

Shed Syndecan-4 and Its Possible Roles in Osteoarthritis


Abstract
1. Introduction
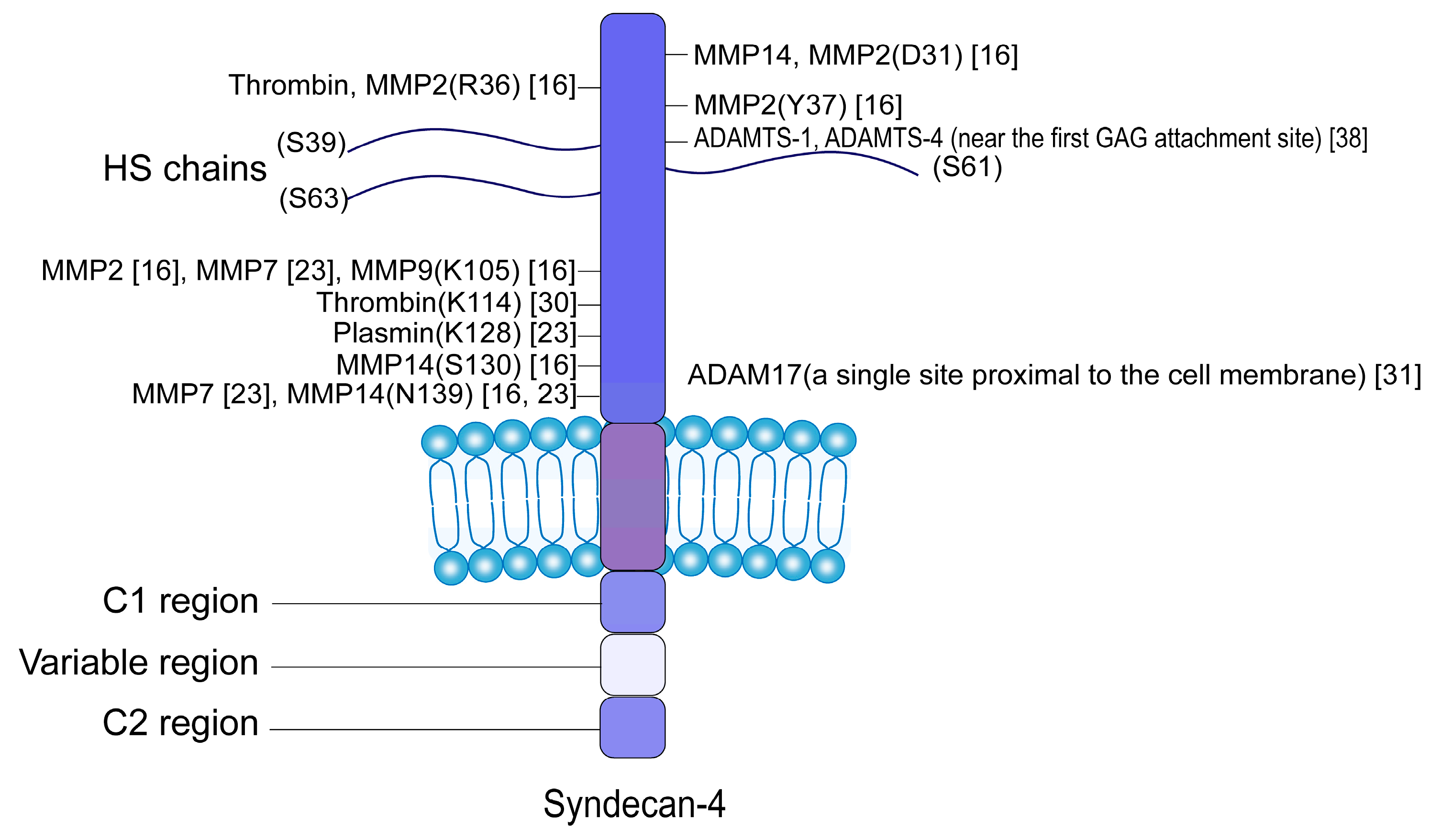
2. Proteolytic Enzymes and Cleavage Sites of SDC4 Shedding
3. Factors Affecting SDC4 Shedding
3.1. Inflammation
3.2. Oxidative Stress
3.3. Mechanical Stress
3.4. Different Specific Cytokines
4. Functional Changes in the Remaining Membrane Domain After SDC4 Shedding
5. Functions of sSDC4
5.1. sSDC4 Can Function as a Decoy Receptor, a Molecular Chaperone, and a Signaling Molecule
5.2. sSDC4 Is Involved in the Pathogenesis of Numerous Diseases
6. The Roles of sSDC4 in OA
6.1. The Level of sSDC4 Is Positively Relative to the Severity of OA
6.2. sSDC4 Can Affect Cartilage Regeneration
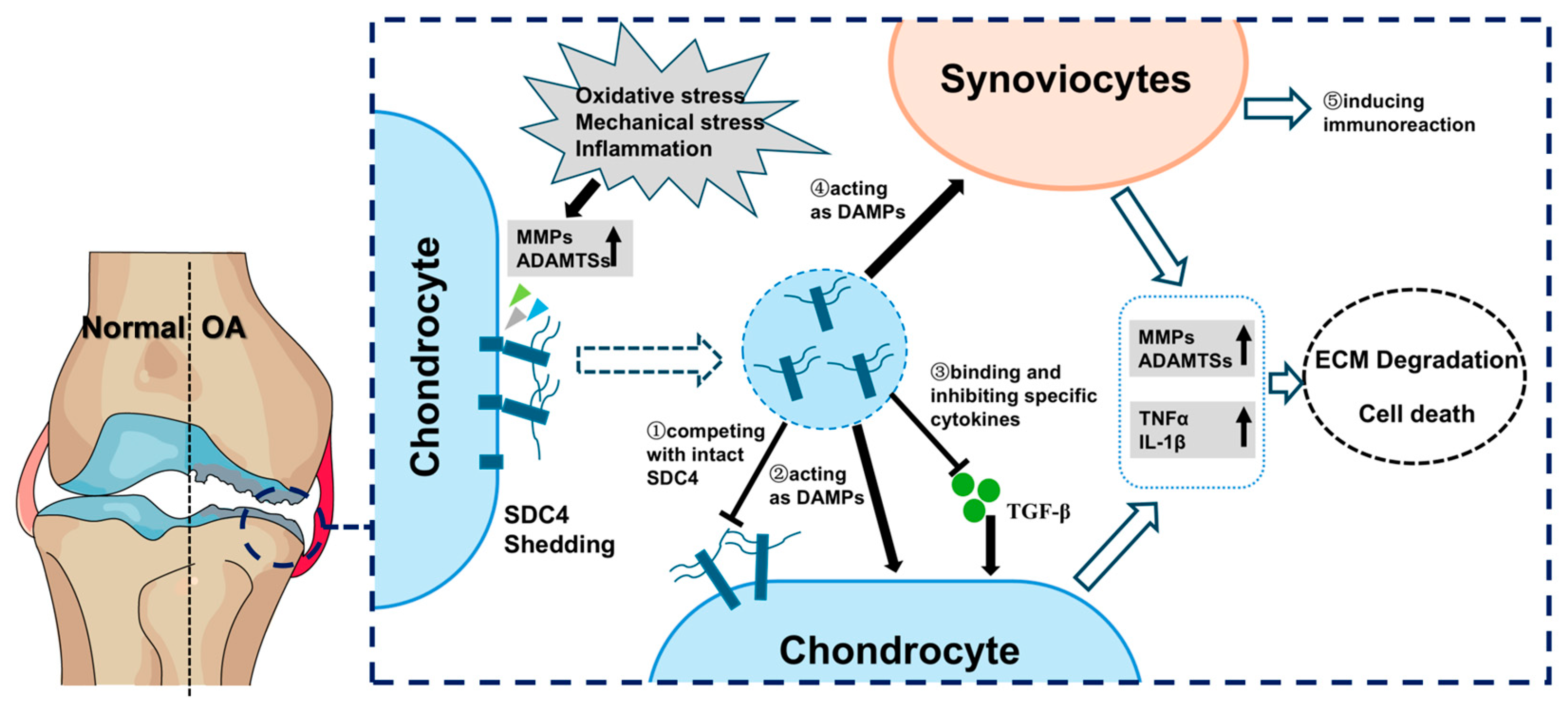
6.3. sSDC4 Can Act as a DAMP to Destroy the Cartilage Homeostasis
7. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADAMs | a disintegrin and metalloproteinases |
| ADAMTS | a disintegrin and metalloproteinase with thrombospondin motifs |
| AGEs | advanced glycation end products |
| ASM | airway smooth muscle |
| ASMC | airway smooth muscle cells |
| AMPK | adenosine monophosphate-activated protein kinase |
| ACSL4 | acyl-coA synthetase long chain family member 4 |
| ACAN | aggrecan |
| CCL5 | C-C motif chemokine ligand 5 |
| CCR5 | C-C chemokine receptor type 5 |
| CXCR4 | C-X-C chemokine receptor type 4 |
| CXCL12 | C-X-C motif chemokine ligand 12 |
| COL2A1 | collagen type II alpha 1 |
| DAMP | damage-associated molecular pattern |
| DRG | dorsal root ganglion |
| ECM | extracellular matrix |
| EPC | endothelial progenitor cell |
| EG | endothelial glycocalyx |
| ERK | extracellular regulated protein kinases |
| FN | fibronectin |
| FAK | focal adhesion kinase |
| FGF | fibroblast growth factor |
| VEGFA | vascular endothelial growth factor A |
| VEGFR | vascular endothelial growth factor receptor |
| GAG | galactosylated glycosaminoglycan |
| GPCR | G protein-coupled receptor |
| GPX4 | glutathione peroxidase 4 |
| HSPGs | heparan sulfate proteoglycans |
| HS | heparan sulfate |
| HIF-2α | hypoxia-inducible factor 2 alpha |
| HIF-1 | hypoxia-inducible factor 1 |
| IL-1β | interleukin-1β |
| IL-1Ra | IL-1 receptor antagonist |
| IL-6 | interleukin-6 |
| LPS | lipopolysaccharides |
| LDL | low-density lipoprotein |
| LPL | lipoprotein lipase |
| LRP | low-density lipoprotein receptor-related protein |
| MMPs | matrix metalloproteinases |
| MAPK | mitogen-activated protein kinase |
| NAC | N-acetylcysteine |
| NF-κB | nuclear factor kappa-B |
| NLRP3 | NOD-like receptor family, pyrin domain containing 3 |
| OA | osteoarthritis |
| OPN | osteopontin |
| PKC | protein kinase C |
| PMA | phorbol myristate acetate |
| PKCε | protein kinase C epsilon |
| PGC-1α | peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| ROS | reactive oxygen species |
| RAGE | receptor for advanced glycation endproducts |
| SDC4 | syndecan-4 |
| SDC | syndecan |
| sSDC4 | shed syndecan-4 |
| SRC | scleroderma renal crisis |
| STAT3 | signal transducer and activator of transcription 3 |
| Sirtuin1 | silent information regulator factor 1 |
| Sirt1endo−/− | endothelial sirtuin1-deficient |
| SASP | senescence-associated secretory phenotype |
| SDF-1 | stromal cell-derived factor-1 |
| SLC7A11 | solute carrier family 7 member 11 |
| TNFα | tumor necrosis factor-α |
| TIMP | tissue inhibitor of metalloproteinase |
| TLR4 | toll-like receptor 4 |
| TGF-β | transforming growth factor beta |
| TMJOA | temporomandibular joint osteoarthritis |
| UUO | Unilateral Ureteral Obstruction |
References
- Hunter, D.J.; Bierma-Zeinstra, S. Osteoarthritis. Lancet 2019, 393, 1745–1759. [Google Scholar] [CrossRef]
- Yao, Q.; Wu, X.; Tao, C.; Gong, W.; Chen, M.; Qu, M.; Zhong, Y.; He, T.; Chen, S.; Xiao, G. Osteoarthritis: Pathogenic Signaling Pathways and Therapeutic Targets. Signal Transduct. Target. Ther. 2023, 8, 56. [Google Scholar] [CrossRef]
- He, F.; Wu, H.; He, B.; Han, Z.; Chen, J.; Huang, L. Antioxidant Hydrogels for the Treatment of Osteoarthritis: Mechanisms and Recent Advances. Front. Pharmacol. 2024, 15, 1488036. [Google Scholar] [CrossRef]
