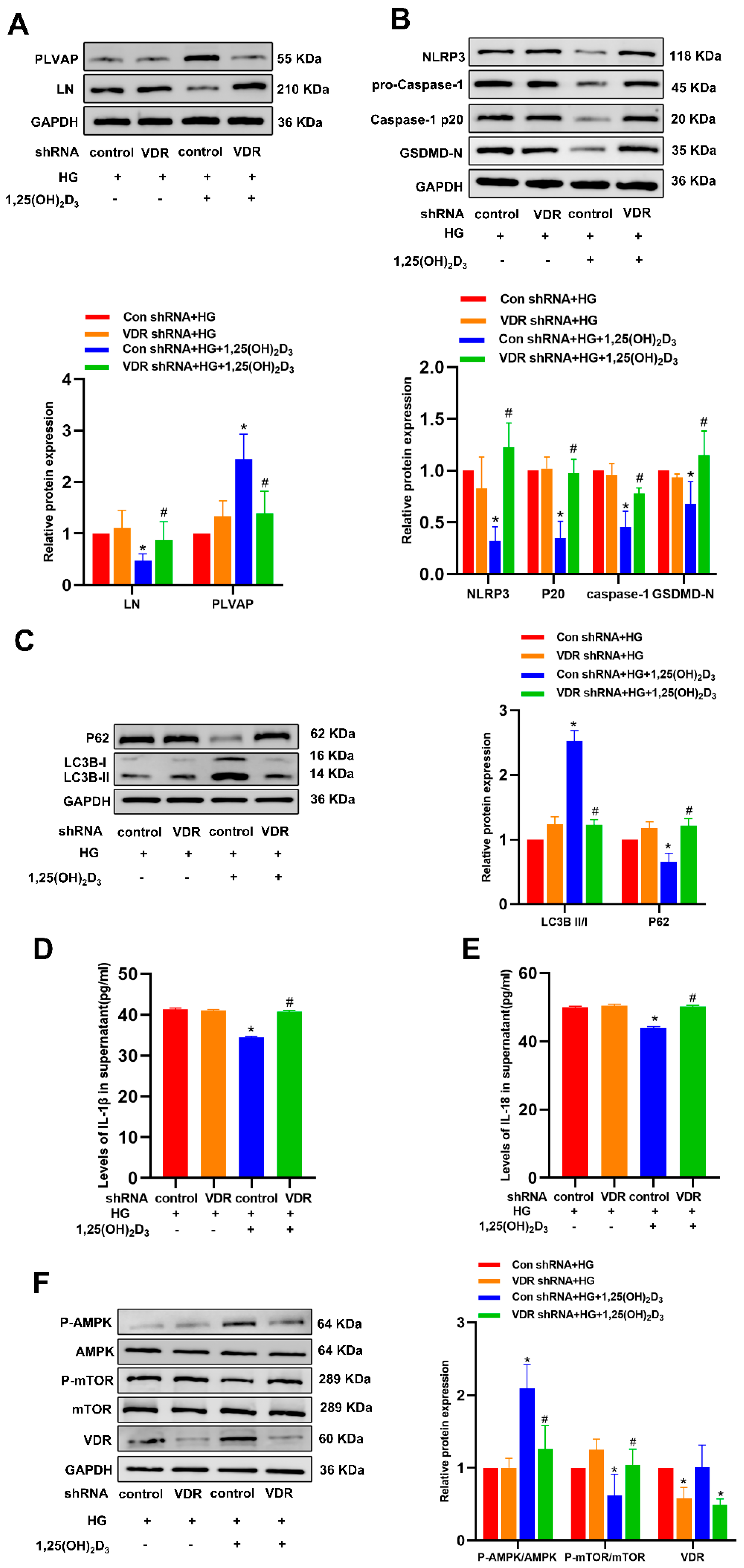
3.1. 1,25(OH)2D3 Protects Against HG-Induced HLSEC Capillarization
3.2. 1,25(OH)2D3 Inhibits HG-Induced Pyroptosis in HLSECs to Protect Against Hepatic Sinusoidal Capillarization
3.3. 1,25(OH)2D3 Promotes Autophagic Activity in HG-Treated HLSECs to Alleviate Hepatic Sinusoidal Capillarization
3.4. 1,25(OH)2D3 Induces Autophagy to Protect Against HLSEC Pyroptosis
3.5. 1,25(OH)2D3 May Control Autophagy Through the AMPK/mTOR Pathway to Prevent Pyroptosis and Hepatic Sinusoidal Capillarization
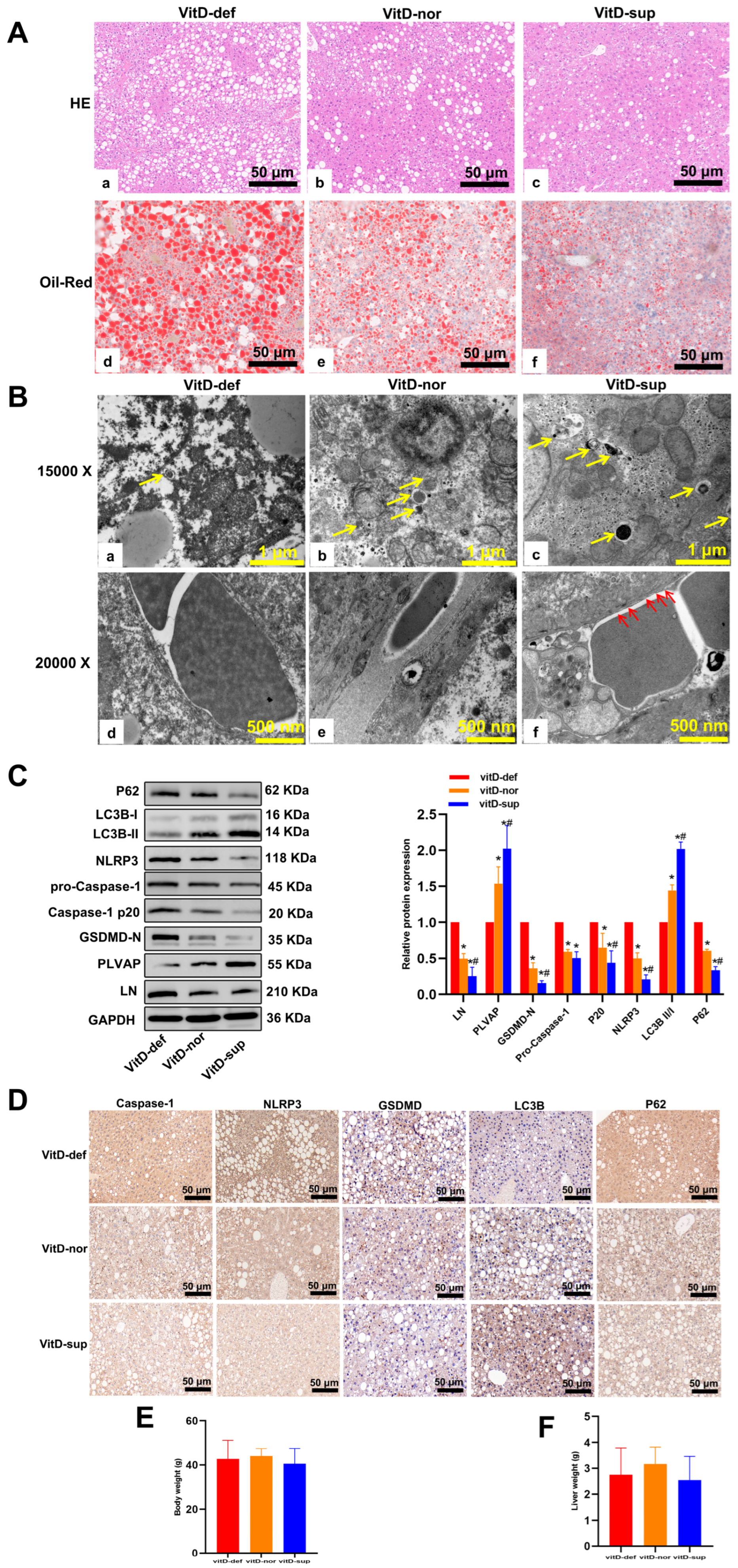
3.6. Vitamin D Supplementation Suppresses the Development of MAFLD in db/db Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
Appendix A
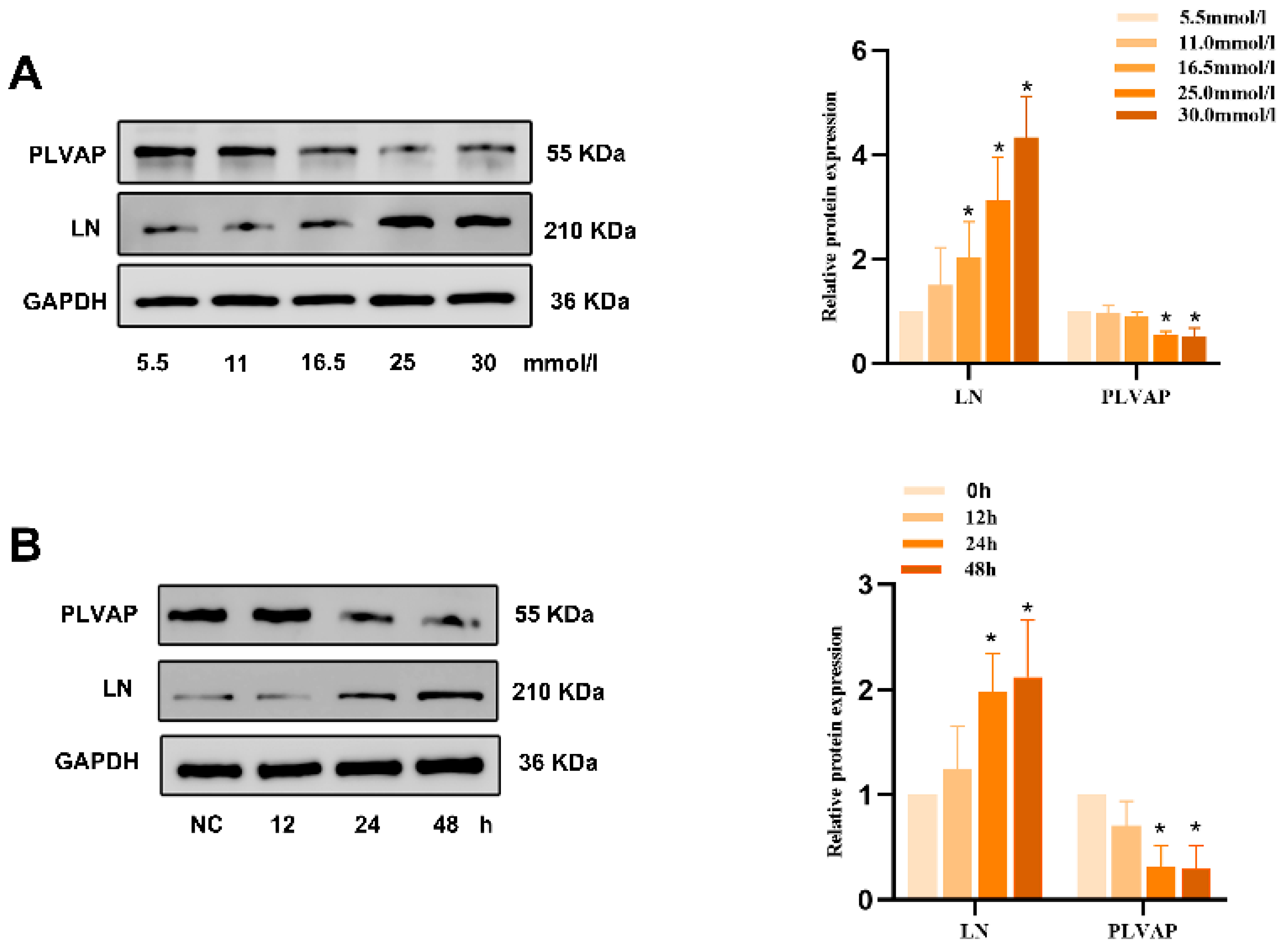
Appendix B

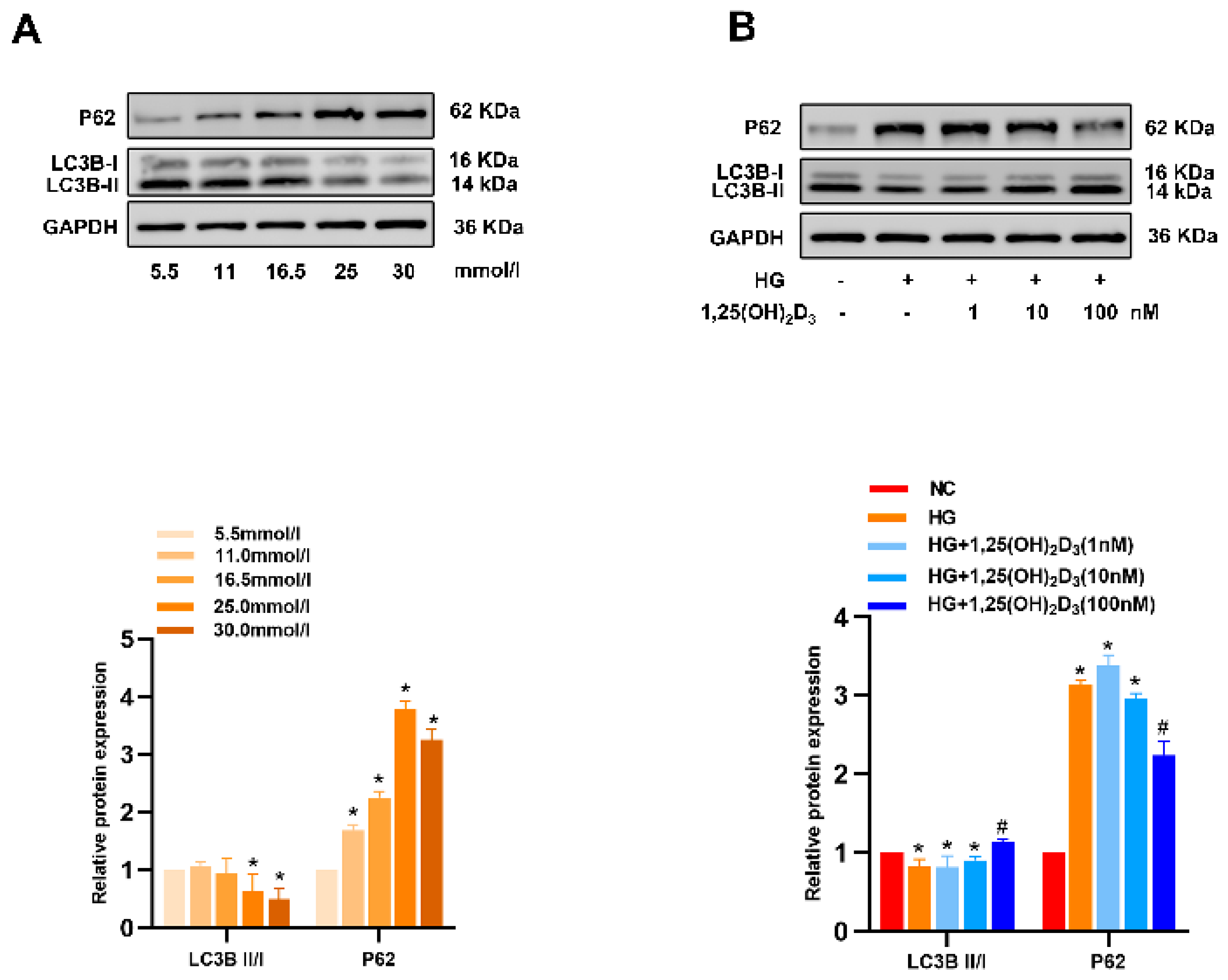
Appendix C
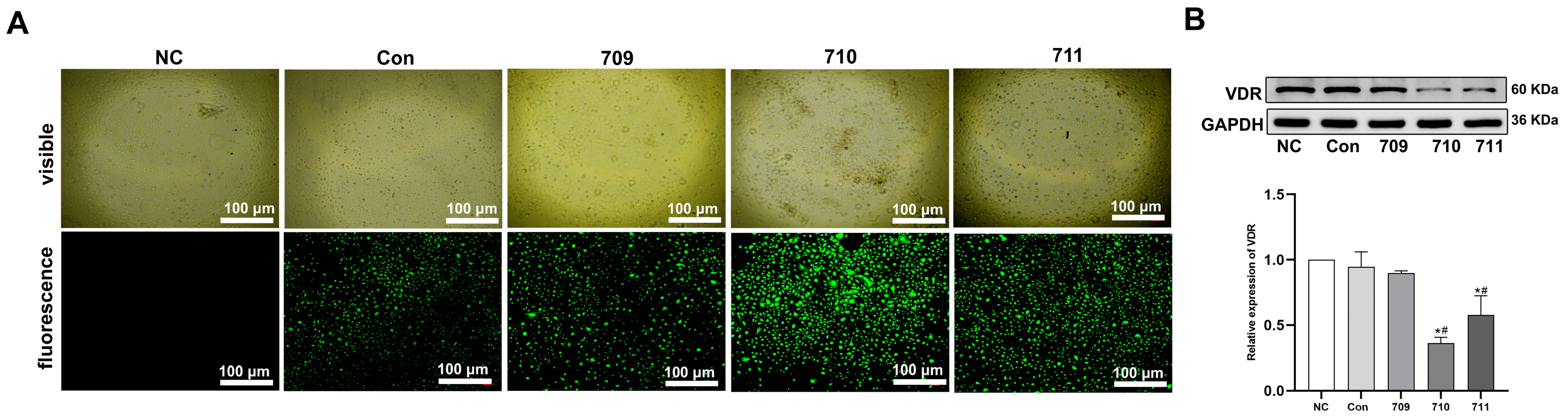
Appendix D
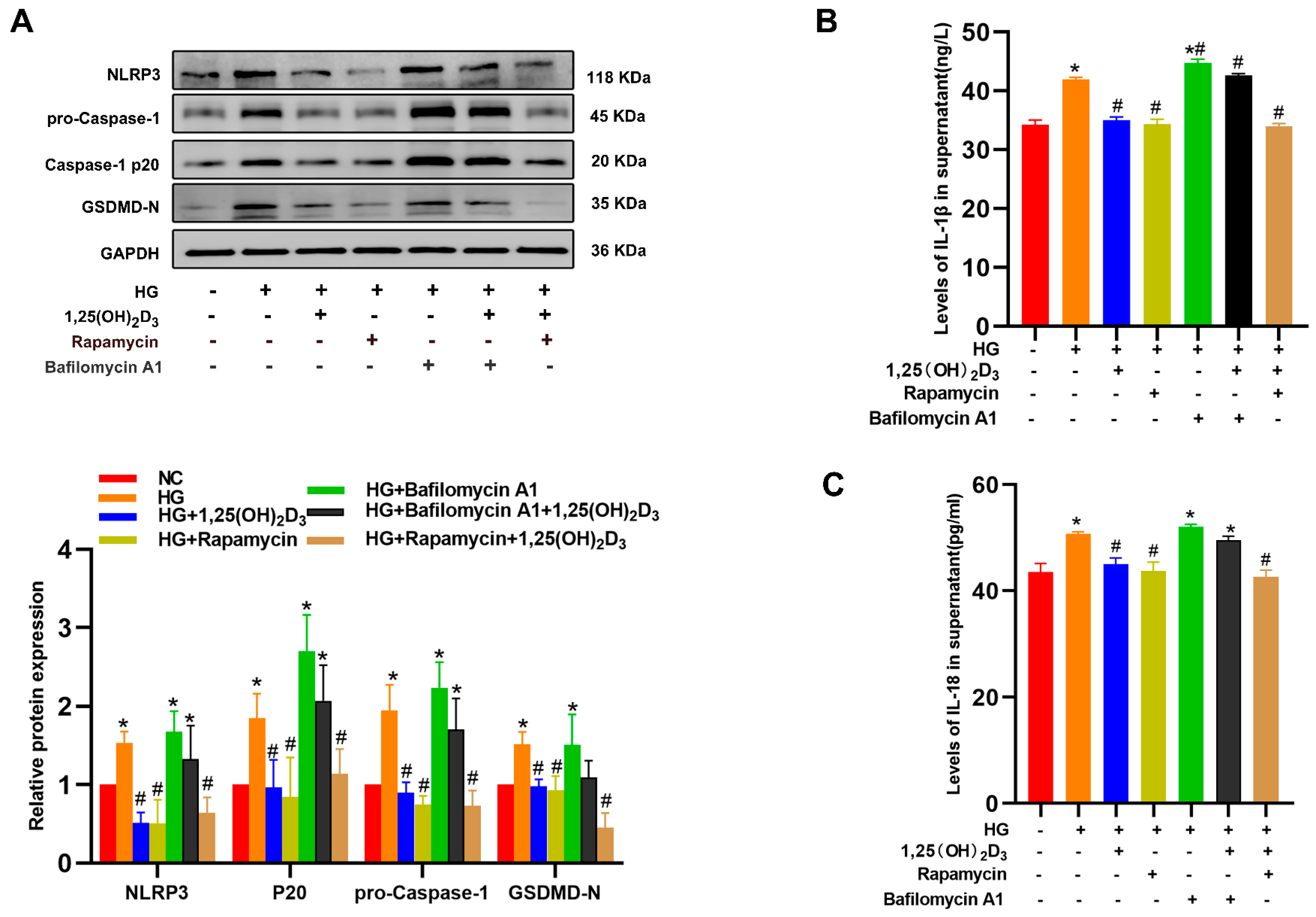
References
- Mantovani, A.; Scorletti, E.; Mosca, A.; Alisi, A.; Byrne, C.D.; Targher, G. Complications morbidity and mortality of nonalcoholic fatty liver disease. Metabolism 2020, 111S, 154170. [Google Scholar] [CrossRef]
- Tanase, D.M.; Gosav, E.M.; Costea, C.F.; Ciocoiu, M.; Lacatusu, C.M.; Maranduca, M.A.; Ouatu, A.; Floria, M. The Intricate Relationship between Type 2 Diabetes Mellitus (T2DM), Insulin Resistance (IR), and Nonalcoholic Fatty Liver Disease (NAFLD). J. Diabetes Res. 2020, 2020, 3920196. [Google Scholar] [CrossRef] [PubMed]
