

Oligodendroglioma: Advances in Molecular Mechanisms and Immunotherapeutic Strategies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
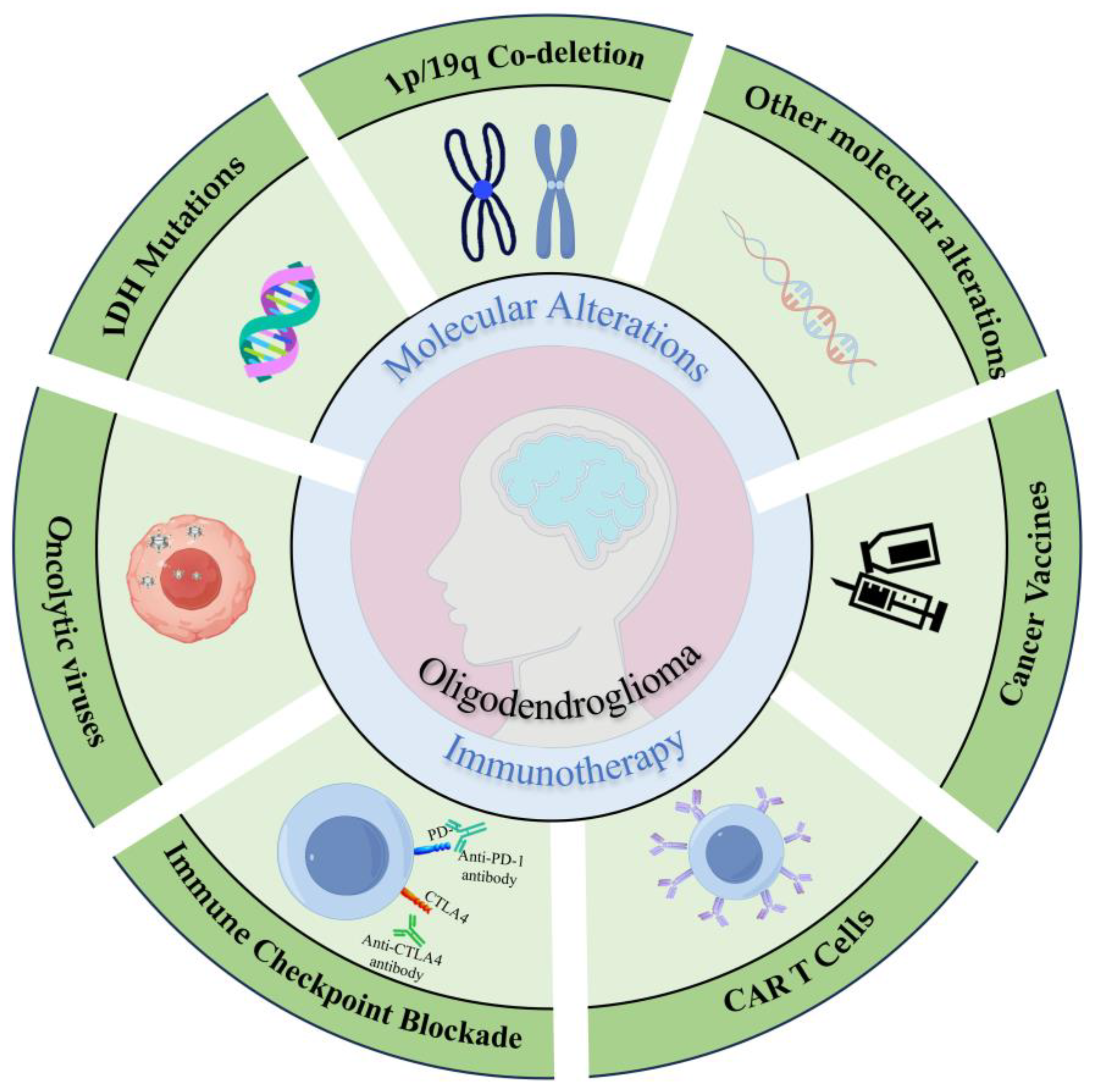
2. Molecular Alterations
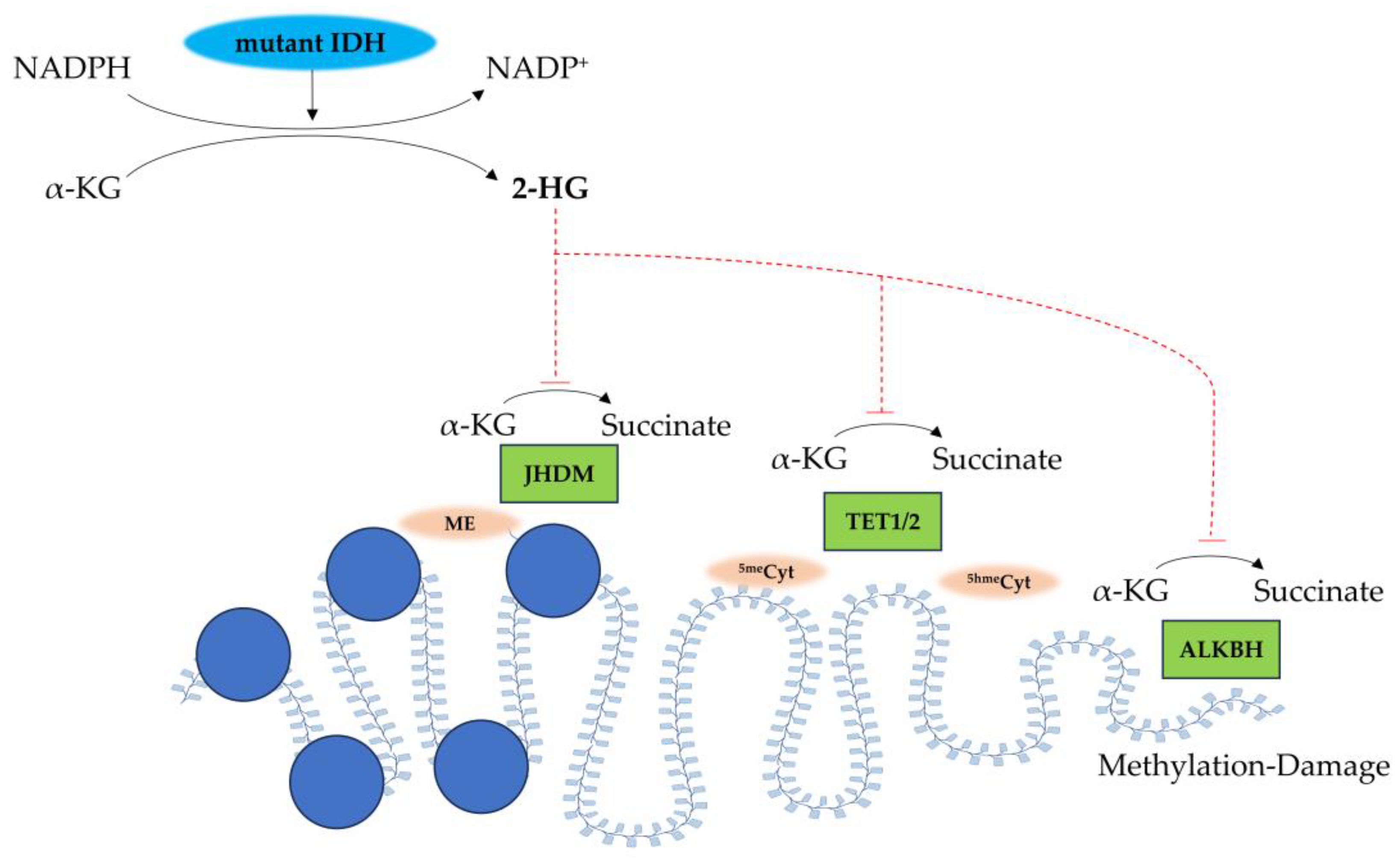
2.1. IDH Mutations
2.2. 1p/19q Co-Deletion
2.3. Other Molecular Alterations
3. Immunotherapy
3.1. Immune Microenvironment
3.2. Immunotherapeutic Approaches in Oligodendrogliomas
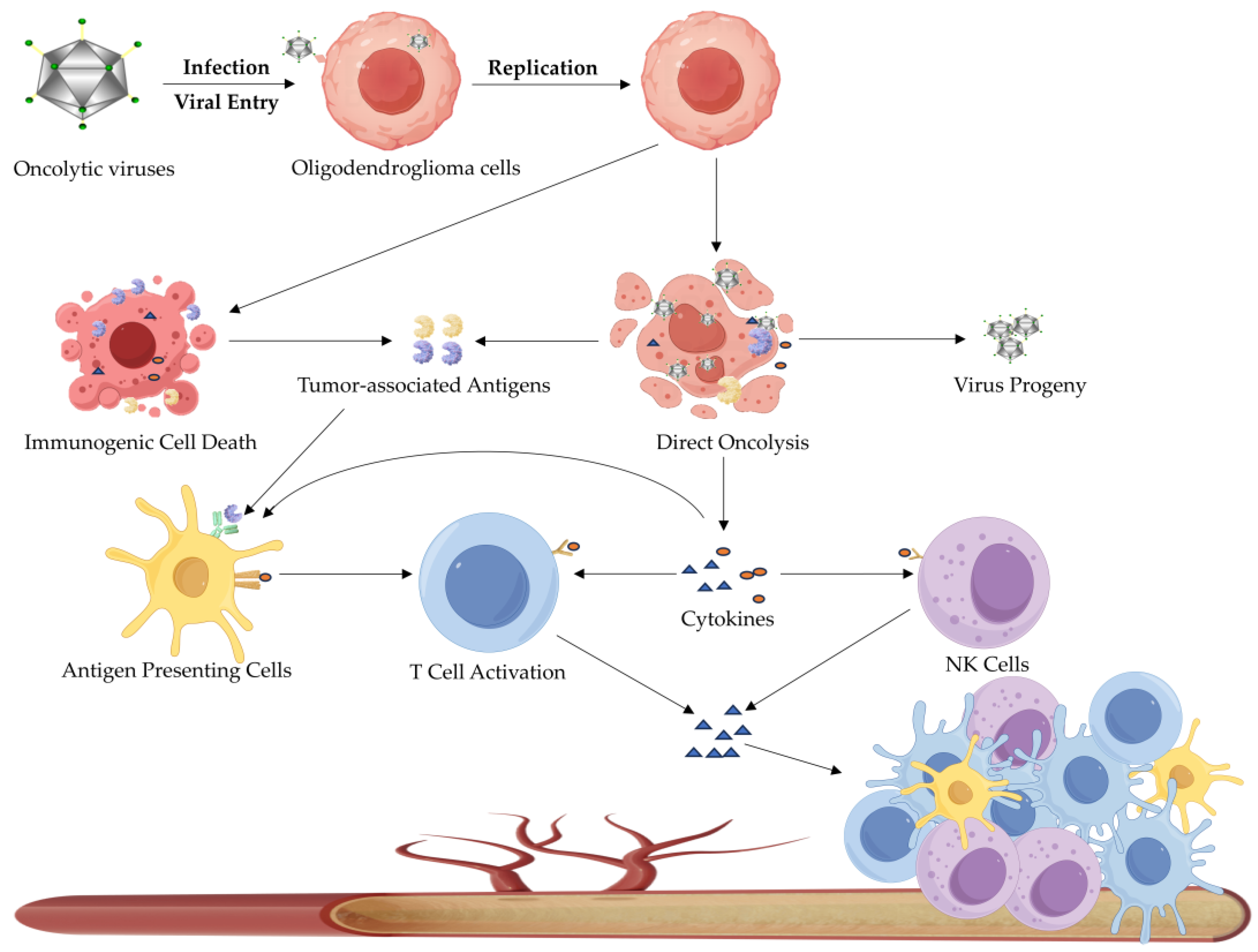
3.2.1. Oncolytic Virotherapy
3.2.2. Immune Checkpoint Blockade Therapy
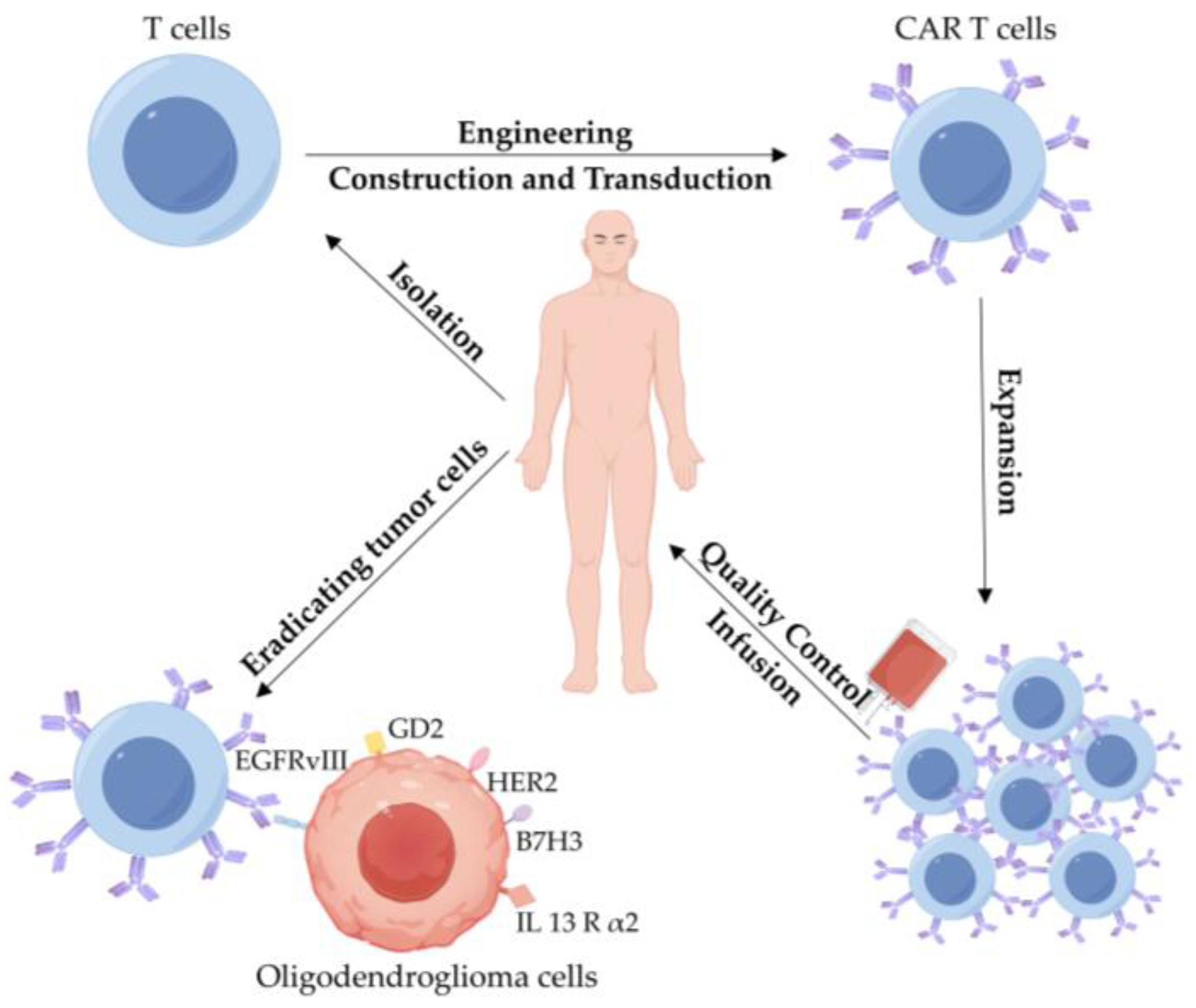
3.2.3. CAR T-Cell Therapy
3.2.4. Cancer Vaccines
4. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Yasinjan, F.; Xing, Y.; Geng, H.; Guo, R.; Yang, L.; Liu, Z.; Wang, H. Immunotherapy: A promising approach for glioma treatment. Front. Immunol. 2023, 14, 1255611. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Saijo, A.; Ogino, H.; Butowski, N.A.; Tedesco, M.R.; Gibson, D.; Watchmaker, P.B.; Okada, K.; Wang, A.S.; Shai, A.; Salazar, A.M.; et al. A combinatory vaccine with IMA950 plus varlilumab promotes effector memory T-cell differentiation in the peripheral blood of patients with low-grade gliomas. Neuro-Oncology 2024, 26, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Bianconi, A.; Prior, A.; Zona, G.; Fiaschi, P. Anticoagulant therapy in high grade gliomas: A systematic review on state of the art and future perspectives. J. Neurosurg. Sci. 2023, 67, 236–240. [Google Scholar] [CrossRef]
- Taylor, J.W.; Warrier, G.; Hansen, H.M.; McCoy, L.; Rice, T.; Guerra, G.; Francis, S.S.; Clarke, J.L.; Bracci, P.M.; Hadad, S.; et al. Oligodendroglioma patient survival is associated with circulating B-cells and age. Neuro-Oncol. Adv. 2024, 6, vdae143. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, P.; van den Bent, M.; Perry, A. Oligodendroglioma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015, 129, 809–827. [Google Scholar] [CrossRef]
- Bou Zerdan, M.; Assi, H.I. Oligodendroglioma: A Review of Management and Pathways. Front. Mol. Neurosci. 2021, 14, 722396. [Google Scholar] [CrossRef]
- Wu, F.; Yin, Y.-Y.; Fan, W.-H.; Zhai, Y.; Yu, M.-C.; Wang, D.; Pan, C.-Q.; Zhao, Z.; Li, G.-Z.; Zhang, W. Immunological profiles of human oligodendrogliomas define two distinct molecular subtypes. EBioMedicine 2023, 87, 104410. [Google Scholar] [CrossRef]
- Kessler, T.; Ito, J.; Wick, W.; Wick, A. Conventional and emerging treatments of astrocytomas and oligodendrogliomas. J. Neuro-Oncol. 2023, 162, 471–478. [Google Scholar] [CrossRef]
- Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; Morozova, O.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef]
- Demirdizen, E.; Al-Ali, R.; Narayanan, A.; Sun, X.; Varga, J.P.; Steffl, B.; Brom, M.; Krunic, D.; Schmidt, C.; Schmidt, G.; et al. TRIM67 drives tumorigenesis in oligodendrogliomas through Rho GTPase-dependent membrane blebbing. Neuro-Oncology 2023, 25, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Huang, Z.; Mei, H.; Hu, Y. Immunotherapy in hematologic malignancies: Achievements, challenges and future prospects. Signal Transduct. Target. Ther. 2023, 8, 306. [Google Scholar] [CrossRef]
