
The Development and Assessment of a Unique Disulfidptosis-Associated lncRNA Profile for Immune Microenvironment Prediction and Personalized Therapy in Gastric Adenocarcinoma

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
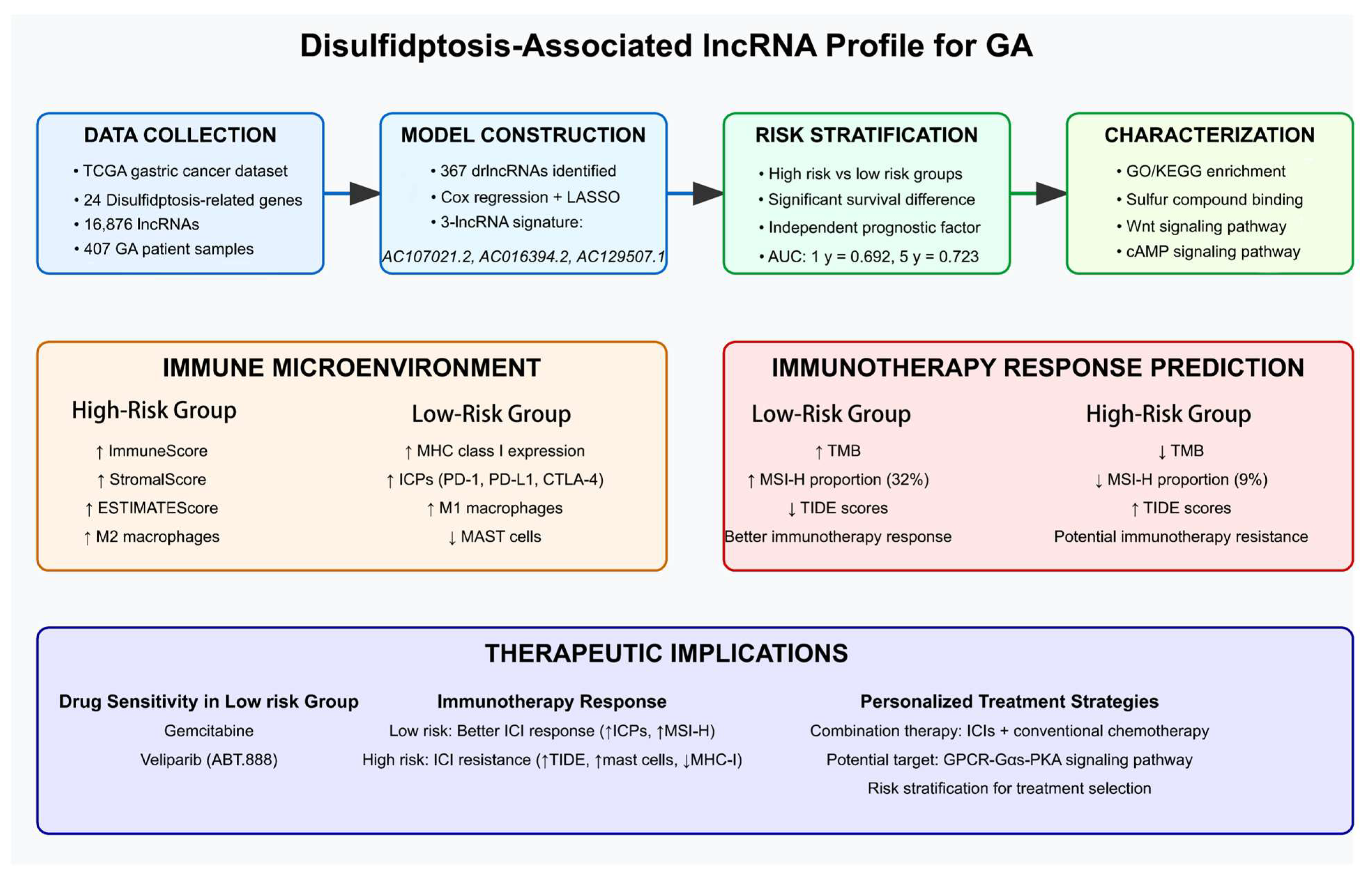
Abstract
1. Introduction
2. Materials and Methods
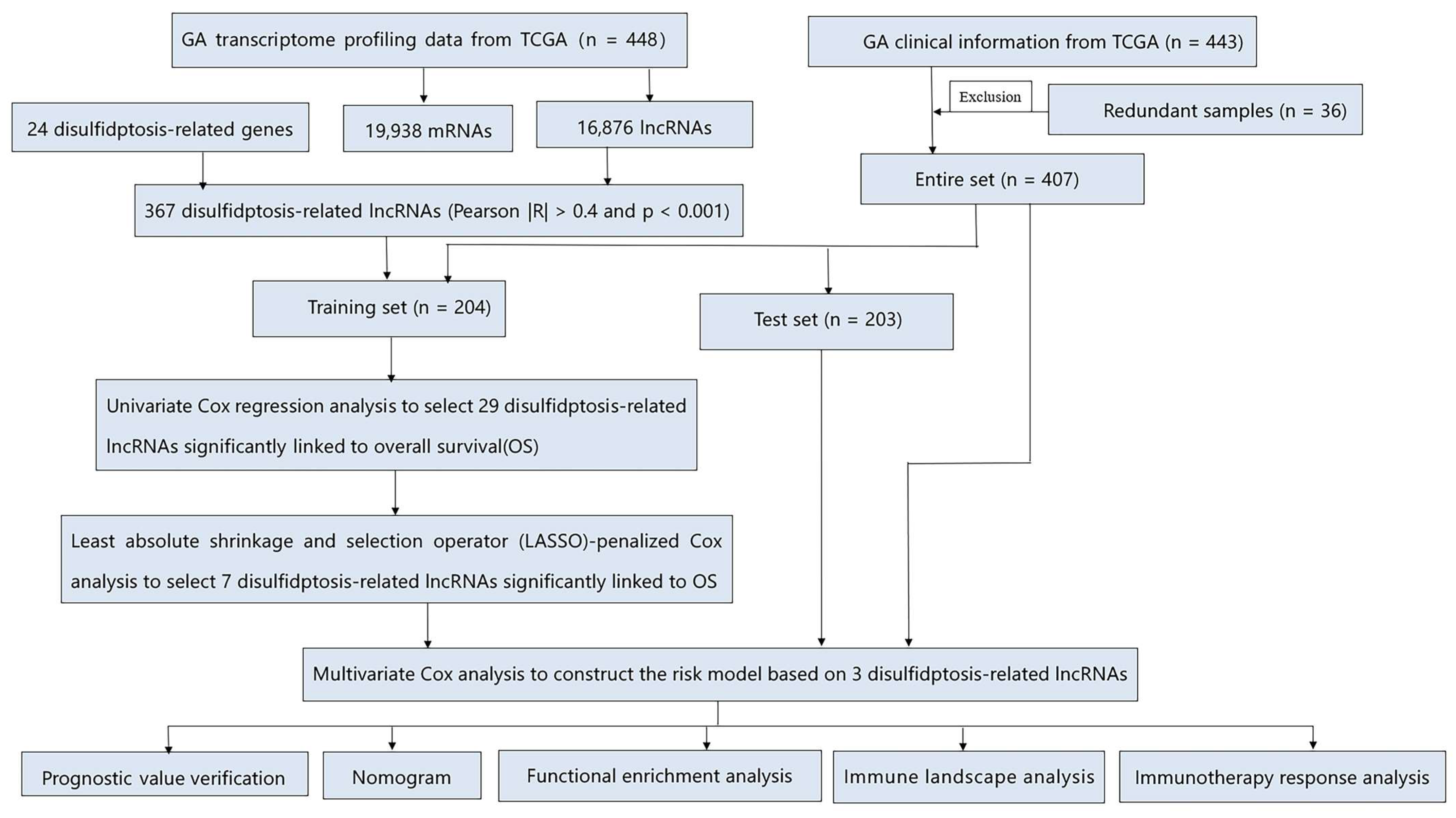
2.1. Data Source
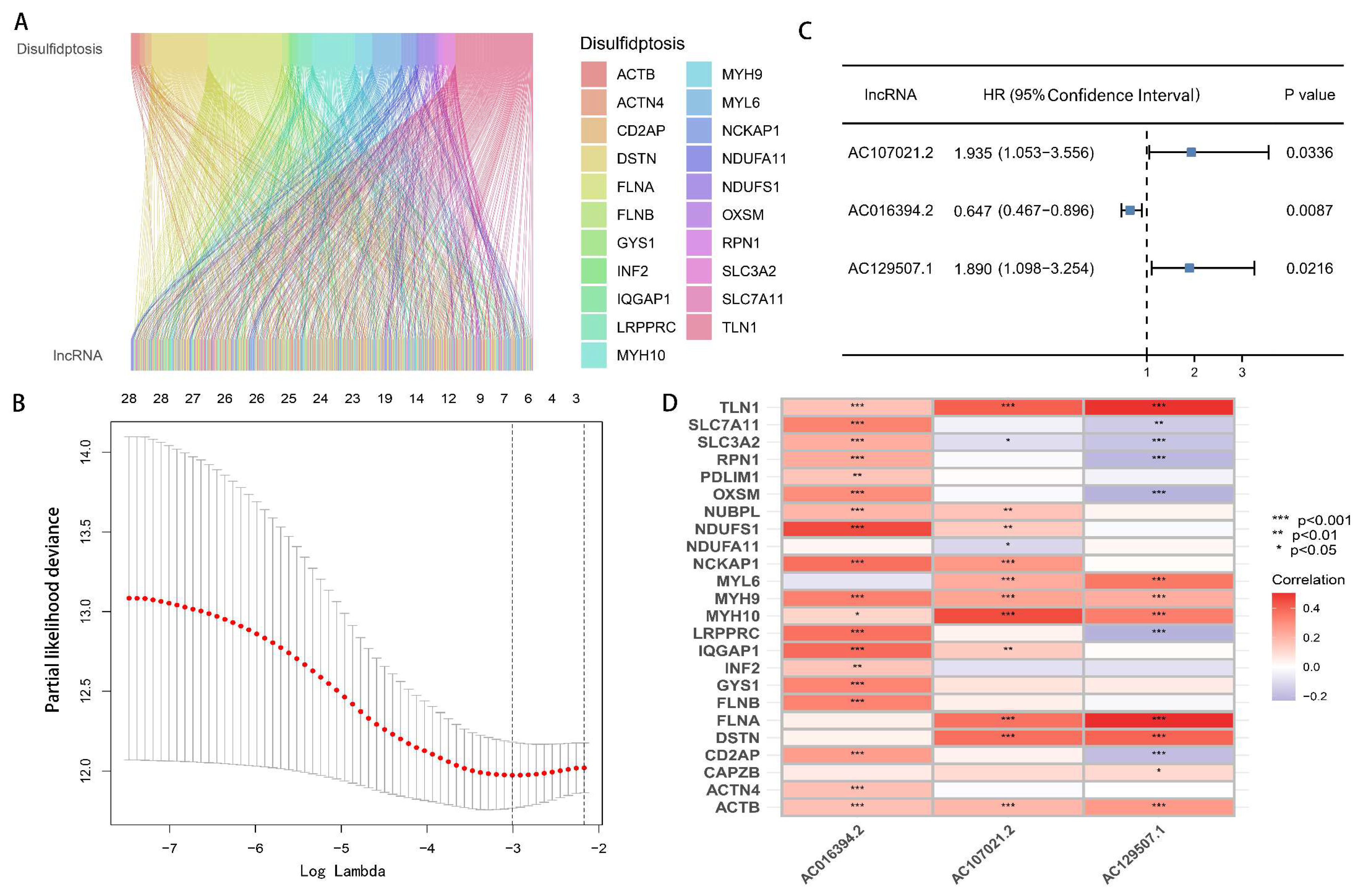
2.2. The Development and Verification of the drlncRNAs Signature
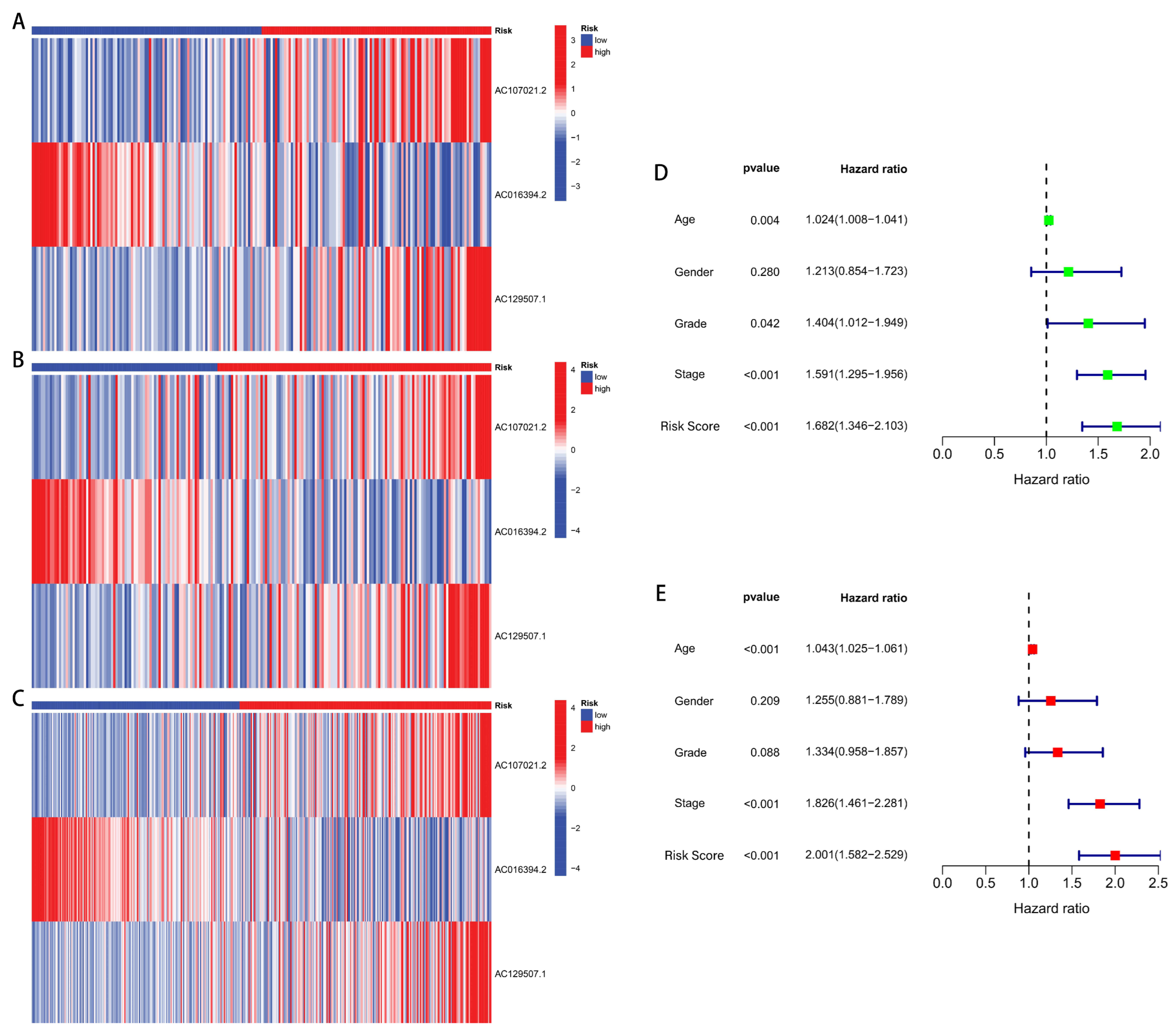
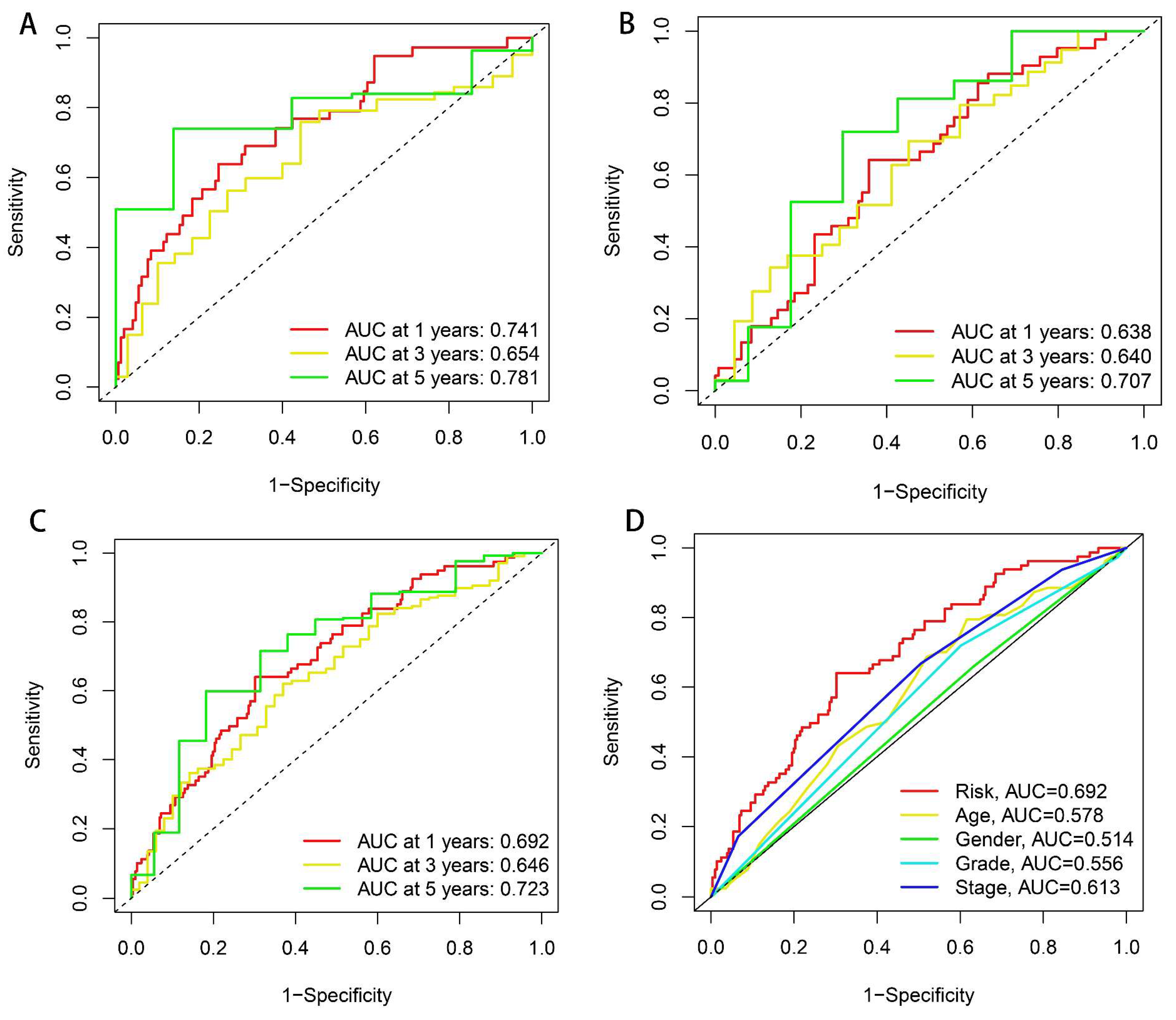
2.3. Independent Prognostic Analysis and ROC Curve Plotting
2.4. Nomogram and Calibration
2.5. GO and KEGG Enrichment Analysis
2.6. Investigation of Tumor Microenvironment, Immune Infiltration, and Immune Checkpoints
2.7. TMB, MSI, and TIDE
2.8. Drug Sensitivity Analysis
3. Results
3.1. Data Source and Processing
3.2. Development and Verification of drlncRNA Predictive Algorithm
3.3. Nomogram and Calibration Curves
3.4. Functional Analysis of Model
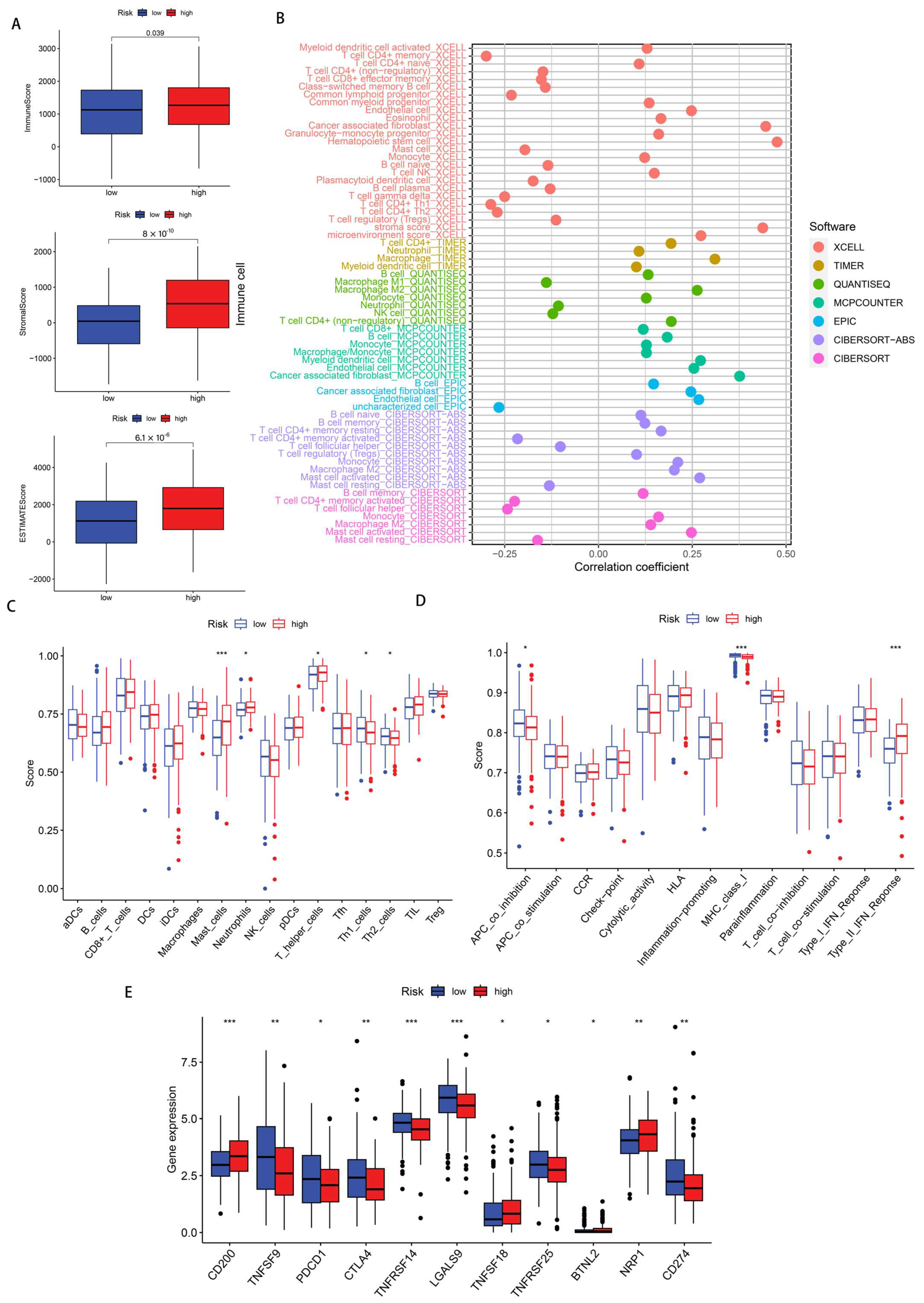
3.5. Immune Infiltration Status
3.6. Immunotherapy Response Analysis
3.7. Drug Sensitivity Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van Cutsem, E.; Sagaert, X.; Topal, B.; Haustermans, K.; Prenen, H. Gastric Cancer. Lancet 2016, 388, 2654–2664. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.; Arnold, M.; Camargo, M.C.; Gini, A.; Kunzmann, A.T.; Matsuda, T.; Meheus, F.; Verhoeven, R.H.A.; Vignat, J.; Laversanne, M.; et al. The Current and Future Incidence and Mortality of Gastric Cancer in 185 Countries, 2020–2040: A Population-Based Modelling Study. eClinicalMedicine 2022, 47, 101404. [Google Scholar] [CrossRef] [PubMed]
- Thrift, A.P.; El-Serag, H.B. Burden of Gastric Cancer. Clin. Gastroenterol. Hepatol. 2020, 18, 534–542. [Google Scholar] [CrossRef]
- Karimi, P.; Islami, F.; Anandasabapathy, S.; Freedman, N.D.; Kamangar, F. Gastric Cancer: Descriptive Epidemiology, Risk Factors, Screening, and Prevention. Cancer Epidemiol. Biomark. Prev. 2014, 23, 700–713. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Meltzer, S.J. Gastric Cancer in the Era of Precision Medicine. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 348–358. [Google Scholar] [CrossRef]
- Jin, X.; Liu, Z.; Yang, D.; Yin, K.; Chang, X. Recent Progress and Future Perspectives of Immunotherapy in Advanced Gastric Cancer. Front. Immunol. 2022, 13, 948647. [Google Scholar] [CrossRef]