- Pafili, K.; Roden, M. Nonalcoholic fatty liver disease (NAFLD) from pathogenesis to treatment concepts in humans. Mol. Metab. 2021, 50, 101122. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic Dysfunction-Associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Caparrós, E.; Fernández-Iglesias, A.; Francés, R. Role of liver sinusoidal endothelial cells in liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 411–431. [Google Scholar] [CrossRef]
- Hammoutene, A.; Rautou, P.E. Role of liver sinusoidal endothelial cells in Non-Alcoholic fatty liver disease. J. Hepatol. 2019, 70, 1278–1291. [Google Scholar] [CrossRef] [PubMed]
- Velliou, R.-I.; Legaki, A.-I.; Nikolakopoulou, P.; Vlachogiannis, N.I.; Chatzigeorgiou, A. Liver endothelial cells in NAFLD and transition to NASH and HCC. Cell Mol. Life Sci. 2023, 80, 314. [Google Scholar] [CrossRef] [PubMed]
- Furuta, K.; Tang, X.; Islam, S.; Tapia, A.; Chen, Z.B.; Ibrahim, S.H. Endotheliopathy in the metabolic syndrome: Mechanisms and clinical implications. Pharmacol. Ther. 2023, 244, 108372. [Google Scholar] [CrossRef]
- Denzer, L.; Muranyi, W.; Schroten, H.; Schwerk, C. The role of PLVAP in endothelial cells. Cell Tissue Res. 2023, 392, 393–412. [Google Scholar] [CrossRef]
- Zhang, Q.; Niu, X.; Tian, L.; Liu, J.; Niu, R.; Quan, J.; Yu, J.; Lin, W.; Qian, Z.; Zeng, P. CTRP13 attenuates the expression of LN and CAV-1 Induced by high glucose via CaMKKβ/AMPK pathway in rLSECs. Aging 2020, 12, 11485–11499. [Google Scholar] [CrossRef]