- McNutt, M. Cancer immunotherapy. Science 2013, 342, 1417. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zheng, L.; Chen, W.; Weng, W.; Song, J.; Ji, J. Delivery strategies of cancer immunotherapy: Recent advances and future perspectives. J. Hematol. Oncol. 2019, 12, 126. [Google Scholar] [CrossRef] [PubMed]
- Uslu, U.; Castelli, S.; June, C.H. CAR T cell combination therapies to treat cancer. Cancer Cell 2024, 42, 1319–1325. [Google Scholar] [CrossRef]
- Ramezani, F.; Panahi Meymandi, A.R.; Akbari, B.; Tamtaji, O.R.; Mirzaei, H.; Brown, C.E.; Mirzaei, H.R. Outsmarting trogocytosis to boost CAR NK/T cell therapy. Mol. Cancer 2023, 22, 183. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef]
- Garfall, A.L.; Cohen, A.D.; Susanibar-Adaniya, S.P.; Hwang, W.-T.; Vogl, D.T.; Waxman, A.J.; Lacey, S.F.; Gonzalez, V.E.; Fraietta, J.A.; Gupta, M.; et al. Anti-BCMA/CD19 CAR T Cells with Early Immunomodulatory Maintenance for Multiple Myeloma Responding to Initial or Later-Line Therapy. Blood Cancer Discov. 2023, 4, 118–133. [Google Scholar] [CrossRef]
- Sharma, P.; Goswami, S.; Raychaudhuri, D.; Siddiqui, B.A.; Singh, P.; Nagarajan, A.; Liu, J.; Subudhi, S.K.; Poon, C.; Gant, K.L.; et al. Immune checkpoint therapy—Current perspectives and future directions. Cell 2023, 186, 1652–1669. [Google Scholar] [CrossRef]
- Uslu, U.; Da, T.; Assenmacher, C.A.; Scholler, J.; Young, R.M.; Tchou, J.; June, C.H. Chimeric antigen receptor T cells as adjuvant therapy for unresectable adenocarcinoma. Sci. Adv. 2023, 9, eade2526. [Google Scholar] [CrossRef]
- Khalil, D.N.; Smith, E.L.; Brentjens, R.J.; Wolchok, J.D. The future of cancer treatment: Immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 273–290. [Google Scholar] [CrossRef] [PubMed]
- Van den Bent, M.J.; Reni, M.; Gatta, G.; Vecht, C. Oligodendroglioma. Crit. Rev. Oncol. Hematol. 2008, 66, 262–272. [Google Scholar] [CrossRef]
- Cahill, D.P.; Louis, D.N.; Cairncross, J.G. Molecular background of oligodendroglioma: 1p/19q, IDH, TERT, CIC and FUBP1. CNS Oncol. 2015, 4, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Richardson, T.E.; Walker, J.M.; Hambardzumyan, D.; Brem, S.; Hatanpaa, K.J.; Viapiano, M.S.; Pai, B.; Umphlett, M.; Becher, O.J.; Snuderl, M.; et al. Genetic and epigenetic instability as an underlying driver of progression and aggressive behavior in IDH-mutant astrocytoma. Acta Neuropathol. 2024, 148, 5. [Google Scholar] [CrossRef]
- Capper, D.; Weißert, S.; Balss, J.; Habel, A.; Meyer, J.; Jäger, D.; Ackermann, U.; Tessmer, C.; Korshunov, A.; Zentgraf, H.; et al. Characterization of R132H Mutation-specific IDH1 Antibody Binding in Brain Tumors. Brain Pathol. 2009, 20, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.M.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Reiter-Brennan, C.; Semmler, L.; Klein, A. The effects of 2-hydroxyglutarate on the tumorigenesis of gliomas. Contemp. Oncol. 2018, 22, 215–222. [Google Scholar] [CrossRef]
- Schvartzman, J.-M.; Forsyth, G.; Walch, H.; Chatila, W.; Taglialatela, A.; Lee, B.J.; Zhu, X.; Gershik, S.; Cimino, F.V.; Santella, A.; et al. Oncogenic IDH mutations increase heterochromatin-related replication stress without impacting homologous recombination. Mol. Cell 2023, 83, 2347–2356. [Google Scholar] [CrossRef]
