
Analysis of Energy Metabolism and Lipid Spatial Distribution in Hypoxic-Ischemic Encephalopathy Revealed by MALDI-MSI


Abstract
1. Introduction
2. Materials and Methods
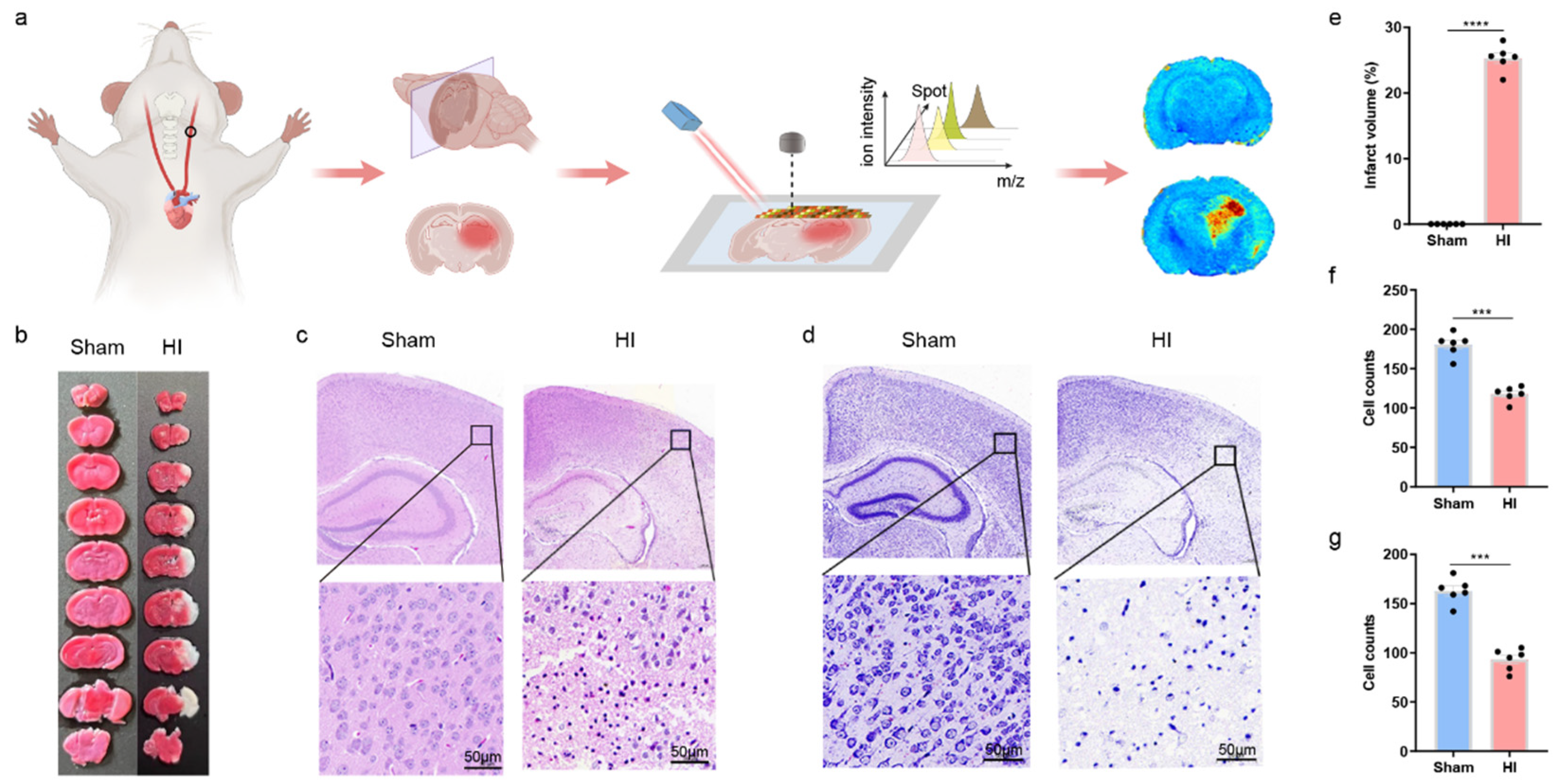
2.1. Animals and HIBD Models
2.2. TTC Staining
2.3. MALDI Mass Spectrometry Imaging and Data Analysis
2.4. Statistical Analysis
3. Results
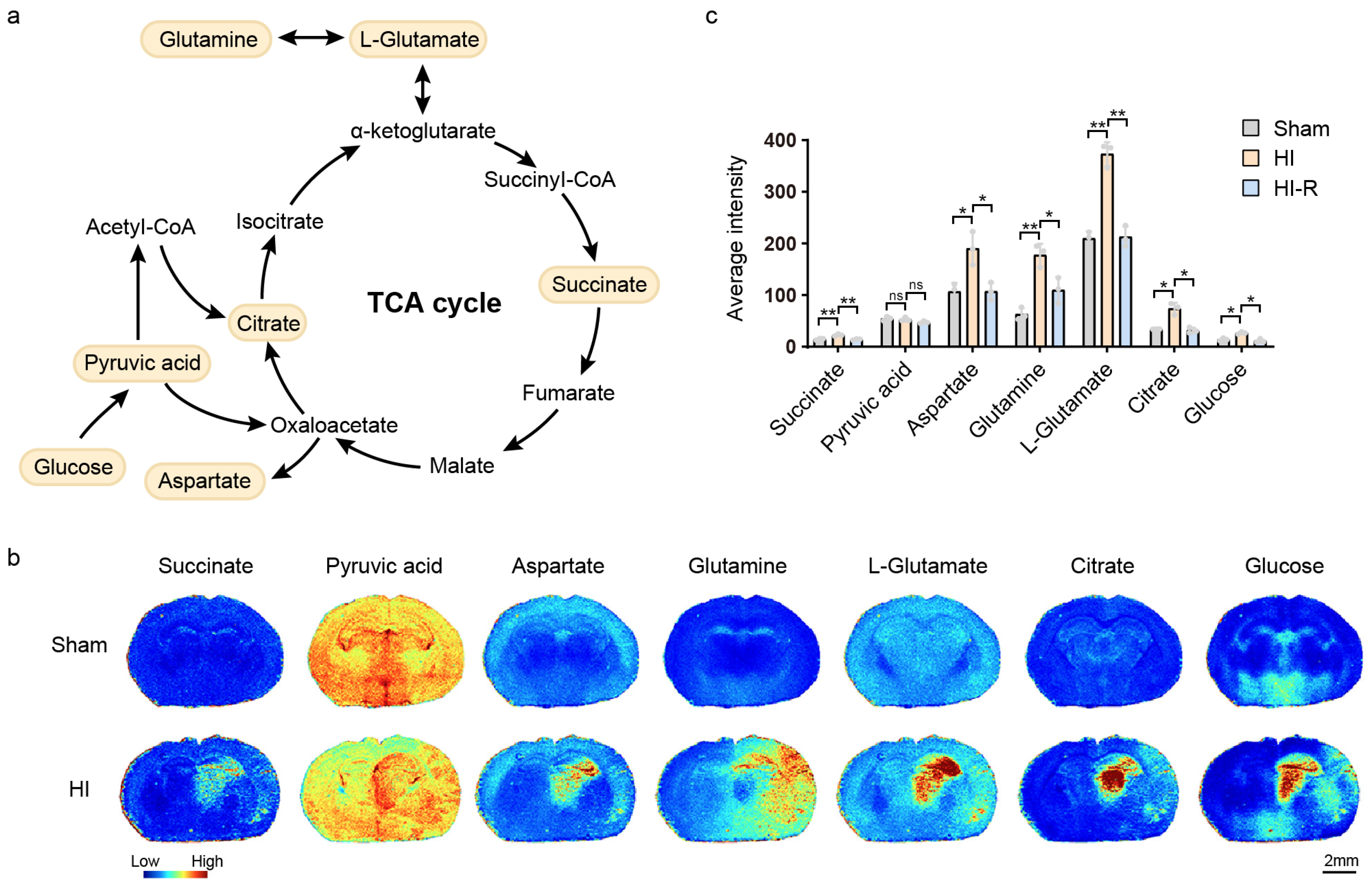
3.1. Blockage of the TCA Cycle
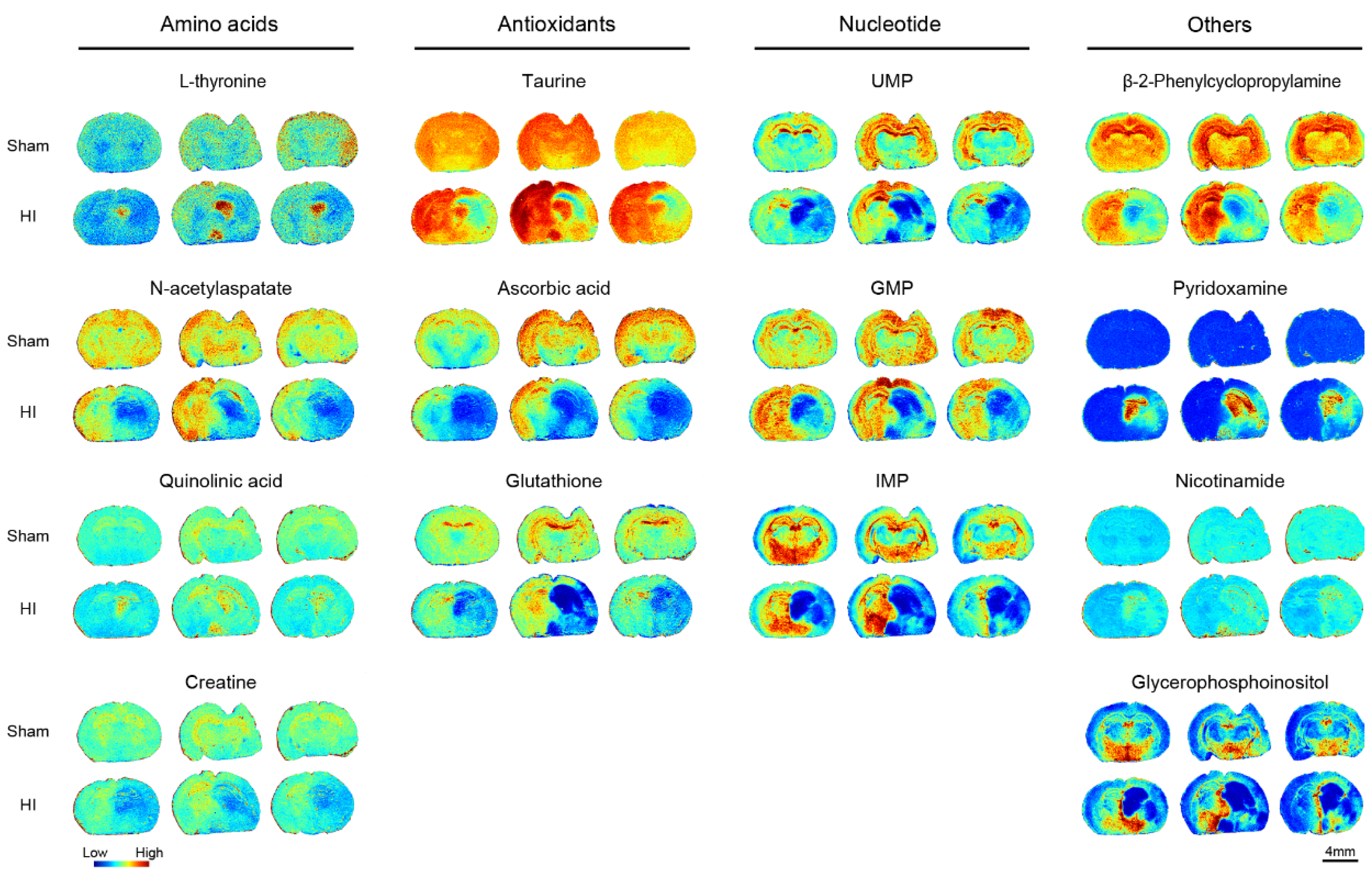
3.2. Impairment of Purine Metabolism
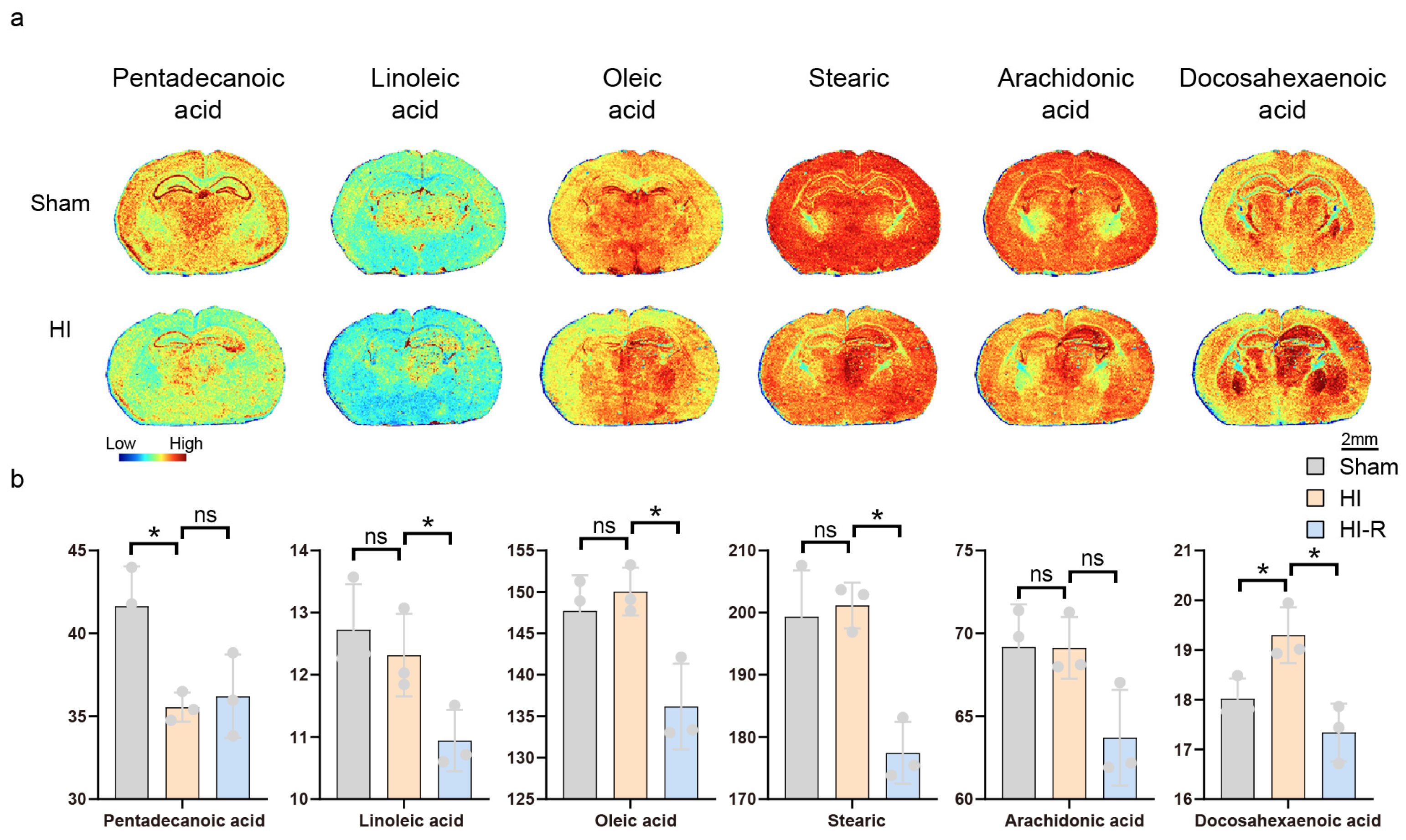
3.3. Alterations in Unsaturated Fatty Acid Metabolism
3.4. Phosphoglyceride and Other Metabolite Alterations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cotten, C.M.; Shankaran, S. Hypothermia for hypoxic–ischemic encephalopathy. Expert Rev. Obstet. Gynecol. 2010, 5, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Cornet, M.-C.; Kuzniewicz, M.; Scheffler, A.; Forquer, H.; Hamilton, E.; Newman, T.B.; Wu, Y.W. Perinatal Hypoxic-Ischemic Encephalopathy: Incidence over Time Within a Modern US Birth Cohort. Pediatr. Neurol. 2023, 149, 145. [Google Scholar] [CrossRef]
- Black, R.E.; Cousens, S.; Johnson, H.L.; Lawn, J.E.; Rudan, I.; Bassani, D.G.; Jha, P.; Campbell, H.; Walker, C.F.; Cibulskis, R.; et al. Global, regional, and national causes of child mortality in 2008: A systematic analysis. Lancet 2010, 375, 1969–1987. [Google Scholar] [CrossRef] [PubMed]
- Blair, E.; Watson, L. Epidemiology of cerebral palsy. Semin. Fetal. Neonatal Med. 2006, 11, 117–125. [Google Scholar] [CrossRef]