- Maleki Vareki, S.; Garrigós, C.; Duran, I. Biomarkers of Response to Pd-1/Pd-L1 Inhibition. Crit. Rev. Oncol. Hematol. 2017, 116, 116–124. [Google Scholar] [CrossRef]
- Liu, X.; Nie, L.; Zhang, Y.; Yan, Y.; Wang, C.; Colic, M.; Olszewski, K.; Horbath, A.; Chen, X.; Lei, G.; et al. Actin Cytoskeleton Vulnerability to Disulfide Stress Mediates Disulfidptosis. Nat. Cell Biol. 2023, 25, 404–414. [Google Scholar] [CrossRef]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine Transporter Slc7a11/Xct in Cancer: Ferroptosis, Nutrient Dependency, and Cancer Therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef]
- Zheng, T.; Liu, Q.; Xing, F.; Zeng, C.; Wang, W. Disulfidptosis: A New Form of Programmed Cell Death. J. Exp. Clin. Cancer Res. 2023, 42, 137. [Google Scholar] [CrossRef]
- Zheng, P.; Zhou, C.; Ding, Y.; Duan, S. Disulfidptosis: A New Target for Metabolic Cancer Therapy. J. Exp. Clin. Cancer Res. 2023, 42, 103. [Google Scholar] [CrossRef] [PubMed]
- Shuai, Y.; Ma, Z.; Liu, W.; Yu, T.; Yan, C.; Jiang, H.; Tian, S.; Xu, T.; Shu, Y. Tead4 Modulated Lncrna Mnx1-As1 Contributes to Gastric Cancer Progression Partly through Suppressing Btg2 and Activating Bcl2. Mol. Cancer 2020, 19, 6. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Gou, H.; Wang, D.; Li, C.; Li, Y.; Su, H.; Wang, X.; Zhang, X.; Yu, J. Lncrna Tnfrsf10a-As1 Promotes Gastric Cancer by Directly Binding to Oncogenic Mpzl1 and Is Associated with Patient Outcome. Int. J. Biol. Sci. 2022, 18, 3156–3166. [Google Scholar] [CrossRef]
- Qi, F.; Liu, X.; Wu, H.; Yu, X.; Wei, C.; Huang, X.; Ji, G.; Nie, F.; Wang, K. Long Noncoding Agap2-As1 Is Activated by Sp1 and Promotes Cell Proliferation and Invasion in Gastric Cancer. J. Hematol. Oncol. 2017, 10, 48. [Google Scholar] [CrossRef]
- Liu, Y.W.; Xia, R.; Lu, K.; Xie, M.; Yang, F.; Sun, M.; De, W.; Wang, C.; Ji, G. Lincrnafezf1-As1 Represses P21 Expression to Promote Gastric Cancer Proliferation through Lsd1-Mediated H3k4me2 Demethylation. Mol. Cancer 2017, 16, 39. [Google Scholar] [CrossRef]
- Mao, M.; Yu, Q.; Huang, R.; Lu, Y.; Wang, Z.; Liao, L. Stromal Score as a Prognostic Factor in Primary Gastric Cancer and Close Association with Tumor Immune Microenvironment. Cancer Med. 2020, 9, 4980–4990. [Google Scholar] [CrossRef] [PubMed]