- Matsui, Y.; Hasegawa, M.; Iino, T.; Imanaka-Yoshida, K.; Yoshida, T.; Sudo, A. Tenascin-C Prevents Articular Cartilage Degeneration in Murine Osteoarthritis Models. Cartilage 2018, 9, 80–88. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, X.; Wang, S.; Jing, Y.; Su, J. Subchondral Bone Microenvironment in Osteoarthritis and Pain. Bone Res. 2021, 9, 20. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, S.; Huang, J.; Guo, W.; Chen, J.; Zhang, L.; Zhao, B.; Peng, J.; Wang, A.; Wang, Y.; et al. The ECM-Cell Interaction of Cartilage Extracellular Matrix on Chondrocytes. Biomed. Res. Int. 2014, 2014, 648459. [Google Scholar] [CrossRef]
- Bertrand, J.; Bollmann, M. Soluble Syndecans: Biomarkers for Diseases and Therapeutic Options. Br. J. Pharmacol. 2019, 176, 67–81. [Google Scholar] [CrossRef]
- Echtermeyer, F.; Bertrand, J.; Dreier, R.; Meinecke, I.; Neugebauer, K.; Fuerst, M.; Lee, Y.J.; Song, Y.W.; Herzog, C.; Theilmeier, G.; et al. Syndecan-4 Regulates ADAMTS-5 Activation and Cartilage Breakdown in Osteoarthritis. Nat. Med. 2009, 15, 1072–1076. [Google Scholar] [CrossRef]
- Brioudes, E.; Alibashe-Ahmed, M.; Lavallard, V.; Berney, T.; Bosco, D. Syndecan-4 Is Regulated by IL-1β in β-Cells and Human Islets. Mol. Cell Endocrinol. 2020, 510, 110815. [Google Scholar] [CrossRef]
- Li, R.; Xie, J.; Wu, H.; Li, G.; Chen, J.; Chen, Q.; Wang, L.; Xu, B. Syndecan-4 Shedding Impairs Macrovascular Angiogenesis in Diabetes Mellitus. Biochem. Biophys. Res. Commun. 2016, 474, 15–21. [Google Scholar] [CrossRef]
- Cao, C.; Shi, Y.; Zhang, X.; Li, Q.; Zhang, J.; Zhao, F.; Meng, Q.; Dai, W.; Liu, Z.; Yan, W.; et al. Cholesterol-Induced LRP3 Downregulation Promotes Cartilage Degeneration in Osteoarthritis by Targeting Syndecan-4. Nat. Commun. 2022, 13, 7139. [Google Scholar] [CrossRef]
- Chronopoulos, A.; Thorpe, S.D.; Cortes, E.; Lachowski, D.; Rice, A.J.; Mykuliak, V.V.; Róg, T.; Lee, D.A.; Hytönen, V.P.; Del Río Hernández, A.E. Syndecan-4 Tunes Cell Mechanics by Activating the Kindlin-Integrin-RhoA Pathway. Nat. Mater. 2020, 19, 669–678. [Google Scholar] [CrossRef]
- Nikaido, T.; Tanino, Y.; Wang, X.; Sato, S.; Misa, K.; Fukuhara, N.; Sato, Y.; Fukuhara, A.; Uematsu, M.; Suzuki, Y.; et al. Serum Syndecan-4 as a Possible Biomarker in Patients With Acute Pneumonia. J. Infect. Dis. 2015, 212, 1500–1508. [Google Scholar] [CrossRef]
- Chen, X.; He, F.; Zhang, H.; Ma, Y.; Yu, J.; Qin, H.; Wu, F.; Wang, Z.; Zhan, Y.; Zhang, J.; et al. Syndecan-4 Inhibition Attenuates Cartilage Degeneration in Temporomandibular Joint Osteoarthritis. J. Oral. Rehabil. 2024, 51, 2324–2335. [Google Scholar] [CrossRef]
- Jalkanen, M.; Rapraeger, A.; Saunders, S.; Bernfield, M. Cell Surface Proteoglycan of Mouse Mammary Epithelial Cells Is Shed by Cleavage of Its Matrix-Binding Ectodomain from Its Membrane-Associated Domain. J. Cell Biol. 1987, 105, 3087–3096. [Google Scholar] [CrossRef]
- Manon-Jensen, T.; Multhaupt, H.A.B.; Couchman, J.R. Mapping of Matrix Metalloproteinase Cleavage Sites on Syndecan-1 and Syndecan-4 Ectodomains. FEBS J. 2013, 280, 2320–2331. [Google Scholar] [CrossRef]
- Bollmann, M.; Pinno, K.; Ehnold, L.I.; Märtens, N.; Märtson, A.; Pap, T.; Stärke, C.; Lohmann, C.H.; Bertrand, J. MMP-9 Mediated Syndecan-4 Shedding Correlates with Osteoarthritis Severity. Osteoarthr. Cartil. 2021, 29, 280–289. [Google Scholar] [CrossRef]
- Strand, M.E.; Vanhaverbeke, M.; Henkens, M.T.H.M.; Sikking, M.A.; Rypdal, K.B.; Braathen, B.; Almaas, V.M.; Tønnessen, T.; Christensen, G.; Heymans, S.; et al. Inflammation and Syndecan-4 Shedding from Cardiac Cells in Ischemic and Non-Ischemic Heart Disease. Biomedicines 2023, 11, 1066. [Google Scholar] [CrossRef]
- Strand, M.E.; Aronsen, J.M.; Braathen, B.; Sjaastad, I.; Kvaløy, H.; Tønnessen, T.; Christensen, G.; Lunde, I.G. Shedding of Syndecan-4 Promotes Immune Cell Recruitment and Mitigates Cardiac Dysfunction after Lipopolysaccharide Challenge in Mice. J. Mol. Cell Cardiol. 2015, 88, 133–144. [Google Scholar] [CrossRef]
- Lee, S.; Kolset, S.O.; Birkeland, K.I.; Drevon, C.A.; Reine, T.M. Acute Exercise Increases Syndecan-1 and -4 Serum Concentrations. Glycoconj. J. 2019, 36, 113–125. [Google Scholar] [CrossRef]
- Solbu, M.D.; Kolset, S.O.; Jenssen, T.G.; Wilsgaard, T.; Løchen, M.-L.; Mathiesen, E.B.; Melsom, T.; Eriksen, B.O.; Reine, T.M. Gender Differences in the Association of Syndecan-4 with Myocardial Infarction: The Population-Based Tromsø Study. Atherosclerosis 2018, 278, 166–173. [Google Scholar] [CrossRef]