- Shang, Y.; Mu, L.; Guo, X.; Li, Y.; Wang, L.; Yang, W.; Li, S.; Shen, Q. Clinical significance of interleukin-6, tumor necrosis factor-α and high-sensitivity C-reactive protein in neonates with hypoxic-ischemic encephalopathy. Exp. Ther. Med. 2014, 8, 1259–1262. [Google Scholar] [CrossRef] [PubMed]
- Locci, E.; Noto, A.; Puddu, M.; Pomero, G.; Demontis, R.; Dalmazzo, C.; Delogu, A.; Fanos, V.; d’ Aloja, E.; Gancia, P. A longitudinal 1H-NMR metabolomics analysis of urine from newborns with hypoxic-ischemic encephalopathy undergoing hypothermia therapy. Clinical and medical legal insights. PLoS ONE 2018, 13, e0194267. [Google Scholar] [CrossRef]
- Massaro, A.N.; Wu, Y.W.; Bammler, T.K.; Comstock, B.; Mathur, A.; McKinstry, R.C.; Chang, T.; Mayock, D.E.; Mulkey, S.B.; Van Meurs, K.; et al. Plasma Biomarkers of Brain Injury in Neonatal Hypoxic-Ischemic Encephalopathy. J. Pediatr. 2018, 194, 67–75.e1. [Google Scholar] [CrossRef]
- Li, R.; Lee, J.K.; Govindan, R.B.; Graham, E.M.; Everett, A.D.; Perin, J.; Vezina, G.; Tekes, A.; Chen, M.W.; Northington, F.; et al. Plasma Biomarkers of Evolving Encephalopathy and Brain Injury in Neonates with Hypoxic-Ischemic Encephalopathy. J. Pediatr. 2023, 252, 146–153.e2. [Google Scholar] [CrossRef]
- Caramelo, I.; Coelho, M.; Rosado, M.; Cardoso, C.M.P.; Dinis, A.; Duarte, C.B.; Grãos, M.; Manadas, B. Biomarkers of hypoxic–ischemic encephalopathy: A systematic review. World J. Pediatr. 2023, 19, 505–548. [Google Scholar] [CrossRef]
- Caprioli, R.M.; Farmer, T.B.; Gile, J. Molecular Imaging of Biological Samples: Localization of Peptides and Proteins Using MALDI-TOF MS. Anal. Chem. 1997, 69, 4751–4760. [Google Scholar] [CrossRef]