- Wei, Y.; Li, G.; Feng, J.; Wu, F.; Zhao, Z.; Bao, Z.; Zhang, W.; Su, X.; Li, J.; Qi, X.; et al. Stalled oligodendrocyte differentiation in IDH-mutant gliomas. Genome Med. 2023, 15, 24. [Google Scholar] [CrossRef]
- Hartmann, C.; Meyer, J.; Balss, J.; Capper, D.; Mueller, W.; Christians, A.; Felsberg, J.; Wolter, M.; Mawrin, C.; Wick, W.; et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: A study of 1,010 diffuse gliomas. Acta Neuropathol. 2009, 118, 469–474. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Cairncross, J.G.; Wang, M.; Jenkins, R.B.; Shaw, E.G.; Giannini, C.; Brachman, D.G.; Buckner, J.C.; Fink, K.L.; Souhami, L.; Laperriere, N.J.; et al. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 783–790. [Google Scholar] [CrossRef]
- Dono, A.; Alfaro-Munoz, K.; Yan, Y.; Lopez-Garcia, C.A.; Soomro, Z.; Williford, G.; Takayasu, T.; Robell, L.; Majd, N.K.; de Groot, J.; et al. Molecular, Histological, and Clinical Characteristics of Oligodendrogliomas: A Multi-Institutional Retrospective Study. Neurosurgery 2022, 90, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Griffin, C.A.; Burger, P.; Morsberger, L.; Yonescu, R.; Swierczynski, S.; Weingart, J.D.; Murphy, K.M. Identification of der (1;19)(q10;p10) in five oligodendrogliomas suggests mechanism of concurrent 1p and 19q loss. J. Neuropathol. Exp. Neurol. 2006, 65, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Thomas, C.; Munoz, F.A.; Alexandrescu, S.; Horbinski, C.M.; Olar, A.; McGuone, D.; Camelo-Piragua, S.; Wang, L.; Pentsova, E.; et al. Polysomy is associated with poor outcome in 1p/19q codeleted oligodendroglial tumors. Neuro-Oncology 2019, 21, 1164–1174. [Google Scholar] [CrossRef]
- Reifenberger, J.; Reifenberger, G.; Liu, L.; James, C.D.; Wechsler, W.; Collins, V.P. Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am. J. Pathol. 1994, 145, 1175–1190. [Google Scholar]
- Zhang, F.; Xiong, Q.; Wang, M.; Cao, X.; Zhou, C. FUBP1 in human cancer: Characteristics, functions, and potential applications. Transl. Oncol. 2024, 48, 102066. [Google Scholar] [CrossRef]
- Brandner, S.; McAleenan, A.; Jones, H.E.; Kernohan, A.; Robinson, T.; Schmidt, L.; Dawson, S.; Kelly, C.; Leal, E.S.; Faulkner, C.L.; et al. Diagnostic accuracy of 1p/19q codeletion tests in oligodendroglioma: A comprehensive meta-analysis based on a Cochrane systematic review. Neuropathol. Appl. Neurobiol. 2022, 48, e12790. [Google Scholar] [CrossRef]
- Branzoli, F.; Pontoizeau, C.; Tchara, L.; Di Stefano, A.L.; Kamoun, A.; Deelchand, D.K.; Valabrègue, R.; Lehéricy, S.; Sanson, M.; Ottolenghi, C.; et al. Cystathionine as a marker for 1p/19q codeleted gliomas by in vivo magnetic resonance spectroscopy. Neuro-Oncology 2019, 21, 765–774. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, J.H.; Quan, J.; Kang, X.; Wang, H.J.; Dai, P.G. Identification of MGMT promoter methylation sites correlating with gene expression and IDH1 mutation in gliomas. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 13571–13579. [Google Scholar] [CrossRef]
- Miller, J.J.; Cahill, D.P. MGMT promoter methylation and hypermutant recurrence in IDH mutant lower-grade glioma. Neuro-Oncology 2020, 22, 1553–1554. [Google Scholar] [CrossRef]