- Herrnberger, L.; Hennig, R.; Kremer, W.; Hellerbrand, C.; Goepferich, A.; Kalbitzer, H.R.; Tamm, E.R. Formation of fenestrae in murine liver sinusoids depends on plasmalemma Vesicle-Associated protein and is required for lipoprotein passage. PLoS ONE 2014, 9, e115005. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef]
- Allaire, M.; Rautou, P.-E.; Codogno, P.; Lotersztajn, S. Autophagy in liver diseases: Time for translation? J. Hepatol. 2019, 70, 985–998. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.-S.; Zhang, S.-F.; Luo, G.; Cheng, B.C.-Y.; Zhang, C.; Wang, Y.-W.; Qiu, X.-Y.; Zhou, X.-H.; Wang, Q.-G.; Song, X.-L.; et al. Schisandrin B mitigates hepatic steatosis and promotes fatty acid oxidation by inducing autophagy through AMPK/mTOR signaling pathway. Metabolism 2022, 131, 155200. [Google Scholar] [CrossRef]
- Ribeiro, M.D.C.; Szabo, G. Role of the Inflammasome in Liver Disease. Annu. Rev. Pathol. 2022, 17, 345–365. [Google Scholar] [CrossRef]
- Mitsuyoshi, H.; Yasui, K.; Hara, T.; Taketani, H.; Ishiba, H.; Okajima, A.; Seko, Y.; Umemura, A.; Nishikawa, T.; Yamaguchi, K.; et al. Hepatic nucleotide binding oligomerization Domain-Like receptors pyrin Domain-Containing 3 inflammasomes are associated with the histologic severity of Non-Alcoholic fatty liver disease. Hepatol. Res. 2017, 47, 1459–1468. [Google Scholar] [CrossRef]