- Shalak, L.; Perlman, J.M. Hypoxic–ischemic brain injury in the term infant-current concepts. Early Hum. Dev. 2004, 80, 125–141. [Google Scholar] [CrossRef]
- Vannucci, R.C.; Vannucci, S.J. Perinatal hypoxic-ischemic brain damage: Evolution of an animal model. Dev. Neurosci. 2005, 27, 81–86. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef]
- Kostić, D.A.; Dimitrijević, D.S.; Stojanović, G.S.; Palić, I.R.; Đorđević, A.S.; Ickovski, J.D. Xanthine Oxidase: Isolation, Assays of Activity, and Inhibition. J. Chem. 2015, 2015, 294858. [Google Scholar] [CrossRef]
- Sculley, D.G.; Dawson, P.A.; Emmerson, B.T.; Gordon, R.B. A review of the molecular basis of hypoxanthine-guanine phosphoribosyltransferase (HPRT) deficiency. Hum. Genet. 1992, 90, 195–207. [Google Scholar] [CrossRef]
- Flood, D.; Lee, E.S.; Taylor, C.T. Intracellular energy production and distribution in hypoxia. J. Biol. Chem. 2023, 299, 105103. [Google Scholar] [CrossRef]
- Golias, T.; Papandreou, I.; Sun, R.; Kumar, B.; Brown, N.V.; Swanson, B.J.; Pai, R.; Jaitin, D.; Le, Q.-T.; Teknos, T.N.; et al. Hypoxic repression of pyruvate dehydrogenase activity is necessary for metabolic reprogramming and growth of model tumours. Sci. Rep. 2016, 6, 31146. [Google Scholar] [CrossRef]
- Chavda, V.; Lu, B. Reverse Electron Transport at Mitochondrial Complex I in Ischemic Stroke, Aging, and Age-Related Diseases. Antioxidants 2023, 12, 895. [Google Scholar] [CrossRef]
- Xie, T.; Wang, J.; Han, J.; Huang, Z.; Nie, Z.; Wang, X.; Liu, H.; Jiang, L. In situ analysis of metabolic changes under hypoxic-ischemic encephalopathy via MALDI mass spectrometry imaging. Talanta 2024, 268, 125306. [Google Scholar] [CrossRef]