- Mair, M.J.; Leibetseder, A.; Heller, G.; Puhr, R.; Tomasich, E.; Goldberger, S.; Hatziioannou, T.; Wöhrer, A.; Widhalm, G.; Dieckmann, K.; et al. Early Postoperative Treatment versus Initial Observation in CNS WHO Grade 2 and 3 Oligodendroglioma: Clinical Outcomes and DNA Methylation Patterns. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 4565–4573. [Google Scholar] [CrossRef] [PubMed]
- Gilhodes, J.; Meola, A.; Cabarrou, B.; Peyraga, G.; Dehais, C.; Figarella-Branger, D.; Ducray, F.; Maurage, C.A.; Loussouarn, D.; Uro-Coste, E.; et al. A Multigene Signature Associated with Progression-Free Survival after Treatment for IDH Mutant and 1p/19q Codeleted Oligodendrogliomas. Cancers 2023, 15, 3067. [Google Scholar] [CrossRef] [PubMed]
- Jovanović, N.; Lazarević, M.; Cvetković, V.J.; Nikolov, V.; Kostić Perić, J.; Ugrin, M.; Pavlović, S.; Mitrović, T. The Significance of MGMT Promoter Methylation Status in Diffuse Glioma. Int. J. Mol. Sci. 2022, 23, 3034. [Google Scholar] [CrossRef] [PubMed]
- Darabi, S.; Xiu, J.; Samec, T.; Kesari, S.; Carrillo, J.; Aulakh, S.; Walsh, K.M.; Sengupta, S.; Sumrall, A.; Spetzler, D.; et al. Capicua (CIC) mutations in gliomas in association with MAPK activation for exposing a potential therapeutic target. Med. Oncol. 2023, 40, 197. [Google Scholar] [CrossRef]
- Pierini, T.; Nardelli, C.; Lema Fernandez, A.G.; Pierini, V.; Pellanera, F.; Nofrini, V.; Gorello, P.; Moretti, M.; Arniani, S.; Roti, G.; et al. New somatic TERT promoter variants enhance the Telomerase activity in Glioblastoma. Acta Neuropathol. Commun. 2020, 8, 1–10. [Google Scholar] [CrossRef]
- Rautajoki, K.J.; Jaatinen, S.; Tiihonen, A.M.; Annala, M.; Vuorinen, E.M.; Kivinen, A.; Rauhala, M.J.; Maass, K.K.; Pajtler, K.W.; Yli-Harja, O.; et al. PTPRD and CNTNAP2 as markers of tumor aggressiveness in oligodendrogliomas. Sci. Rep. 2022, 12, 14083. [Google Scholar] [CrossRef]
- Fomchenko, E.I.; Holland, E.C. Mouse models of brain tumors and their applications in preclinical trials. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 5288–5297. [Google Scholar] [CrossRef]
- Bralten, L.B.; Gravendeel, A.M.; Kloosterhof, N.K.; Sacchetti, A.; Vrijenhoek, T.; Veltman, J.A.; van den Bent, M.J.; Kros, J.M.; Hoogenraad, C.C.; Sillevis Smitt, P.A.; et al. The CASPR2 cell adhesion molecule functions as a tumor suppressor gene in glioma. Oncogene 2010, 29, 6138–6148. [Google Scholar] [CrossRef]
- Zhu, Q.; Jiang, H.; Cui, Y.; Ren, X.; Li, M.; Zhang, X.; Li, H.; Shen, S.; Li, M.; Lin, S. Intratumoral calcification: Not only a diagnostic but also a prognostic indicator in oligodendrogliomas. Eur. Radiol. 2024, 34, 3674–3685. [Google Scholar] [CrossRef]
- Broderick, D.K.; Di, C.; Parrett, T.J.; Samuels, Y.R.; Cummins, J.M.; McLendon, R.E.; Fults, D.W.; Velculescu, V.E.; Bigner, D.D.; Yan, H. Mutations of PIK3CA in anaplastic oligodendrogliomas, high-grade astrocytomas, and medulloblastomas. Cancer Res. 2004, 64, 5048–5050. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, K.; Nakamura, T.; Juratli, T.A.; Williams, E.A.; Matsushita, Y.; Miyake, S.; Nishi, M.; Miller, J.J.; Tummala, S.S.; Fink, A.L.; et al. PI3K/AKT/mTOR Pathway Alterations Promote Malignant Progression and Xenograft Formation in Oligodendroglial Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 4375–4387. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Jiang, T.; Liu, Y.; Zhao, Z.; Huang, L.; Li, G. FXYD2 mRNA expression represents a new independent factor that affects survival of glioma patients and predicts chemosensitivity of patients to temozolomide. BMC Neurol. 2021, 21, 438. [Google Scholar] [CrossRef]