- Smart, L.; Macdonald, S.P.J.; Burrows, S.; Bosio, E.; Arendts, G.; Fatovich, D.M. Endothelial Glycocalyx Biomarkers Increase in Patients with Infection during Emergency Department Treatment. J. Crit. Care 2017, 42, 304–309. [Google Scholar] [CrossRef]
- Gondelaud, F.; Ricard-Blum, S. Structures and Interactions of Syndecans. FEBS J. 2019, 286, 2994–3007. [Google Scholar] [CrossRef]
- Iozzo, R.V.; Schaefer, L. Proteoglycan Form and Function: A Comprehensive Nomenclature of Proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef]
- Qin, Y.; Zhu, Y.; Luo, F.; Chen, C.; Chen, X.; Wu, M. Killing Two Birds with One Stone: Dual Blockade of Integrin and FGF Signaling through Targeting Syndecan-4 in Postoperative Capsular Opacification. Cell Death Dis. 2017, 8, e2920. [Google Scholar] [CrossRef]
- McFall, A.J.; Rapraeger, A.C. Identification of an Adhesion Site within the Syndecan-4 Extracellular Protein Domain. J. Biol. Chem. 1997, 272, 12901–12904. [Google Scholar] [CrossRef]
- Ramani, V.C.; Pruett, P.S.; Thompson, C.A.; DeLucas, L.D.; Sanderson, R.D. Heparan Sulfate Chains of Syndecan-1 Regulate Ectodomain Shedding. J. Biol. Chem. 2012, 287, 9952–9961. [Google Scholar] [CrossRef]
- Ramnath, R.; Foster, R.R.; Qiu, Y.; Cope, G.; Butler, M.J.; Salmon, A.H.; Mathieson, P.W.; Coward, R.J.; Welsh, G.I.; Satchell, S.C. Matrix Metalloproteinase 9-Mediated Shedding of Syndecan 4 in Response to Tumor Necrosis Factor α: A Contributor to Endothelial Cell Glycocalyx Dysfunction. FASEB J. 2014, 28, 4686–4699. [Google Scholar] [CrossRef]
- Fitzgerald, M.L.; Wang, Z.; Park, P.W.; Murphy, G.; Bernfield, M. Shedding of Syndecan-1 and -4 Ectodomains Is Regulated by Multiple Signaling Pathways and Mediated by a TIMP-3-Sensitive Metalloproteinase. J. Cell Biol. 2000, 148, 811–824. [Google Scholar] [CrossRef]
- Schmidt, A.; Echtermeyer, F.; Alozie, A.; Brands, K.; Buddecke, E. Plasmin- and Thrombin-Accelerated Shedding of Syndecan-4 Ectodomain Generates Cleavage Sites at Lys(114)-Arg(115) and Lys(129)-Val(130) Bonds. J. Biol. Chem. 2005, 280, 34441–34446. [Google Scholar] [CrossRef]
- Pruessmeyer, J.; Martin, C.; Hess, F.M.; Schwarz, N.; Schmidt, S.; Kogel, T.; Hoettecke, N.; Schmidt, B.; Sechi, A.; Uhlig, S.; et al. A Disintegrin and Metalloproteinase 17 (ADAM17) Mediates Inflammation-Induced Shedding of Syndecan-1 and -4 by Lung Epithelial Cells. J. Biol. Chem. 2010, 285, 555–564. [Google Scholar] [CrossRef]
- Arner, E.C.; Pratta, M.A.; Trzaskos, J.M.; Decicco, C.P.; Tortorella, M.D. Generation and Characterization of Aggrecanase. A Soluble, Cartilage-Derived Aggrecan-Degrading Activity. J. Biol. Chem. 1999, 274, 6594–6601. [Google Scholar] [CrossRef]
- Tortorella, M.D.; Pratta, M.; Liu, R.Q.; Austin, J.; Ross, O.H.; Abbaszade, I.; Burn, T.; Arner, E. Sites of Aggrecan Cleavage by Recombinant Human Aggrecanase-1 (ADAMTS-4). J. Biol. Chem. 2000, 275, 18566–18573. [Google Scholar] [CrossRef]
- Fears, C.Y.; Gladson, C.L.; Woods, A. Syndecan-2 Is Expressed in the Microvasculature of Gliomas and Regulates Angiogenic Processes in Microvascular Endothelial Cells. J. Biol. Chem. 2006, 281, 14533–14536. [Google Scholar] [CrossRef]
- Ali, M.F.; Dasari, H.; Van Keulen, V.P.; Cornec, D.; Vasmatzis, G.; Peikert, T.; Carmona, E.M. Microbial Antigens Stimulate Metalloprotease-7 Secretion in Human B-Lymphocytes Using mTOR-Dependent and Independent Pathways. Sci. Rep. 2017, 7, 3869. [Google Scholar] [CrossRef]
- Reine, T.M.; Lanzalaco, F.; Kristiansen, O.; Enget, A.R.; Satchell, S.; Jenssen, T.G.; Kolset, S.O. Matrix Metalloproteinase-9 Mediated Shedding of Syndecan-4 in Glomerular Endothelial Cells. Microcirculation 2019, 26, e12534. [Google Scholar] [CrossRef]
- Brule, S.; Charnaux, N.; Sutton, A.; Ledoux, D.; Chaigneau, T.; Saffar, L.; Gattegno, L. The Shedding of Syndecan-4 and Syndecan-1 from HeLa Cells and Human Primary Macrophages Is Accelerated by SDF-1/CXCL12 and Mediated by the Matrix Metalloproteinase-9. Glycobiology 2006, 16, 488–501. [Google Scholar] [CrossRef]
- Rodríguez-Manzaneque, J.C.; Carpizo, D.; Plaza-Calonge, M.d.C.; Torres-Collado, A.X.; Thai, S.N.-M.; Simons, M.; Horowitz, A.; Iruela-Arispe, M.L. Cleavage of Syndecan-4 by ADAMTS1 Provokes Defects in Adhesion. Int. J. Biochem. Cell Biol. 2009, 41, 800–810. [Google Scholar] [CrossRef]
- Subramanian, S.V.; Fitzgerald, M.L.; Bernfield, M. Regulated Shedding of Syndecan-1 and -4 Ectodomains by Thrombin and Growth Factor Receptor Activation. J. Biol. Chem. 1997, 272, 14713–14720. [Google Scholar] [CrossRef]