- Qiu, T.; Pei, P.; Yao, X.; Jiang, L.; Wei, S.; Wang, Z.; Bai, J.; Yang, G.; Gao, N.; Yang, L.; et al. Taurine attenuates arsenic-induced pyroptosis and nonalcoholic steatohepatitis by inhibiting the autophagic-inflammasomal pathway. Cell Death Dis. 2018, 9, 946. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, J.W.; Huang, J.; Tang, L.; Xu, Y.H.; Sun, H.; Tang, J.; Wang, G. Pyroptosis, a target for cancer treatment? Apoptosis 2022, 27, 1–13. [Google Scholar] [CrossRef]
- Knorr, J.; Wree, A.; Feldstein, A.E. Pyroptosis in Steatohepatitis and Liver Diseases. J. Mol. Biol. 2022, 434, 167271. [Google Scholar] [CrossRef]
- Colak, Y.; Hasan, B.; Erkalma, B.; Tandon, K.; Zervos, X.; Menzo, E.L.; Erim, T. Pathogenetic mechanisms of nonalcoholic fatty liver disease and inhibition of the inflammasome as a new therapeutic target. Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101710. [Google Scholar] [CrossRef]
- Biasizzo, M.; Kopitar-Jerala, N. Interplay Between NLRP3 Inflammasome and Autophagy. Front. Immunol. 2020, 11, 591803. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Cimini, F.A.; Cavallo, M.G. Vitamin D and Metabolic Dysfunction-Associated Fatty Liver Disease (MAFLD): An Update. Nutrients 2020, 12, 3302. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, H.; Lim, Y. Vitamin D3 improves Lipophagy-Associated renal lipid metabolism and tissue damage in diabetic mice. Nutr. Res. 2020, 80, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shang, X.; Jin, S.; Ma, Z.; Wang, H.; Ao, N.; Yang, J.; Du, J. Vitamin D ameliorates high-fat-diet-induced hepatic injury via inhibiting pyroptosis and alters gut microbiota in rats. Arch. Biochem. Biophys. 