- Nuechterlein, N.; Cimino, S.; Shelbourn, A.; Ha, V.; Arora, S.; Rajan, S.; Shapiro, L.G.; Holland, E.C.; Aldape, K.; McGranahan, T.; et al. HOXD12 defines an age-related aggressive subtype of oligodendroglioma. Acta Neuropathol. 2024, 148, 41. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Altamimi, A.S.A.; Babu, M.A.; Goyal, K.; Kaur, I.; Kumar, S.; Sharma, N.; Kumar, M.R.; Alanazi, F.J.; Alruwaili, A.N.; et al. Non-coding RNAs (ncRNAs) as therapeutic targets and biomarkers in oligodendroglioma. Pathol.-Res. Pract. 2024, 264, 155708. [Google Scholar] [CrossRef]
- de la Cruz-Ojeda, P.; Flores-Campos, R.; Dios-Barbeito, S.; Navarro-Villarán, E.; Muntané, J. Role of Nitric Oxide in Gene Expression Regulation during Cancer: Epigenetic Modifications and Non-Coding RNAs. Int. J. Mol. Sci. 2021, 22, 6264. [Google Scholar] [CrossRef]
- Miyata, K.; Takahashi, A. Pericentromeric repetitive ncRNA regulates chromatin interaction and inflammatory gene expression. Nucleus 2022, 13, 74–78. [Google Scholar] [CrossRef]
- Mair, M.J.; Geurts, M.; van den Bent, M.J.; Berghoff, A.S. A basic review on systemic treatment options in WHO grade II-III gliomas. Cancer Treat. Rev. 2021, 92, 102124. [Google Scholar] [CrossRef]
- Chiorazzi, M.; Martinek, J.; Krasnick, B.; Zheng, Y.; Robbins, K.J.; Qu, R.; Kaufmann, G.; Skidmore, Z.; Juric, M.; Henze, L.A.; et al. Autologous humanized PDX modeling for immuno-oncology recapitulates features of the human tumor microenvironment. J. Immunother. Cancer 2023, 11, e006921. [Google Scholar] [CrossRef]
- Zhou, J.; Li, L.; Jia, M.; Liao, Q.; Peng, G.; Luo, G.; Zhou, Y. Dendritic cell vaccines improve the glioma microenvironment: Influence, challenges, and future directions. Cancer Med. 2023, 12, 7207–7221. [Google Scholar] [CrossRef]
- Sokratous, G.; Polyzoidis, S.; Ashkan, K. Immune infiltration of tumor microenvironment following immunotherapy for glioblastoma multiforme. Hum. Vaccines Immunother. 2017, 13, 2575–2582. [Google Scholar] [CrossRef]
- Dang, N.-N.; Li, X.-B.; Zhang, M.; Han, C.; Fan, X.-Y.; Huang, S.-H. NLGN3 Upregulates Expression of ADAM10 to Promote the Cleavage of NLGN3 via Activating the LYN Pathway in Human Gliomas. Front. Cell Dev. Biol. 2021, 9, 662763. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Jackson, C.; Kim, T.; Choi, J.; Lim, M. A Characterization of Dendritic Cells and Their Role in Immunotherapy in Glioblastoma: From Preclinical Studies to Clinical Trials. Cancers 2019, 11, 537. [Google Scholar] [CrossRef]
- Terstappen, G.C.; Meyer, A.H.; Bell, R.D.; Zhang, W. Strategies for delivering therapeutics across the blood-brain barrier. Nat. reviews. Drug Discov. 2021, 20, 362–383. [Google Scholar] [CrossRef] [PubMed]