- Park, P.W.; Foster, T.J.; Nishi, E.; Duncan, S.J.; Klagsbrun, M.; Chen, Y. Activation of Syndecan-1 Ectodomain Shedding by Staphylococcus Aureus Alpha-Toxin and Beta-Toxin. J. Biol. Chem. 2004, 279, 251–258. [Google Scholar] [CrossRef]
- Manon-Jensen, T.; Itoh, Y.; Couchman, J.R. Proteoglycans in Health and Disease: The Multiple Roles of Syndecan Shedding. FEBS J. 2010, 277, 3876–3889. [Google Scholar] [CrossRef]
- Strand, M.E.; Herum, K.M.; Rana, Z.A.; Skrbic, B.; Askevold, E.T.; Dahl, C.P.; Vistnes, M.; Hasic, A.; Kvaløy, H.; Sjaastad, I.; et al. Innate Immune Signaling Induces Expression and Shedding of the Heparan Sulfate Proteoglycan Syndecan-4 in Cardiac Fibroblasts and Myocytes, Affecting Inflammation in the Pressure-Overloaded Heart. FEBS J. 2013, 280, 2228–2247. [Google Scholar] [CrossRef]
- Houston, M.; Julien, M.A.; Parthasarathy, S.; Chaikof, E.L. Oxidized Linoleic Acid Regulates Expression and Shedding of Syndecan-4. Am. J. Physiol. Cell Physiol. 2005, 288, C458–C466. [Google Scholar] [CrossRef]
- Sato, Y.; Tanino, Y.; Wang, X.; Nikaido, T.; Sato, S.; Misa, K.; Togawa, R.; Frevert, C.W.; Munakata, M. Baseline Serum Syndecan-4 Predicts Prognosis after the Onset of Acute Exacerbation of Idiopathic Interstitial Pneumonia. PLoS ONE 2017, 12, e0176789. [Google Scholar] [CrossRef]
- Luo, Q.; Ning, P.; Zheng, Y.; Shang, Y.; Zhou, B.; Gao, Z. Serum suPAR and Syndecan-4 Levels Predict Severity of Community-Acquired Pneumonia: A Prospective, Multi-Centre Study. Crit. Care 2018, 22, 15. [Google Scholar] [CrossRef]
- Nakao, M.; Sugaya, M.; Takahashi, N.; Otobe, S.; Nakajima, R.; Oka, T.; Kabasawa, M.; Suga, H.; Morimura, S.; Miyagaki, T.; et al. Increased Syndecan-4 Expression in Sera and Skin of Patients with Atopic Dermatitis. Arch. Dermatol. Res. 2016, 308, 655–660. [Google Scholar] [CrossRef]
- Shaik, F.; Balderstone, M.J.M.; Arokiasamy, S.; Whiteford, J.R. Roles of Syndecan-4 in Cardiac Injury and Repair. Int. J. Biochem. Cell Biol. 2022, 146, 106196. [Google Scholar] [CrossRef]
- Ansari, M.Y.; Ahmad, N.; Haqqi, T.M. Oxidative Stress and Inflammation in Osteoarthritis Pathogenesis: Role of Polyphenols. Biomed. Pharmacother. 2020, 129, 110452. [Google Scholar] [CrossRef]
- Wang, Y.; Herrera, A.H.; Li, Y.; Belani, K.K.; Walcheck, B. Regulation of Mature ADAM17 by Redox Agents for L-Selectin Shedding. J. Immunol. 2009, 182, 2449–2457. [Google Scholar] [CrossRef]
- Lipphardt, M.; Song, J.W.; Ratliff, B.B.; Dihazi, H.; Müller, G.A.; Goligorsky, M.S. Endothelial Dysfunction Is a Superinducer of Syndecan-4: Fibrogenic Role of Its Ectodomain. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H484–H496. [Google Scholar] [CrossRef]
- Wu, H.; Chen, Z.; Chen, J.-Z.; Xie, J.; Xu, B. Resveratrol Improves Tube Formation in AGE-Induced Late Endothelial Progenitor Cells by Suppressing Syndecan-4 Shedding. Oxid. Med. Cell Longev. 2018, 2018, 9045976. [Google Scholar] [CrossRef]
- Xie, J.; Li, R.; Wu, H.; Chen, J.; Li, G.; Chen, Q.; Wei, Z.; He, G.; Wang, L.; Ferro, A.; et al. Advanced Glycation Endproducts Impair Endothelial Progenitor Cell Migration and Homing via Syndecan 4 Shedding. Stem Cells 2017, 35, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhou, Q.; Xie, J.; Li, G.-N.; Chen, Q.-H.; Kang, L.-N.; Xu, B. Syndecan-4 Shedding Is Involved in the Oxidative Stress and Inflammatory Responses in Left Atrial Tissue with Valvular Atrial Fibrillation. Int. J. Clin. Exp. Pathol. 2015, 8, 6387–6396. [Google Scholar] [PubMed]
- Altindag, O.; Erel, O.; Aksoy, N.; Selek, S.; Celik, H.; Karaoglanoglu, M. Increased Oxidative Stress and Its Relation with Collagen Metabolism in Knee Osteoarthritis. Rheumatol. Int. 2007, 27, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, J.A.; Collins, J.A.; Loeser, R.F. Reactive Oxygen Species, Aging and Articular Cartilage Homeostasis. Free Radic. Biol. Med. 2019, 132, 73–82. [Google Scholar] [CrossRef]
- Jiang, X.Z.; Luo, K.H.; Ventikos, Y. Principal Mode of Syndecan-4 Mechanotransduction for the Endothelial Glycocalyx Is a Scissor-like Dimer Motion. Acta Physiol. 2020, 228, e13376. [Google Scholar] [CrossRef]
- Sanchez-Lopez, E.; Coras, R.; Torres, A.; Lane, N.E.; Guma, M. Synovial Inflammation in Osteoarthritis Progression. Nat. Rev. Rheumatol. 2022, 18, 258–275. [Google Scholar] [CrossRef]