2021, 705, 108894. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.E.; Wong, V.W.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Wilkinson, A.L.; Qurashi, M.; Shetty, S. The Role of Sinusoidal Endothelial Cells in the Axis of Inflammation and Cancer Within the Liver. Front. Physiol. 2020, 11, 990. [Google Scholar] [CrossRef]
- Ramos-Tovar, E.; Muriel, P. NLRP3 inflammasome in hepatic diseases: A pharmacological target. Biochem. Pharmacol. 2023, 217, 115861. [Google Scholar] [CrossRef]
- Fantauzzi, C.B.; Menini, S.; Iacobini, C.; Rossi, C.; Santini, E.; Solini, A.; Pugliese, G. Deficiency of the Purinergic Receptor 2X7 Attenuates Nonalcoholic Steatohepatitis Induced by High-Fat Diet: Possible Role of the NLRP3 Inflammasome. Oxid. Med. Cell Longev. 2017, 2017, 8962458. [Google Scholar] [CrossRef]
- Cimini, F.A.; Barchetta, I.; Carotti, S.; Morini, S.; Cavallo, M.G. Overview of studies of the vitamin D/vitamin D receptor system in the development of non-alcoholic fatty liver disease. World J. Gastrointest. Pathophysiol. 2019, 10, 11–16. [Google Scholar] [CrossRef]
- Yuan, S.; Larsson, S.C. Inverse Association Between Serum 25-Hydroxyvitamin D and Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2023, 21, 398–405.e4. [Google Scholar] [CrossRef]