- Desai, R.; Suryadevara, C.M.; Batich, K.A.; Farber, S.H.; Sanchez-Perez, L.; Sampson, J.H. Emerging immunotherapies for glioblastoma. Expert. Opin. Emerg. Drugs 2016, 21, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Adhikaree, J.; Franks, H.A.; Televantos, C.; Vaghela, P.; Kaur, A.P.; Walker, D.; Schmitz, M.; Jackson, A.M.; Patel, P.M. Impaired circulating myeloid CD1c+ dendritic cell function in human glioblastoma is restored by p38 inhibition-implications for the next generation of DC vaccines. OncoImmunology 2019, 8, e1593803. [Google Scholar] [CrossRef]
- Gieryng, A.; Pszczolkowska, D.; Walentynowicz, K.A.; Rajan, W.D.; Kaminska, B. Immune microenvironment of gliomas. Lab. Investig. 2017, 97, 498–518. [Google Scholar] [CrossRef]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 2017, 355, eaai8478. [Google Scholar] [CrossRef]
- Fu, W.; Wang, W.; Li, H.; Jiao, Y.; Weng, J.; Huo, R.; Yan, Z.; Wang, J.; Xu, H.; Wang, S.; et al. CyTOF Analysis Reveals a Distinct Immunosuppressive Microenvironment in IDH Mutant Anaplastic Gliomas. Front. Oncol. 2021, 10, 560211. [Google Scholar] [CrossRef]
- Qiao, Q.; Wang, Y.; Zhang, R.; Pang, Q. Autophagy related DNA methylation signature predict clinical prognosis and immune microenvironment in low-grade glioma. Transl. Cancer Res. 2022, 11, 2157–2174. [Google Scholar] [CrossRef]
- Kim, W.; Lee, S.; Seo, D.; Kim, D.; Kim, K.; Kim, E.; Kang, J.; Seong, K.M.; Youn, H.; Youn, B. Cellular Stress Responses in Radiotherapy. Cells 2019, 8, 1105. [Google Scholar] [CrossRef]
- Nisar, M.; Paracha, R.Z.; Adil, S.; Qureshi, S.N.; Janjua, H.A. An Extensive Review on Preclinical and Clinical Trials of Oncolytic Viruses Therapy for Pancreatic Cancer. Front. Oncol. 2022, 12, 875188. [Google Scholar] [CrossRef] [PubMed]
- Melcher, A.; Harrington, K.; Vile, R. Oncolytic virotherapy as immunotherapy. Science 2021, 374, 1325–1326. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Shen, Y.; Liang, T. Oncolytic virotherapy: Basic principles, recent advances and future directions. Signal Transduct. Target. Ther. 2023, 8, 156. [Google Scholar] [CrossRef] [PubMed]
- Romanishin, A.; Vasilev, A.; Khasanshin, E.; Evtekhov, A.; Pusynin, E.; Rubina, K.; Kakotkin, V.; Agapov, M.; Semina, E. Oncolytic viral therapy for gliomas: Advances in the mechanisms and approaches to delivery. Virology 2024, 593, 110033. [Google Scholar] [CrossRef]
- Pol, J.G.; Workenhe, S.T.; Konda, P.; Gujar, S.; Kroemer, G. Cytokines in oncolytic virotherapy. Cytokine Growth Factor. Rev. 2020, 56, 4–27. [Google Scholar] [CrossRef]
- Friedman, G.K.; Johnston, J.M.; Bag, A.K.; Bernstock, J.D.; Li, R.; Aban, I.; Kachurak, K.; Nan, L.; Kang, K.-D.; Totsch, S.; et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. N. Engl. J. Med. 2021, 384, 1613–1622. [Google Scholar] [CrossRef]
- Chen, X.; Liu, J.; Li, Y.; Zeng, Y.; Wang, F.; Cheng, Z.; Duan, H.; Pan, G.; Yang, S.; Chen, Y.; et al. IDH1 mutation impairs antiviral response and potentiates oncolytic virotherapy in glioma. Nat. Commun. 2023, 14, 6781. [Google Scholar] [CrossRef]
- Drake, C.G.; Jaffee, E.; Pardoll, D.M. Mechanisms of Immune Evasion by Tumors. Adv. Immunol. 2006, 90, 51–81. [Google Scholar]