- Ringvold, H.C.; Khalil, R.A. Protein Kinase C as Regulator of Vascular Smooth Muscle Function and Potential Target in Vascular Disorders. Adv. Pharmacol. 2017, 78, 203–301. [Google Scholar] [CrossRef]
- Charnaux, N.; Brule, S.; Chaigneau, T.; Saffar, L.; Sutton, A.; Hamon, M.; Prost, C.; Lievre, N.; Vita, C.; Gattegno, L. RANTES (CCL5) Induces a CCR5-Dependent Accelerated Shedding of Syndecan-1 (CD138) and Syndecan-4 from HeLa Cells and Forms Complexes with the Shed Ectodomains of These Proteoglycans as Well as with Those of CD44. Glycobiology 2005, 15, 119–130. [Google Scholar] [CrossRef]
- Wang, S.; Mobasheri, A.; Zhang, Y.; Wang, Y.; Dai, T.; Zhang, Z. Exogenous Stromal Cell-Derived Factor-1 (SDF-1) Suppresses the NLRP3 Inflammasome and Inhibits Pyroptosis in Synoviocytes from Osteoarthritic Joints via Activation of the AMPK Signaling Pathway. Inflammopharmacology 2021, 29, 695–704. [Google Scholar] [CrossRef]
- Zhao, Y.; Lin, D.; Zhu, X.; Yan, J.; Liang, Y.; Wang, Y.; Dai, T.; Zhang, Z.; Wang, S. SDF-1 Alleviates Osteoarthritis by Resolving Mitochondrial Dysfunction through the Activation of the Sirt3/PGC-1α Signalling Pathway. Arthritis Res. Ther. 2025, 27, 51. [Google Scholar] [CrossRef]
- Li, J.; Chen, H.; Cai, L.; Guo, D.; Zhang, D.; Zhou, X.; Xie, J. SDF-1α Promotes Chondrocyte Autophagy through CXCR4/mTOR Signaling Axis. Int. J. Mol. Sci. 2023, 24, 1710. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, H.; Zhang, D.; Xie, J.; Zhou, X. The Role of Stromal Cell-Derived Factor 1 on Cartilage Development and Disease. Osteoarthr. Cartil. 2021, 29, 313–322. [Google Scholar] [CrossRef]
- Yang, T.; Yang, X.; Wang, G.; Jia, D.; Li, Y. Unraveling the Crucial Role of SDF-1 in Osteoarthritis Progression: IL6/HIF-1α Positive Feedback and Chondrocyte Ferroptosis. Int. Immunopharmacol. 2025, 152, 114400. [Google Scholar] [CrossRef] [PubMed]
- Herum, K.M.; Romaine, A.; Wang, A.; Melleby, A.O.; Strand, M.E.; Pacheco, J.; Braathen, B.; Dunér, P.; Tønnessen, T.; Lunde, I.G.; et al. Syndecan-4 Protects the Heart From the Profibrotic Effects of Thrombin-Cleaved Osteopontin. J. Am. Heart Assoc. 2020, 9, e013518. [Google Scholar] [CrossRef]
- Kim, E.Y.; Roshanravan, H.; Dryer, S.E. Syndecan-4 Ectodomain Evokes Mobilization of Podocyte TRPC6 Channels and Their Associated Pathways: An Essential Role for Integrin Signaling. Biochim. Biophys. Acta 2015, 1853, 2610–2620. [Google Scholar] [CrossRef]
- Hayashida, K.; Chen, Y.; Bartlett, A.H.; Park, P.W. Syndecan-1 Is an In Vivo Suppressor of Gram-Positive Toxic Shock*. J. Biol. Chem. 2008, 283, 19895–19903. [Google Scholar] [CrossRef] [PubMed]
- Reizes, O.; Goldberger, O.; Smith, A.C.; Xu, Z.; Bernfield, M.; Bickel, P.E. Insulin Promotes Shedding of Syndecan Ectodomains from 3T3-L1 Adipocytes: A Proposed Mechanism for Stabilization of Extracellular Lipoprotein Lipase. Biochemistry 2006, 45, 5703–5711. [Google Scholar] [CrossRef]
- Lambert, J.; Makin, K.; Akbareian, S.; Johnson, R.; Alghamdi, A.A.A.; Robinson, S.D.; Edwards, D.R. ADAMTS-1 and Syndecan-4 Intersect in the Regulation of Cell Migration and Angiogenesis. J. Cell Sci. 2020, 133, jcs235762. [Google Scholar] [CrossRef]
- Endo, K.; Takino, T.; Miyamori, H.; Kinsen, H.; Yoshizaki, T.; Furukawa, M.; Sato, H. Cleavage of Syndecan-1 by Membrane Type Matrix Metalloproteinase-1 Stimulates Cell Migration. J. Biol. Chem. 2003, 278, 40764–40770. [Google Scholar] [CrossRef]
- Nikolova, V.; Koo, C.-Y.; Ibrahim, S.A.; Wang, Z.; Spillmann, D.; Dreier, R.; Kelsch, R.; Fischgräbe, J.; Smollich, M.; Rossi, L.H.; et al. Differential Roles for Membrane-Bound and Soluble Syndecan-1 (CD138) in Breast Cancer Progression. Carcinogenesis 2009, 30, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Seidel, C.; Sundan, A.; Hjorth, M.; Turesson, I.; Dahl, I.M.; Abildgaard, N.; Waage, A.; Borset, M. Serum Syndecan-1: A New Independent Prognostic Marker in Multiple Myeloma. Blood 2000, 95, 388–392. [Google Scholar] [CrossRef]
- Su, G.; Blaine, S.A.; Qiao, D.; Friedl, A. Membrane Type 1 Matrix Metalloproteinase-Mediated Stromal Syndecan-1 Shedding Stimulates Breast Carcinoma Cell Proliferation. Cancer Res. 2008, 68, 9558–9565. [Google Scholar] [CrossRef] [PubMed]