- Francioso, A.; Conrado, A.B.; Mosca, L.; Fontana, M. Chemistry and Biochemistry of Sulfur Natural Compounds: Key Intermediates of Metabolism and Redox Biology. Oxid. Med. Cell. Longev. 2020, 2020, 8294158. [Google Scholar] [CrossRef]
- Barayeu, U.; Sawa, T.; Nishida, M.; Wei, F.Y.; Motohashi, H.; Akaike, T. Supersulfide Biology and Translational Medicine for Disease Control. Br. J. Pharmacol. 2023. [Google Scholar] [CrossRef]
- Jiang, Y.; Sun, M. Slc7a11: The Achilles Heel of Tumor? Front. Immunol. 2024, 15, 1438807. [Google Scholar] [CrossRef]
- Zhou, Q.; Yu, H.; Chen, Y.; Ren, J.; Lu, Y.; Sun, Y. The Crl3(Kctd10) Ubiquitin Ligase-Usp18 Axis Coordinately Regulates Cystine Uptake and Ferroptosis by Modulating Slc7a11. Proc. Natl. Acad. Sci. USA 2024, 121, e2320655121. [Google Scholar] [CrossRef]
- Shi, W.; Sun, S.; Liu, H.; Meng, Y.; Ren, K.; Wang, G.; Liu, M.; Wu, J.; Zhang, Y.; Huang, H.; et al. Guiding Bar Motif of Thioredoxin Reductase 1 Modulates Enzymatic Activity and Inhibitor Binding by Communicating with the Co-Factor Fad and Regulating the Flexible C-Terminal Redox Motif. Redox Biol. 2024, 70, 103050. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.S.; Ling, H.H.; Setiawan, S.A.; Hardianti, M.S.; Fong, I.H.; Yeh, C.T.; Chen, J.H. Therapeutic Targeting of Thioredoxin Reductase 1 Causes Ferroptosis While Potentiating Anti-Pd-1 Efficacy in Head and Neck Cancer. Chem. Biol. Interact. 2024, 395, 111004. [Google Scholar] [CrossRef] [PubMed]
- Georgiou-Siafis, S.K.; Tsiftsoglou, A.S. The Key Role of Gsh in Keeping the Redox Balance in Mammalian Cells: Mechanisms and Significance of Gsh in Detoxification Via Formation of Conjugates. Antioxidants 2023, 12, 1953. [Google Scholar] [CrossRef]
- Liang, Q.; Yan, J.; Zhang, S.; Yang, N.; Li, M.; Jin, Y.; Bai, F.; Wu, W.; Cheng, Z. Ctra Activates the Expression of Glutathione S-Transferase Conferring Oxidative Stress Resistance to Ehrlichia Chaffeensis. Front. Cell. Infect. Microbiol. 2022, 12, 1081614. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhuang, L.; Gan, B. Disulfidptosis: Disulfide Stress-Induced Cell Death. Trends Cell Biol. 2024, 34, 327–337. [Google Scholar] [CrossRef]
- Yan, Y.; Teng, H.; Hang, Q.; Kondiparthi, L.; Lei, G.; Horbath, A.; Liu, X.; Mao, C.; Wu, S.; Zhuang, L.; et al. Slc7a11 Expression Level Dictates Differential Responses to Oxidative Stress in Cancer Cells. Nat. Commun. 2023, 14, 3673. [Google Scholar] [CrossRef]