- Wu, M.; Lu, L.; Guo, K.; Lu, J.; Chen, H. Vitamin D protects against high Glucose-Induced pancreatic β-cell dysfunction via AMPK-NLRP3 inflammasome pathway. Mol. Cell Endocrinol. 2022, 547, 111596. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Sun, Q.; Fu, J. Dysfunction of autophagy in High-Fat Diet-Induced Non-Alcoholic fatty liver disease. Autophagy 2024, 20, 221–241. [Google Scholar] [CrossRef]
- Song, Y.M.; Lee, Y.-H.; Kim, J.-W.; Ham, D.-S.; Kang, E.-S.; Cha, B.S.; Lee, H.C.; Lee, B.-W. Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway. Autophagy 2015, 11, 46–59. [Google Scholar] [CrossRef]
- Lim, H.; Lee, H.; Lim, Y. Effect of vitamin D supplementation on hepatic lipid dysregulation associated with autophagy regulatory AMPK/Akt-mTOR signaling in type 2 diabetic mice. Exp. Biol. Med. 2021, 246, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Lin, H.; Liu, J.; Wang, D.; Li, D.; Jiang, C.; Tang, Y.; Wang, J.; Zhang, T.; Li, Y.; et al. 1,25-Dihydroxyvitamin D attenuates diabetic cardiac autophagy and damage by vitamin D receptor-mediated suppression of FoxO1 translocation. J. Nutr. Biochem. 2020, 80, 108380. [Google Scholar] [CrossRef]
- Bristol, M.L.; Di, X.; Beckman, M.J.; Wilson, E.N.; Henderson, S.C.; Maiti, A.; Fan, Z.; Gewirtz, D.A. Dual functions of autophagy in the response of breast tumor cells to radiation: Cytoprotective autophagy with radiation alone and cytotoxic autophagy in radiosensitization by vitamin D3. Autophagy 2012, 8, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Li, M.-Y.; Zhu, X.-L.; Zhao, B.-X.; Shi, L.; Wang, W.; Hu, W.; Qin, S.-L.; Chen, B.-H.; Zhou, P.-H.; Qiu, B.; et al. Adrenomedullin alleviates the pyroptosis of Leydig cells by promoting autophagy via the ROS-AMPK-mTOR axis. Cell Death Dis. 2019, 10, 489. [Google Scholar] [CrossRef]