- Mácha, H.; Luptáková, D.; Juránek, I.; Andrén, P.E.; Havlíček, V. Hypoxic-Ischemic Insult Alters Polyamine and Neurotransmitter Abundance in the Specific Neonatal Rat Brain Subregions. ACS Chem. Neurosci. 2024, 15, 2811–2821. [Google Scholar] [CrossRef]
- Qin, X.; Cheng, J.; Zhong, Y.; Mahgoub, O.K.; Akter, F.; Fan, Y.; Aldughaim, M.; Xie, Q.; Qin, L.; Gu, L.; et al. Mechanism and Treatment Related to Oxidative Stress in Neonatal Hypoxic-Ischemic Encephalopathy. Front. Mol. Neurosci. 2019, 12, 447078. [Google Scholar] [CrossRef]
- Rüdiger, M.; Derks, J.B.; Annink, K.V.; Benders, M.J.; van Bel, F.; Franz, A.R. Allopurinol: Old Drug, New Indication in Neonates? Curr. Pharm. Des. 2017, 23, 5935–5942. [Google Scholar] [CrossRef]
- Engel, C.; Rüdiger, M.; Benders, M.J.N.L.; Bel, F.; Allegaert, K.; Naulaers, G.; Bassler, D.; Klebermaß-Schrehof, K.; Vento, M.; Vilan, A.; et al. Detailed statistical analysis plan for ALBINO: Effect of Allopurinol in addition to hypothermia for hypoxic-ischemic Brain Injury on Neurocognitive Outcome—A blinded randomized placebo-controlled parallel group multicenter trial for superiority (phase III). Trials 2024, 25, 81. [Google Scholar] [CrossRef]
- Carneiro-Freire, N.; Fernández-Fernández, C.; Mouriño-Bayolo, D.; Seco-Filgueira, M.; Adeva-Andany, M.M. Mitochondrial β-oxidation of saturated fatty acids in humans. Mitochondrion 2019, 46, 73–90. [Google Scholar] [CrossRef]
- Homaidan, F.R.; Chakroun, I.; Haidar, H.; El-Sabban, M.E. Protein Regulators of Eicosanoid Synthesis: Role in Inflammation. Curr. Protein Pept. Sci. 2002, 3, 467–484. [Google Scholar] [CrossRef]