- Aran, D.; Hu, Z.; Butte, A.J. Xcell: Digitally Portraying the Tissue Cellular Heterogeneity Landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating The population Abundance of Tissue-Infiltrating Immune and Stromal Cell Populations Using Gene Expression. Genome Biol. 2016, 17, 218. [Google Scholar]
- Racle, J.; de Jonge, K.; Baumgaertner, P.; Speiser, D.E.; Gfeller, D. Simultaneous Enumeration of Cancer and Immune Cell Types from Bulk Tumor Gene Expression Data. eLife 2017, 6, e26476. [Google Scholar] [CrossRef]
- Finotello, F.; Mayer, C.; Plattner, C.; Laschober, G.; Rieder, D.; Hackl, H.; Krogsdam, A.; Loncova, Z.; Posch, W.; Wilflingseder, D.; et al. Molecular and Pharmacological Modulators of the Tumor Immune Contexture Revealed by Deconvolution of Rna-Seq Data. Genome Med. 2019, 11, 34. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust Enumeration of Cell Subsets from Tissue Expression Profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Wang, Y.; Niu, R. Identification of the Three Subtypes and the Prognostic Characteristics of Stomach Adenocarcinoma: Analysis of the Hypoxia-Related Long Non-Coding Rnas. Funct. Integr. Genom. 2022, 22, 919–936. [Google Scholar] [CrossRef] [PubMed]
- Mao, F.; Li, Z.; Li, Y.; Huang, H.; Shi, Z.; Li, X.; Wu, D.; Liu, H.; Chen, J. Necroptosis-Related Lncrna in Lung Adenocarcinoma: A Comprehensive Analysis Based on a Prognosis Model and a Competing Endogenous Rna Network. Front. Genet. 2022, 13, 940167. [Google Scholar] [CrossRef]
- Lu, Y.; Wu, J.; Li, X.; Leng, Q.; Tan, J.; Huang, H.; Zhong, R.; Chen, Z.; Zhang, Y. Cuproptosis-Related Lncrnas Emerge as a Novel Signature for Predicting Prognosis in Prostate Carcinoma and Functional Experimental Validation. Front. Immunol. 2024, 15, 1471198. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Li, X.; Peng, Y.; Zhou, Y. Comprehensive Analysis of Disulfidptosis-Related Lncrnas in Molecular Classification, Immune Microenvironment Characterization and Prognosis of Gastric Cancer. Biomedicines 2023, 11, 3165. [Google Scholar] [CrossRef] [PubMed]
- Wu, V.H.; Yung, B.S.; Faraji, F.; Saddawi-Konefka, R.; Wang, Z.; Wenzel, A.T.; Song, M.J.; Pagadala, M.S.; Clubb, L.M.; Chiou, J.; et al. The Gpcr-Gα(S)-Pka Signaling Axis Promotes T Cell Dysfunction and Cancer Immunotherapy Failure. Nat. Immunol. 2023, 24, 1318–1330. [Google Scholar] [CrossRef]