- Ghouzlani, A.; Kandoussi, S.; Tall, M.; Reddy, K.P.; Rafii, S.; Badou, A. Immune Checkpoint Inhibitors in Human Glioma Microenvironment. Front. Immunol. 2021, 12, 679425. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Dvir, K.; Giordano, S.; Leone, J.P. Immunotherapy in Breast Cancer. Int. J. Mol. Sci. 2024, 25, 7517. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Liu, B.; Sun, Q.; Xiong, X.; Chen, Q. Immune Checkpoint Targeted Therapy in Glioma: Status and Hopes. Front. Immunol. 2020, 11, 578877. [Google Scholar] [CrossRef]
- Li, X.; Gulati, M.; Larson, A.C.; Solheim, J.C.; Jain, M.; Kumar, S.; Batra, S.K. Immune checkpoint blockade in pancreatic cancer: Trudging through the immune desert. Semin. Cancer Biol. 2022, 86, 14–27. [Google Scholar] [CrossRef]
- Reardon, D.A.; Gokhale, P.C.; Klein, S.R.; Ligon, K.L.; Rodig, S.J.; Ramkissoon, S.H.; Jones, K.L.; Conway, A.S.; Liao, X.; Zhou, J.; et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol. Res. 2016, 4, 124–135. [Google Scholar] [CrossRef]
- Zeng, Z.; Hu, C.; Ruan, W.; Zhang, J.; Lei, S.; Yang, Y.; Peng, P.; Pan, F.; Chen, T. A specific immune signature for predicting the prognosis of glioma patients with IDH1-mutation and guiding immune checkpoint blockade therapy. Front. Immunol. 2022, 13, 1001381. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, Q.; Tan, L.; Wu, X.; Huang, R.; Zuo, Y.; Chen, L.; Yang, J.; Zhang, Z.-X.; Ruan, W.; et al. Neutralizing IL-8 potentiates immune checkpoint blockade efficacy for glioma. Cancer Cell 2023, 41, 693–710. [Google Scholar] [CrossRef]
- Kadiyala, P.; Carney, S.V.; Gauss, J.C.; Garcia-Fabiani, M.B.; Haase, S.; Alghamri, M.S.; Núñez, F.J.; Liu, Y.; Yu, M.; Taher, A.; et al. Inhibition of 2-hydroxyglutarate elicits metabolic reprogramming and mutant IDH1 glioma immunity in mice. J. Clin. Investig. 2021, 131, e139542. [Google Scholar] [CrossRef]
- Noor, H.; Zaman, A.; Teo, C.; Sughrue, M.E. PODNL1 Methylation Serves as a Prognostic Biomarker and Associates with Immune Cell Infiltration and Immune Checkpoint Blockade Response in Lower-Grade Glioma. Int. J. Mol. Sci. 2021, 22, 2572. [Google Scholar] [CrossRef] [PubMed]
- Farhangnia, P.; Khorramdelazad, H.; Nickho, H.; Delbandi, A.-A. Current and future immunotherapeutic approaches in pancreatic cancer treatment. J. Hematol. Oncol. 2024, 17, 40. [Google Scholar] [CrossRef] [PubMed]
- Song, K.-W.; Scott, B.J. CAR T-cell therapy for gliomas. Curr. Opin. Neurol. 2024, 37, 672–681. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Flugel, C.L.; Majzner, R.G.; Krenciute, G.; Dotti, G.; Riddell, S.R.; Wagner, D.L.; Abou-el-Enein, M. Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours. Nat. Rev. Clin. Oncol. 2022, 20, 49–62. [Google Scholar] [CrossRef]
- Pollack, I.F.; Jakacki, R.I.; Butterfield, L.H.; Hamilton, R.L.; Panigrahy, A.; Normolle, D.P.; Connelly, A.K.; Dibridge, S.; Mason, G.; Whiteside, T.L.; et al. Immune responses and outcome after vaccination with glioma-associated antigen peptides and poly-ICLC in a pilot study for pediatric recurrent low-grade gliomas. Neuro-Oncology 2016, 18, 1157–1168. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Swartz, A.M.; Batich, K.A.; Fecci, P.E.; Sampson, J.H. Peptide vaccines for the treatment of glioblastoma. J. Neuro-Oncol. 2015, 123, 433–440. [Google Scholar] [CrossRef]