- Zhao, M.; Zhu, P.; Fujino, M.; Zhuang, J.; Guo, H.; Sheikh, I.; Zhao, L.; Li, X.-K. Oxidative Stress in Hypoxic-Ischemic Encephalopathy: Molecular Mechanisms and Therapeutic Strategies. Int. J. Mol. Sci. 2016, 17, 2078. [Google Scholar] [CrossRef]
- Madrigal, J.L.; Olivenza, R.; Moro, M.A.; Lizasoain, I.; Lorenzo, P.; Rodrigo, J.; Leza, J.C. Glutathione Depletion, Lipid Peroxidation and Mitochondrial Dysfunction Are Induced by Chronic Stress in Rat Brain. Neuropsychopharmacology 2001, 24, 420–429. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A.; Iordache, F.; Stanca, L.; Geicu, O.I.; Bilteanu, L.; Serban, A.I. Antioxidant, anti-inflammatory and immunomodulatory roles of vitamins in COVID-19 therapy. Eur. J. Med. Chem. 2022, 232, 114175. [Google Scholar] [CrossRef]
- Vannucci, S.J.; Hagberg, H. Hypoxia-ischemia in the immature brain. J. Exp. Biol. 2004, 207, 3149–3154. [Google Scholar] [CrossRef]
- Wylezinska, M.; Delpy, D.T.; Lorek, A.; Penrice, J.; Peebles, D.; Reynolds, E.O.R.; Roth, S.C.; Wyatt, J.S.; Cady, E.B.; Owen-Reece, H.; et al. Delayed (“secondary”) cerebral energy failure after acute hypoxia-ischemia in the newborn piglet: Continuous 48-hour studies by phosphorus magnetic resonance spectroscopy. Pediatr. Res. 1994, 36, 699–706. [Google Scholar] [CrossRef]
- Bennet, L.; Gunn, A.J. Fetal hypoxia insults and patterns of brain injury: Insights from animal models. Clin. Perinatol. 2009, 36, 579–593. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Theoretical | Parent Ion | Experimental m/z | Delta (Da) | ppm |
|---|---|---|---|---|---|
| Succinate | 117.018 | [M−H]− | 117.018 | 0.000 | −2.008 |