- Wu, J.J.; Yang, Y.; Peng, W.T.; Sun, J.C.; Sun, W.Y.; Wei, W. G Protein-Coupled Receptor Kinase 2 Regulating Β2-Adrenergic Receptor Signaling in M2-Polarized Macrophages Contributes to Hepatocellular Carcinoma Progression. Onco Targets Ther. 2019, 12, 5499–5513. [Google Scholar] [CrossRef]
- Yano, S.; Ghosh, P.; Kusaba, H.; Buchholz, M.; Longo, D.L. Effect of Promoter Methylation on the Regulation of Ifn-Gamma Gene During in Vitro Differentiation of Human Peripheral Blood T Cells into a Th2 Population. J. Immunol. 2003, 171, 2510–2516. [Google Scholar] [CrossRef]
- Qian, X.; Gu, L.; Ning, H.; Zhang, Y.; Hsueh, E.C.; Fu, M.; Hu, X.; Wei, L.; Hoft, D.F.; Liu, J. Increased Th17 Cells in the Tumor Microenvironment Is Mediated by Il-23 Via Tumor-Secreted Prostaglandin E2. J. Immunol. 2013, 190, 5894–5902. [Google Scholar] [CrossRef]
- Ji, Y.; Zhang, W. Th17 Cells: Positive or Negative Role in Tumor? Cancer Immunol. Immunother. 2010, 59, 979–987. [Google Scholar] [CrossRef]
- Asadzadeh, Z.; Mohammadi, H.; Safarzadeh, E.; Hemmatzadeh, M.; Mahdian-Shakib, A.; Jadidi-Niaragh, F.; Azizi, G.; Baradaran, B. The Paradox of Th17 Cell Functions in Tumor Immunity. Cell. Immunol. 2017, 322, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Y.; Liu, J.; Chen, J.; Wang, J.; Hua, H.; Jiang, Y. Camp-Pka/Epac Signaling and Cancer: The Interplay in Tumor Microenvironment. J. Hematol. Oncol. 2024, 17, 5. [Google Scholar] [CrossRef]
- Huang, Y.C.; Feng, Z.P. The Good and Bad of Microglia/Macrophages: New Hope in Stroke Therapeutics. Acta Pharmacol. Sin. 2013, 34, 6–7. [Google Scholar] [CrossRef]
- Aras, S.; Zaidi, M.R. Tameless Traitors: Macrophages in Cancer Progression and Metastasis. Br. J. Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Toledo, B.; Chen, L.Z.; Paniagua-Sancho, M.; Marchal, J.A.; Perán, M.; Giovannetti, E. Deciphering the Performance of Macrophages in Tumour Microenvironment: A Call for Precision Immunotherapy. J. Hematol. Oncol. 2024, 17, 44. [Google Scholar] [CrossRef]
- Somasundaram, R.; Connelly, T.; Choi, R.; Choi, H.; Samarkina, A.; Li, L.; Gregorio, E.; Chen, Y.; Thakur, R.; Abdel-Mohsen, M.; et al. Tumor-Infiltrating Mast Cells Are Associated with Resistance to Anti-Pd-1 Therapy. Nat. Commun. 2021, 12, 346. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.C.; Balko, J.M. Mechanisms of Mhc-I Downregulation and Role in Immunotherapy Response. Front. Immunol. 2022, 13, 844866. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Shitara, K.; Moehler, M.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Bragagnoli, A.C.; et al. First-Line Nivolumab Plus Chemotherapy Versus Chemotherapy Alone for Advanced Gastric, Gastro-Oesophageal Junction, and Oesophageal Adenocarcinoma (Checkmate 649): A Randomised, Open-Label, Phase 3 Trial. Lancet 2021, 398, 27–40. [Google Scholar] [CrossRef]