- You, B.; Xia, T.; Gu, M.; Zhang, Z.; Zhang, Q.; Shen, J.; Fan, Y.; Yao, H.; Pan, S.; Lu, Y.; et al. AMPK-mTOR-Mediated Activation of Autophagy Promotes Formation of Dormant Polyploid Giant Cancer Cells. Cancer Res. 2022, 82, 846–858. [Google Scholar] [CrossRef]
- Kong, C.; Wang, C.; Shi, Y.; Yan, L.; Xu, J.; Qi, W. Active vitamin D activates chondrocyte autophagy to reduce osteoarthritis via mediating the AMPK-mTOR signaling pathway. Biochem. Cell Biol. 2020, 98, 434–442. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | vitD-def | vitD-nor | vitD-sup | p Value |
|---|---|---|---|---|
| 25(OH)D3 (ng/mL) | 19.12 ± 0.25 | 33.02 ± 1.19 * | 59.74 ± 2.11 *# | <0.01 |
| FBG (mmol/L) | 31.17 ± 2.86 | 32.39 ± 2.65 | 33.43 ± 2.94 | >0.01 |
| TG (mmol/L) | 2.28 ± 0.10 | 1.74 ± 0.08 * | 1.17 ± 0.05 *# | <0.01 |
| TC (mmol/L) | 7.59 ± 0.47 | 6.07 ± 0.27 * | 4.44 ± 0.39 *# | <0.01 |
| HDL-C (mmol/L) | 1.18 ± 0.13 | 2.30 ± 0.13 * | 2.43 ± 0.17 *# | <0.01 |
| LDL-C (mmol/L) | 0.41 ± 0.04 | 0.50 ± 0.07 | 0.60 ± 0.12 | >0.01 |
| ALT (U/L) | 236.10 ± 8.97 | 180.70 ± 10.05 * | 141.8 ± 10.89 *# | <0.01 |
| AST (U/L) | 418.80 ± 16.64 | 340.40 ± 10.42 * | 281.60 ± 11.38 *# | <0.01 |
| IL-1β (pg/mL) | 14.41 ± 0.60 | 6.42 ± 0.51 * | 3.81 ± 0.23 *# | <0.01 |