- Beauvais, D.M.; Ell, B.J.; McWhorter, A.R.; Rapraeger, A.C. Syndecan-1 Regulates Alphavbeta3 and Alphavbeta5 Integrin Activation during Angiogenesis and Is Blocked by Synstatin, a Novel Peptide Inhibitor. J. Exp. Med. 2009, 206, 691–705. [Google Scholar] [CrossRef]
- Pap, T.; Bertrand, J. Syndecans in Cartilage Breakdown and Synovial Inflammation. Nat. Rev. Rheumatol. 2013, 9, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Onyeisi, J.O.S.; Lopes, C.C.; Götte, M. Role of Syndecan-4 in Breast Cancer Pathophysiology. Am. J. Physiol. Cell Physiol. 2022, 323, C1345–C1354. [Google Scholar] [CrossRef]
- Pham, S.H.; Pratt, K.; Okolicsanyi, R.K.; Oikari, L.E.; Yu, C.; Peall, I.W.; Arif, K.T.; Chalmers, T.-A.; Gyimesi, M.; Griffiths, L.R.; et al. Syndecan-1 and -4 Influence Wnt Signaling and Cell Migration in Human Breast Cancers. Biochimie 2022, 198, 60–75. [Google Scholar] [CrossRef]
- Tan, X.; Khalil, N.; Tesarik, C.; Vanapalli, K.; Yaputra, V.; Alkhouri, H.; Oliver, B.G.G.; Armour, C.L.; Hughes, J.M. Th1 Cytokine-Induced Syndecan-4 Shedding by Airway Smooth Muscle Cells Is Dependent on Mitogen-Activated Protein Kinases. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, L700–L710. [Google Scholar] [CrossRef]
- Kunnathattil, M.; Rahul, P.; Skaria, T. Soluble Vascular Endothelial Glycocalyx Proteoglycans as Potential Therapeutic Targets in Inflammatory Diseases. Immunol. Cell Biol. 2024, 102, 97–116. [Google Scholar] [CrossRef]
- Jannaway, M.; Yang, X.; Meegan, J.E.; Coleman, D.C.; Yuan, S.Y. Thrombin-Cleaved Syndecan-3/-4 Ectodomain Fragments Mediate Endothelial Barrier Dysfunction. PLoS ONE 2019, 14, e0214737. [Google Scholar] [CrossRef]
- Milford, E.M.; Meital, L.; Kuballa, A.; Reade, M.C.; Russell, F.D. Fingolimod Does Not Prevent Syndecan-4 Shedding from the Endothelial Glycocalyx in a Cultured Human Umbilical Vein Endothelial Cell Model of Vascular Injury. Intensive Care Med. Exp. 2022, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Brooks, R.; Wei, X.; Lei, M.L.; Cid, F.C.; Roper, J.A.; Williamson, R.C.; Bass, M.D. Inhibition of EphA2 by Syndecan-4 in Wounded Skin Regulates Clustering of Fibroblasts. J. Mol. Cell Biol. 2024, mjae054. [Google Scholar] [CrossRef]
- Couchman, J.R.; Gopal, S.; Lim, H.C.; Nørgaard, S.; Multhaupt, H.A.B. Fell-Muir Lecture: Syndecans: From Peripheral Coreceptors to Mainstream Regulators of Cell Behaviour. Int. J. Exp. Pathol. 2015, 96, 1–10. [Google Scholar] [CrossRef]
- Scarpellini, A.; Huang, L.; Burhan, I.; Schroeder, N.; Funck, M.; Johnson, T.S.; Verderio, E.A.M. Syndecan-4 Knockout Leads to Reduced Extracellular Transglutaminase-2 and Protects against Tubulointerstitial Fibrosis. J. Am. Soc. Nephrol. 2014, 25, 1013–1027. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Yang, Z.; Li, P.; Lu, Y.; Lyu, T.; Liu, Y.; Zhou, X.; Zhao, P.; Chen, J. Duhuo Jisheng Decoction in Reduction of Inflammatory Response via Transforming Growth Factor-β/Smad Signaling Pathway for Repairing Rabbit Articular Cartilage Injury: A Randomized Controlled Trial. Int. Immunopharmacol. 2024, 144, 113646. [Google Scholar] [CrossRef] [PubMed]
- Hlubek, R.; Kušnierová, P.; Walder, P.; Bystroňová, I.; Douša, P. Biomarkers and Their Role in Understanding Osteoarthritis. Acta Chir. Orthop. Traumatol. Cech. 2025, 92, 36–43. [Google Scholar] [CrossRef]
- Hattori, Y.; Hasegawa, M.; Iino, T.; Imanaka-Yoshida, K.; Sudo, A. Role of Syndecan-4 in the Inhibition of Articular Cartilage Degeneration in Osteoarthritis. Biomedicines 2023, 11, 2257. [Google Scholar] [CrossRef]
- Zhou, K.; He, S.; Yu, H.; Pei, F.; Zhou, Z. Inhibition of Syndecan-4 Reduces Cartilage Degradation in Murine Models of Osteoarthritis through the Downregulation of HIF-2α by miR-96-5p. Lab. Investig. 2021, 101, 1060–1070. [Google Scholar] [CrossRef]
- Wang, L.; He, C. Nrf2-Mediated Anti-Inflammatory Polarization of Macrophages as Therapeutic Targets for Osteoarthritis. Front. Immunol. 2022, 13, 967193. [Google Scholar] [CrossRef]
- Zhai, G.; Doré, J.; Rahman, P. TGF-β Signal Transduction Pathways and Osteoarthritis. Rheumatol. Int. 2015, 35, 1283–1292. [Google Scholar] [CrossRef]
- Yu, J.-X.; Zhang, X.-T.; Liao, Y.-Q.; Zhang, Q.-Y.; Chen, H.; Lin, M.; Kumar, S. Relationship between Expression of CD105 and Growth Factors in Malignant Tumors of Gastrointestinal Tract and Its Significance. World J. Gastroenterol. 2003, 9, 2866–2869. [Google Scholar] [CrossRef] [PubMed]