- Pennock, G.K.; Chow, L.Q. The Evolving Role of Immune Checkpoint Inhibitors in Cancer Treatment. Oncologist 2015, 20, 812–822. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer Immunotherapy Using Checkpoint Blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Gaikwad, S.; Agrawal, M.Y.; Kaushik, I.; Ramachandran, S.; Srivastava, S.K. Immune Checkpoint Proteins: Signaling Mechanisms and Molecular Interactions in Cancer Immunotherapy. Semin. Cancer Biol. 2022, 86, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Ionov, Y.; Peinado, M.A.; Malkhosyan, S.; Shibata, D.; Perucho, M. Ubiquitous Somatic Mutations in Simple Repeated Sequences Reveal a New Mechanism for Colonic Carcinogenesis. Nature 1993, 363, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lamberti, G.; Di Federico, A.; Alessi, J.; Ferrara, R.; Sholl, M.L.; Awad, M.M.; Vokes, N.; Ricciuti, B. Tumor Mutational Burden for the Prediction of Pd-(L)1 Blockade Efficacy in Cancer: Challenges and Opportunities. Ann. Oncol. 2024, 35, 508–522. [Google Scholar] [CrossRef]
- Wang, F.; Wei, X.L.; Wang, F.H.; Xu, N.; Shen, L.; Dai, G.H.; Yuan, X.L.; Chen, Y.; Yang, S.J.; Shi, J.H.; et al. Safety, Efficacy and Tumor Mutational Burden as a Biomarker of Overall Survival Benefit in Chemo-Refractory Gastric Cancer Treated with Toripalimab, a Pd-1 Antibody in Phase Ib/Ii Clinical Trial Nct02915432. Ann. Oncol. 2019, 30, 1479–1486. [Google Scholar] [CrossRef]
- Robert, C.; Lewis, K.D.; Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Biomarkers of Treatment Benefit with Atezolizumab Plus Vemurafenib Plus Cobimetinib in Braf(V600) Mutation-Positive Melanoma. Ann. Oncol. 2022, 33, 544–555. [Google Scholar] [CrossRef]
- Budczies, J.; Kazdal, D.; Menzel, M.; Beck, S.; Kluck, K.; Altbürger, C.; Schwab, C.; Allgäuer, M.; Ahadova, A.; Kloor, M.; et al. Tumour Mutational Burden: Clinical Utility, Challenges and Emerging Improvements. Nat. Rev. Clin. Oncol. 2024, 21, 725–742. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting P53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef]