| Pyruvic acid | 122.984 | [M+Cl]− | 122.984 | 0.000 | −2.830 |
| Taurine | 124.007 | [M−H]− | 124.008 | 0.001 | 5.403 |
| Creatine | 130.061 | [M−H]− | 130.061 | 0.000 | −0.792 |
| Aspartate | 132.030 | [M−H]− | 132.031 | 0.001 | 6.211 |
| Hypoxanthine | 135.031 | [M−H]− | 135.032 | 0.001 | 6.073 |
| Glutamine | 145.062 | [M−H]− | 145.063 | 0.001 | 8.203 |
| L-Glutamate | 146.046 | [M−H]− | 146.047 | 0.001 | 8.011 |
| Xanthine | 151.026 | [M−H]− | 151.026 | 0.000 | −0.662 |
| Nicotinamide | 157.016 | [M+Cl]− | 156.952 | −0.064 | −409.620 |
| Quinolinic acid | 166.013 | [M−H]− | 166.015 | 0.002 | 9.132 |
| Pyridoxamine | 167.082 | [M−H]− | 167.080 | −0.002 | −9.002 |
| β-Glycerophosphoric acid | 171.005 | [M−H]− | 170.935 | −0.070 | −411.104 |
| N-acetylaspartate | 174.041 | [M−H]− | 174.042 | 0.001 | 7.240 |
| Ascorbic acid | 175.025 | [M−H]− | 175.026 | 0.001 | 7.085 |
| Citrate | 191.020 | [M−H]− | 191.021 | 0.001 | 6.963 |
| Glucose | 215.032 | [M+Cl]− | 215.033 | 0.001 | 6.083 |
| Pentadecanoic acid | 241.216 | [M−H]− | 241.218 | 0.002 | 7.433 |
| Adenosine | 266.088 | [M−H]− | 265.957 | −0.131 | −493.746 |
| Inosine | 267.073 | [M−H]− | 267.100 | 0.027 | 99.448 |
| L-Thyronine | 272.092 | [M−H]− | 272.031 | −0.061 | −223.211 |
| Linoleic acid | 279.232 | [M−H]− | 279.243 | 0.011 | 39.906 |
| Oleic acid | 281.248 | [M−H]− | 281.258 | 0.010 | 37.309 |
| Stearic acid | 283.263 | [M−H]− | 283.280 | 0.017 | 59.461 |
| Arachidonic acid | 303.232 | [M−H]− | 303.261 | 0.029 | 96.108 |
| Glutathione | 306.076 | [M−H]− | 306.136 | 0.060 | 194.461 |
| Uridine monophosphate (UMP) | 323.027 | [M−H]− | 323.055 | 0.028 | 85.154 |