- Piestrzeniewicz-Ulanska, D.; Brys, M.; Semczuk, A.; Jakowicki, J.A.; Krajewska, W.M. Expression of TGF-β Type I and II Receptors in Normal and Cancerous Human Endometrium. Cancer Lett. 2002, 186, 231–239. [Google Scholar] [CrossRef]
- Tanino, Y.; Wang, X.; Nikaido, T.; Misa, K.; Sato, Y.; Togawa, R.; Kawamata, T.; Kikuchi, M.; Frevert, C.W.; Tanino, M.; et al. Syndecan-4 Inhibits the Development of Pulmonary Fibrosis by Attenuating TGF-β Signaling. Int. J. Mol. Sci. 2019, 20, 4989. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ma, Y.; Li, S.; Ren, H.; Liu, Q.; Chen, X.; Miao, H.; Ye, T.; Lu, Q.; Yang, Z.; et al. Necroptotic TNFα-Syndecan 4-TNFα Vicious Cycle as a Therapeutic Target for Preventing Temporomandibular Joint Osteoarthritis. J. Bone Miner. Res. 2022, 37, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Wan, Q.; Qin, W.; Qin, W.; Yan, J.; Zhu, Y.; Wang, Y.; Ma, Y.; Wan, M.; Han, X.; et al. Effect of Regional Crosstalk between Sympathetic Nerves and Sensory Nerves on Temporomandibular Joint Osteoarthritic Pain. Int. J. Oral. Sci. 2025, 17, 3. [Google Scholar] [CrossRef]
- Lin, T.-J.; Lu, K.-W.; Chen, W.-H.; Cheng, C.-M.; Lin, Y.-W. Roles of Syndecan-4 and Relative Kinases in Dorsal Root Ganglion Neuron Adhesion and Mechanotransduction. Neurosci. Lett. 2015, 592, 88–93. [Google Scholar] [CrossRef]
- Zhu, S.; Zhu, J.; Zhen, G.; Hu, Y.; An, S.; Li, Y.; Zheng, Q.; Chen, Z.; Yang, Y.; Wan, M.; et al. Subchondral Bone Osteoclasts Induce Sensory Innervation and Osteoarthritis Pain. J. Clin. Investig. 2019, 129, 1076–1093. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Sheddases | Cleavage Sites | Cell Types | Diseases |
|---|---|---|---|
| MMP2 | Asp(31)-Leu(32) [16] Arg(36)-Tyr(37) [16] Tyr(37)-Phe(38) [16] Lys(105)-Leu(106) [16] | Mouse brain microvascular endothelial cells | Gliomas [34] |
| MMP7 | Lys(105)-Leu(106) [23] Asn(139)-Ile(140) [23] | B-lymphocytes | Inflammation caused by infection [35] |
| MMP9 | Lys(105)-Leu(106) [16] | Chondrocytes, glomerular endothelial cells, pancreatic β-cells, HeLa cells and human primary macrophages, mouse brain microvascular endothelial cells | Osteoarthritis [17] Diabetic nephropathy [28,36] Type 1 diabetes [9] Tumor [37] Gliomas [34] |
| MMP14 (MT1-MMP) | Asp(31)-Leu(32) [16] Ser(130)-Met(131) [16] Asn(139)-Ile(140) [16,23] | -- | -- |
| Plasmin | Lys(128)-Val(129) [23] | Human umbilical vein endothelial cells | Atheriosclerosis [30] |
| Thrombin | Arg(36)-Tyr(37) [16] Lys(114)-Arg(115) [30] | Human umbilical vein endothelial cells | Atheriosclerosis [30] |
| ADAMTS-1 and -4 | N-terminus near the first GAG attachment site | Mouse lung endothelial cells | Atheriosclerosis [38] |
| ADAM17 | A single site proximal to the cell membrane | the bladder carcinoma epithelial cell line ECV304, the lung epithelial cell line A459, primary alveolar epithelial cells | Acute lung injury [31] |
| Disease | Health Controls | Numbers | Age (Years) | Sex (M/F) | Patients | Numbers | Age (Years) | Sex (M/F) | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Acute pneumonia | 15.10 ± 2.60 ng/mL | 11 | 50.1 ± 4.8 | 6/5 | 20.3 ± 8.9 ng/mL | 30 | 67.1 ± 3.1 | 20/10 | [13] |
| Idiopathic interstitial pneumonia | 16.05 ± 0.77 ng/mL | 45 | 43 ± 2 | 29/16 | Clinically stable idiopathic interstitial pneumonia: 25.22 ± 3.72 ng/mL | 62 | 68 ± 1 | 47/15 | [44] |
| Acute exacerbation of idiopathic interstitial pneumonia: 10.65 ± 0.73 ng/mL | 56 | 70 ± 1 | 47/9 | ||||||
| Community-acquired pneumonia | 14.30 ± 5.34 ng/mL | 30 | -- | -- | Non-severe community-acquired pneumonia: 10.15 ± 4.37 ng/mL | 149 | 50 (33–65) | 92/57 | [45] |
| Severe community-acquired pneumonia: 9.54 ± 5.92 ng/mL | 103 | 56.14 ± 17.32 | 73/30 | ||||||
| Atopic dermatitis | 0.71 ± 0.67 ng/mL | 56 | 33.8 ± 13.13 | 29/27 | 1.41 ± 1.06 ng/mL | 59 | 32.1 ± 12.56 | 32/27 | [46] |
| Osteoarthritis | 0.019 ± 1.36 ng/mL | 16 | -- | -- | 19.23 ± 0.92 ng/mL | 29 | -- | -- | [17] |
| Disease/Condition | Involvement of sSDC4 | Reason for Involvement | Organ/System Affected | Reference |
|---|---|---|---|---|
| Unilateral Ureteral Obstruction (UUO) | Increased sSDC4 levels in the extracellular matrix, contributing to renal fibrosis. | 1. Enhanced NF-κB signaling leading to increased SDC4 expression. 2. Oxidative-stress-induced shedding of sSDC4. 3. sSDC4 acts as a chemoattractant for monocytes/macrophages. 4. The ectodomain of SDC4 promotes the transition of renal fibroblasts to myofibroblasts and enhances the synthesis of the extracellular matrix. 5. Injection of the SDC4 ectodomain leads to an increase in renal fibrosis. | Kidney | [50] |