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting Mutant P53 for Cancer Therapy: Direct and Indirect Strategies. J. Hematol. Oncol. 2021, 14, 157. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. Tp53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef]
- Okamura, R.; Kato, S.; Lee, S.; Jimenez, R.E.; Sicklick, J.K.; Kurzrock, R. Arid1a Alterations Function as a Biomarker for Longer Progression-Free Survival after Anti-Pd-1/Pd-L1 Immunotherapy. J. Immunother. Cancer 2020, 8, e000438. [Google Scholar] [CrossRef]
- Goswami, S.; Chen, Y.; Anandhan, S.; Szabo, P.M.; Basu, S.; Blando, J.M.; Liu, W.; Zhang, J.; Natarajan, S.M.; Xiong, L.; et al. Arid1a Mutation Plus Cxcl13 Expression Act as Combinatorial Biomarkers to Predict Responses to Immune Checkpoint Therapy in Mucc. Sci. Transl. Med. 2020, 12, eabc4220. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, M.; Jiang, Z.; Wang, X. Arid1a Mutations Are Associated with Increased Immune Activity in Gastrointestinal Cancer. Cells 2019, 8, 678. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, M.S.; Sonpavde, G.; Powles, T.; Necchi, A.; Burotto, M.; Schenker, M.; Sade, J.P.; Bamias, A.; Beuzeboc, P.; Bedke, J.; et al. Nivolumab Plus Gemcitabine-Cisplatin in Advanced Urothelial Carcinoma. N. Engl. J. Med. 2023, 389, 1778–1789. [Google Scholar] [CrossRef] [PubMed]
- Tempero, M.A.; Pelzer, U.; O’Reilly, E.M.; Winter, J.; Oh, D.Y.; Li, C.P.; Tortora, G.; Chang, H.M.; Lopez, C.D.; Bekaii-Saab, T.; et al. Adjuvant Nab-Paclitaxel + Gemcitabine in Resected Pancreatic Ductal Adenocarcinoma: Results from a Randomized, Open-Label, Phase Iii Trial. J. Clin. Oncol. 2023, 41, 2007–2019. [Google Scholar] [CrossRef]
- Malhotra, M.K.; Pahuja, S.; Kiesel, B.F.; Appleman, L.J.; Ding, F.; Lin, Y.; Tawbi, H.A.; Stoller, R.G.; Lee, J.J.; Belani, C.P.; et al. A Phase 1 Study of Veliparib (Abt-888) Plus Weekly Carboplatin and Paclitaxel in Advanced Solid Malignancies, with an Expansion Cohort in Triple Negative Breast Cancer (Tnbc) (Etctn 8620). Breast Cancer Res. Treat. 2023, 198, 487–498. [Google Scholar] [CrossRef]
- Cai, Z.; Wuri, Q.; Song, Y.; Qu, X.; Hu, H.; Cao, S.; Wu, H.; Wu, J.; Wang, C.; Yu, X.; et al. Circrna-Loaded Dc Vaccine in Combination with Low-Dose Gemcitabine Induced Potent Anti-Tumor Immunity in Pancreatic Cancer Model. Cancer Immunol. Immunother. 2025, 74, 68. [Google Scholar] [CrossRef]