| Docosahexaenoic acid | 327.232 | [M−H]− | 327.270 | 0.038 | 116.563 |
| AMP | 346.056 | [M−H]− | 346.079 | 0.023 | 67.186 |
| Inosine-5′-monophosphate (IMP) | 347.039 | [M−H]− | 347.110 | 0.071 | 205.378 |
| GMP | 362.051 | [M−H]− | 362.084 | 0.033 | 92.059 |
| Glycerophosphoinositol | 369.035 | [M+Cl]− | 369.065 | 0.030 | 81.830 |
| ADP | 426.022 | [M−H]− | 426.088 | 0.066 | 154.734 |
| PE (18:0) | 480.310 | [M−H]− | 480.382 | 0.072 | 150.924 |
| ATP | 505.988 | [M−H]− | 505.983 | −0.005 | −10.712 |
| PI (18:0) | 599.320 | [M−H]− | 599.369 | 0.049 | 81.542 |
| NADH | 664.116 | [M−H]− | 664.414 | 0.298 | 448.117 |
| PA (34:2) | 671.465 | [M−H]− | 671.501 | 0.036 | 54.162 |
| PA (34:1) | 673.481 | [M−H]− | 673.515 | 0.034 | 49.994 |
| PC (P-16:0/P-16:0) | 700.564 | [M−H]− | 700.570 | 0.006 | 8.633 |
| PA (36:1) | 701.513 | [M−H]− | 701.527 | 0.014 | 20.484 |
| PA (22:1/15:0) | 715.527 | [M−H]− | 715.627 | 0.100 | 139.433 |
| PE (34:1) | 716.524 | [M−H]− | 716.546 | 0.022 | 31.374 |
| PA (15:0/20:4) | 717.426 | [M+Cl]− | 717.569 | 0.143 | 199.798 |
| PA (38:4) | 723.497 | [M−H]− | 723.506 | 0.009 | 12.467 |
| PE (36:3p) | 724.528 | [M−H]− | 724.563 | 0.035 | 48.905 |
| PE (36:1) | 744.554 | [M−H]− | 744.635 | 0.081 | 109.083 |
| PG 34:2 (18:2_16:0) | 745.501 | [M−H]− | 745.606 | 0.105 | 140.292 |
| PE (38:6p) | 746.512 | [M−H]− | 746.576 | 0.064 | 85.843 |
| PA (40:6) | 747.497 | [M−H]− | 747.521 | 0.024 | 32.134 |
| PE (38:4p) | 750.543 | [M−H]− | 750.606 | 0.063 | 83.650 |
| PE (38:3p) | 752.559 | [M−H]− | 752.583 | 0.024 | 32.068 |
| PE (38:7) | 760.491 | [M−H]− | 760.582 | 0.091 | 119.422 |
| PE (38:6) | 762.508 | [M−H]− | 762.531 | 0.023 | 30.334 |