| IL-18 (pg/mL) | 1149 ± 57.54 | 787.0 ± 28.46 * | 610.3 ± 37.49 *# | <0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, P.; Liu, Y.; Liu, J.; Quan, J. Vitamin D Attenuates Hepatic Sinusoidal Capillarization in Type 2 Diabetes Mellitus– Metabolic Dysfunction-Associated Fatty Liver Disease via Dual Autophagy Activation and Pyroptosis Suppression in Liver Sinusoidal Endothelial Cells. Biomedicines 2025, 13, 1459. https://doi.org/10.3390/biomedicines13061459
Jiang P, Liu Y, Liu J, Quan J. Vitamin D Attenuates Hepatic Sinusoidal Capillarization in Type 2 Diabetes Mellitus– Metabolic Dysfunction-Associated Fatty Liver Disease via Dual Autophagy Activation and Pyroptosis Suppression in Liver Sinusoidal Endothelial Cells. Biomedicines. 2025; 13(6):1459. https://doi.org/10.3390/biomedicines13061459
Chicago/Turabian StyleJiang, Panpan, Yang Liu, Juxiang Liu, and Jinxing Quan. 2025. "Vitamin D Attenuates Hepatic Sinusoidal Capillarization in Type 2 Diabetes Mellitus– Metabolic Dysfunction-Associated Fatty Liver Disease via Dual Autophagy Activation and Pyroptosis Suppression in Liver Sinusoidal Endothelial Cells" Biomedicines 13, no. 6: 1459. https://doi.org/10.3390/biomedicines13061459
APA StyleJiang, P., Liu, Y., Liu, J., & Quan, J. (2025). Vitamin D Attenuates Hepatic Sinusoidal Capillarization in Type 2 Diabetes Mellitus– Metabolic Dysfunction-Associated Fatty Liver Disease via Dual Autophagy Activation and Pyroptosis Suppression in Liver Sinusoidal Endothelial Cells. Biomedicines, 13(6), 1459. https://doi.org/10.3390/biomedicines13061459