- Okada, H.; Butterfield, L.H.; Hamilton, R.L.; Hoji, A.; Sakaki, M.; Ahn, B.J.; Kohanbash, G.; Drappatz, J.; Engh, J.; Amankulor, N.; et al. Induction of Robust Type-I CD8+ T-cell Responses in WHO Grade 2 Low-Grade Glioma Patients Receiving Peptide-Based Vaccines in Combination with Poly-ICLC. Clin. Cancer Res. 2015, 21, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef]
- Dutoit, V.; Herold-Mende, C.; Hilf, N.; Schoor, O.; Beckhove, P.; Bucher, J.; Dorsch, K.; Flohr, S.; Fritsche, J.; Lewandrowski, P.; et al. Exploiting the glioblastoma peptidome to discover novel tumour-associated antigens for immunotherapy. Brain A J. Neurol. 2012, 135, 1042–1054. [Google Scholar] [CrossRef]
- Vitale, L.A.; He, L.-Z.; Thomas, L.J.; Widger, J.; Weidlick, J.; Crocker, A.; O’Neill, T.; Storey, J.; Glennie, M.J.; Grote, D.M.; et al. Development of a Human Monoclonal Antibody for Potential Therapy of CD27-Expressing Lymphoma and Leukemia. Clin. Cancer Res. 2012, 18, 3812–3821. [Google Scholar] [CrossRef]
- Ogino, H.; Taylor, J.W.; Nejo, T.; Gibson, D.; Watchmaker, P.B.; Okada, K.; Saijo, A.; Tedesco, M.R.; Shai, A.; Wong, C.M.; et al. Randomized trial of neoadjuvant vaccination with tumor-cell lysate induces T cell response in low-grade gliomas. J. Clin. Investig. 2022, 132, e151239. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, F.; Ali, H.; Lathia, J.D.; Chen, P. Immunotherapy for glioblastoma: Current state, challenges, and future perspectives. Cell. Mol. Immunol. 2024, 21, 1354–1375. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Raphael, I.; Olin, M.; Okada, H.; Li, X.; Kohanbash, G. Glioblastoma vaccines: Past, present, and opportunities. EBioMedicine 2024, 100, 104963. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Kitamura, Y.; Toda, M.; Hirose, Y.; Yoshida, K. Oligodendroglioma, IDH-mutant and 1p/19q-codeleted-prognostic factors, standard of care and chemotherapy, and future perspectives with neoadjuvant strategy. Brain Tumor Pathol. 2024, 41, 43–49. [Google Scholar] [CrossRef]
- Bianconi, A.; Palmieri, G.; Aruta, G.; Monticelli, M.; Zeppa, P.; Tartara, F.; Melcarne, A.; Garbossa, D.; Cofano, F. Updates in Glioblastoma Immunotherapy: An Overview of the Current Clinical and Translational Scenario. Biomedicines 2023, 11, 1520. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Yu, Y.; Chen, W.; Zhang, X.; Lv, J.; Zhao, H. Oligodendroglioma: Advances in Molecular Mechanisms and Immunotherapeutic Strategies. Biomedicines 2025, 13, 1133. https://doi.org/10.3390/biomedicines13051133
Zhao Y, Yu Y, Chen W, Zhang X, Lv J, Zhao H. Oligodendroglioma: Advances in Molecular Mechanisms and Immunotherapeutic Strategies. Biomedicines. 2025; 13(5):1133. https://doi.org/10.3390/biomedicines13051133
Chicago/Turabian StyleZhao, Yongxin, Yan Yu, Weizhi Chen, Xiaojun Zhang, Jing Lv, and Heping Zhao. 2025. "Oligodendroglioma: Advances in Molecular Mechanisms and Immunotherapeutic Strategies" Biomedicines 13, no. 5: 1133. https://doi.org/10.3390/biomedicines13051133
APA StyleZhao, Y., Yu, Y., Chen, W., Zhang, X., Lv, J., & Zhao, H. (2025). Oligodendroglioma: Advances in Molecular Mechanisms and Immunotherapeutic Strategies. Biomedicines, 13(5), 1133. https://doi.org/10.3390/biomedicines13051133