| PE (38:4) | 766.538 | [M−H]− | 766.542 | 0.004 | 5.047 |
| PE (P-40:6) | 774.544 | [M−H]− | 774.570 | 0.026 | 33.232 |
| PE (40:6) | 790.539 | [M−H]− | 790.563 | 0.024 | 30.144 |
| PG (40:6) | 821.533 | [M−H]− | 821.536 | 0.003 | 4.002 |
| PS (40:6) | 834.529 | [M−H]− | 834.563 | 0.034 | 40.742 |
| PI (34:1) | 835.533 | [M−H]− | 835.547 | 0.014 | 16.629 |
| PI (36:4) | 857.519 | [M−H]− | 857.545 | 0.026 | 30.903 |
| PI (38:4) | 885.550 | [M−H]− | 885.570 | 0.020 | 22.811 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Chen, P.; Yu, L.; Gao, C.; Wang, S.; Yang, Z.; Feng, Z. Analysis of Energy Metabolism and Lipid Spatial Distribution in Hypoxic-Ischemic Encephalopathy Revealed by MALDI-MSI. Biomedicines 2025, 13, 1431. https://doi.org/10.3390/biomedicines13061431
Zhao X, Chen P, Yu L, Gao C, Wang S, Yang Z, Feng Z. Analysis of Energy Metabolism and Lipid Spatial Distribution in Hypoxic-Ischemic Encephalopathy Revealed by MALDI-MSI. Biomedicines. 2025; 13(6):1431. https://doi.org/10.3390/biomedicines13061431
Chicago/Turabian StyleZhao, Xingxing, Peipei Chen, Lun Yu, Chuchu Gao, Sannan Wang, Zuming Yang, and Zongtai Feng. 2025. "Analysis of Energy Metabolism and Lipid Spatial Distribution in Hypoxic-Ischemic Encephalopathy Revealed by MALDI-MSI" Biomedicines 13, no. 6: 1431. https://doi.org/10.3390/biomedicines13061431
APA StyleZhao, X., Chen, P., Yu, L., Gao, C., Wang, S., Yang, Z., & Feng, Z. (2025). Analysis of Energy Metabolism and Lipid Spatial Distribution in Hypoxic-Ischemic Encephalopathy Revealed by MALDI-MSI. Biomedicines, 13(6), 1431. https://doi.org/10.3390/biomedicines13061431