| Tubulointerstitial Fibrosis | SDC4 knockout reduces fibrosis, indicating a pro-fibrotic role of sSDC4. | 1. SDC4 ectodomain promotes fibroblast-to-myofibroblast transition. 2. SDC4 ectodomain enhances collagen cross-linking. | Kidney | [50] |
| Diabetic Nephropathy | TNF-α-induced SDC4 shedding leads to increased glomerular permeability and proteinuria. | 1. TNF-α activates MMP9, leading to SDC4 shedding. 2. Loss of sSDC4 disrupts the glomerular endothelial glycocalyx, increasing protein permeability. | Kidney | [28] |
| Endothelial Dysfunction | Elevated sSDC4 in the secretome of endothelial cells, contributing to fibrogenesis. | 1. Sirt1 deficiency leading to increased NF-κB activity and SDC4 expression. 2. Oxidative stress causing increased shedding of SDC4. | Vascular system | [50] |
| Myocardial Infarction | sSDC4 plays a role in wound healing and fibrosis following myocardial infarction. | sSDC4 promotes collagen cross-linking and immune cell infiltration. | Heart | [84] |
| Asthma | Increased expression and shedding of SDC4 in airway smooth muscle cells (ASMCs). | 1. IL-1β and TNF-α enhance SDC4 shedding, which may regulate chemokine activity and mast cell recruitment. 2. sSDC4 binds chemokines like CXCL 10, potentially modulating their activity and contributing to airway hyper-responsiveness and remodeling. 3. Asthmatic ASMC may concentrate more SDC4 on their surface, leading to increased local chemokine accumulation. | Lungs (specifically airway smooth muscle) | [78] |
| Septic Cardiac Dysfunction | Increased expression and shedding of SDC4 in cardiac myocytes and fibroblasts. | 1. LPS challenge induces NF-κB-dependent upregulation of SDC4 expression and shedding. 2. Shed SDC4 ectodomains promote the expression of adhesion molecules and cytokines, facilitating immune cell recruitment to the myocardium. 3. Shed SDC4 regulates the expression of ECM components, including collagen synthesis and cross-linking enzymes, affecting cardiac stiffness and function. | Heart | [19] |
| Heart Failure | Elevated circulating levels of sSDC4 in patients. | 1. Chronic inflammation in heart failure leads to increased SDC4 shedding, which may reflect ongoing cardiac remodeling. 2. sSDC4 levels correlate with cardiac remodeling parameters and could serve as a biomarker for disease severity. | Heart | [19] |
| Acute Myocardial Infarction | Increased plasma levels of sSDC4; sSDC4 deficiency exacerbates cardiac dysfunction and impairs immune cell recruitment. | 1. SDC4 shedding is part of the acute inflammatory response following myocardial infarction. 2. sSDC4 may play a role in the recruitment of immune cells and ECM remodeling during the healing process. | Heart | [19,79] |
| Thrombotic Diseases | Increased shedding of SDC4. | Elevated thrombin activity during thrombotic events leads to SDC4 cleavage, contributing to endothelial barrier dysfunction. | Cardiovascular system | [80] |
| Sepsis | Increased shedding of SDC4. | Elevated levels of inflammatory mediators lead to endothelial glycocalyx damage and SDC4 shedding. | Multiple organs (systemic involvement) | [81] |
| Wound | Increased shedding of SDC4. | Unknown. | Skin | [82] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, K.; Ren, H.; Chen, X.; He, F.; Zhang, Y.; Zhang, H.; Li, F.; Yu, S. Shed Syndecan-4 and Its Possible Roles in Osteoarthritis. Biomedicines 2025, 13, 1037. https://doi.org/10.3390/biomedicines13051037
He K, Ren H, Chen X, He F, Zhang Y, Zhang H, Li F, Yu S. Shed Syndecan-4 and Its Possible Roles in Osteoarthritis. Biomedicines. 2025; 13(5):1037. https://doi.org/10.3390/biomedicines13051037
Chicago/Turabian StyleHe, Kangping, Haozhe Ren, Xiaohua Chen, Feng He, Yueying Zhang, Hongyun Zhang, Feifei Li, and Shibin Yu. 2025. "Shed Syndecan-4 and Its Possible Roles in Osteoarthritis" Biomedicines 13, no. 5: 1037. https://doi.org/10.3390/biomedicines13051037
APA StyleHe, K., Ren, H., Chen, X., He, F., Zhang, Y., Zhang, H., Li, F., & Yu, S. (2025). Shed Syndecan-4 and Its Possible Roles in Osteoarthritis. Biomedicines, 13(5), 1037. https://doi.org/10.3390/biomedicines13051037