- Wang, J.; Ma, J.; Xie, F.; Miao, F.; Lv, L.; Huang, Y.; Zhang, X.; Yu, J.; Tai, Z.; Zhu, Q.; et al. Immunogenic Cell Death-Based Cancer Vaccines: Promising Prospect in Cancer Therapy. Front. Immunol. 2024, 15, 1389173. [Google Scholar] [CrossRef]
- Paul, S.; Chatterjee, S.; Sinha, S.; Dash, S.R.; Pradhan, R.; Das, B.; Goutam, K.; Kundu, C.N. Veliparib (Abt-888), a Parp Inhibitor Potentiates the Cytotoxic Activity of 5-Fluorouracil by Inhibiting Mmr Pathway through Deregulation of Msh6 in Colorectal Cancer Stem Cells. Expert Opin. Ther. Targets 2023, 27, 999–1015. [Google Scholar] [CrossRef]
- Hindson, J. Gemcitabine and Cisplatin Plus Immunotherapy in Advanced Biliary Tract Cancer: A Phase Ii Study. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 280. [Google Scholar] [CrossRef]
- Clarke, J.M.; Patel, J.D.; Robert, F.; Kio, E.A.; Thara, E.; Camidge, D.R.; Dunbar, M.; Nuthalapati, S.; Dinh, M.H.; Bach, B.A. Veliparib and Nivolumab in Combination with Platinum Doublet Chemotherapy in Patients with Metastatic or Advanced Non-Small Cell Lung Cancer: A Phase 1 Dose Escalation Study. Lung Cancer 2021, 161, 180–188. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, J.; Zhu, X.; Su, T.; Zhou, H.; Wang, S.; Shi, W. The Development and Assessment of a Unique Disulfidptosis-Associated lncRNA Profile for Immune Microenvironment Prediction and Personalized Therapy in Gastric Adenocarcinoma. Biomedicines 2025, 13, 1224. https://doi.org/10.3390/biomedicines13051224
Zhu J, Zhu X, Su T, Zhou H, Wang S, Shi W. The Development and Assessment of a Unique Disulfidptosis-Associated lncRNA Profile for Immune Microenvironment Prediction and Personalized Therapy in Gastric Adenocarcinoma. Biomedicines. 2025; 13(5):1224. https://doi.org/10.3390/biomedicines13051224
Chicago/Turabian StyleZhu, Jiyue, Xiang Zhu, Tingting Su, Huiqing Zhou, Shouhua Wang, and Weibin Shi. 2025. "The Development and Assessment of a Unique Disulfidptosis-Associated lncRNA Profile for Immune Microenvironment Prediction and Personalized Therapy in Gastric Adenocarcinoma" Biomedicines 13, no. 5: 1224. https://doi.org/10.3390/biomedicines13051224
APA StyleZhu, J., Zhu, X., Su, T., Zhou, H., Wang, S., & Shi, W. (2025). The Development and Assessment of a Unique Disulfidptosis-Associated lncRNA Profile for Immune Microenvironment Prediction and Personalized Therapy in Gastric Adenocarcinoma. Biomedicines, 13(5), 1224. https://doi.org/10.3390/biomedicines13051224