
Modelling Population Genetic Screening in Rare Neurodegenerative Diseases

 , ,
, ,
Abstract
1. Introduction
2. Methods
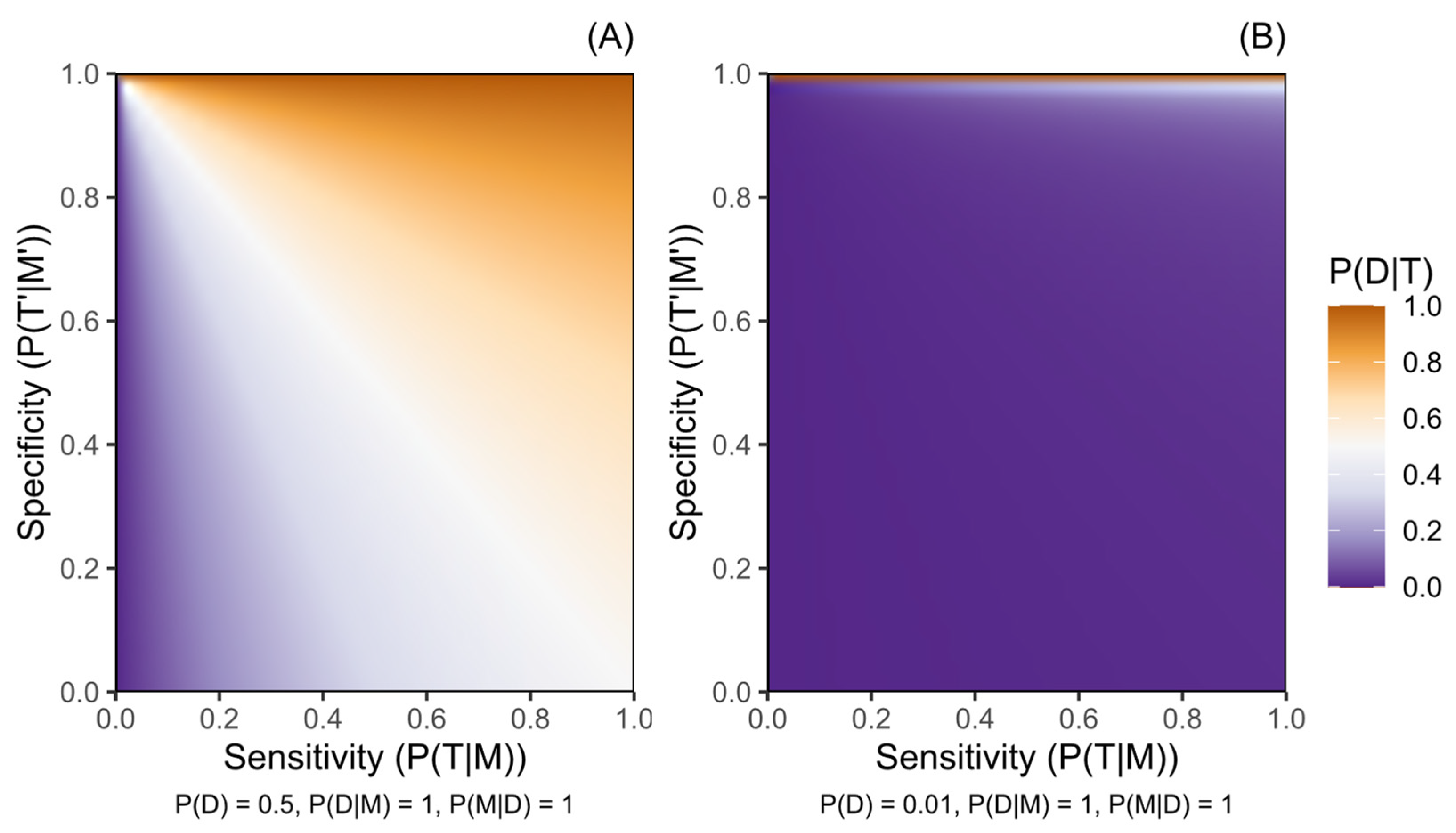
Definition of Probability in the Bayesian Framework
- P(D), probability of a person having or later manifesting disease D prior to testing;
- P(M|D), frequency of marker M among those affected by D;
- P(D|M), penetrance, probability of having or later manifesting D for people harbouring M;
- P(T|M), sensitivity (true positive rate) of the testing procedure for detecting M;
- P(T′|M′), specificity (true negative rate) of the testing procedure for identifying the absence of M.
3. Case Studies
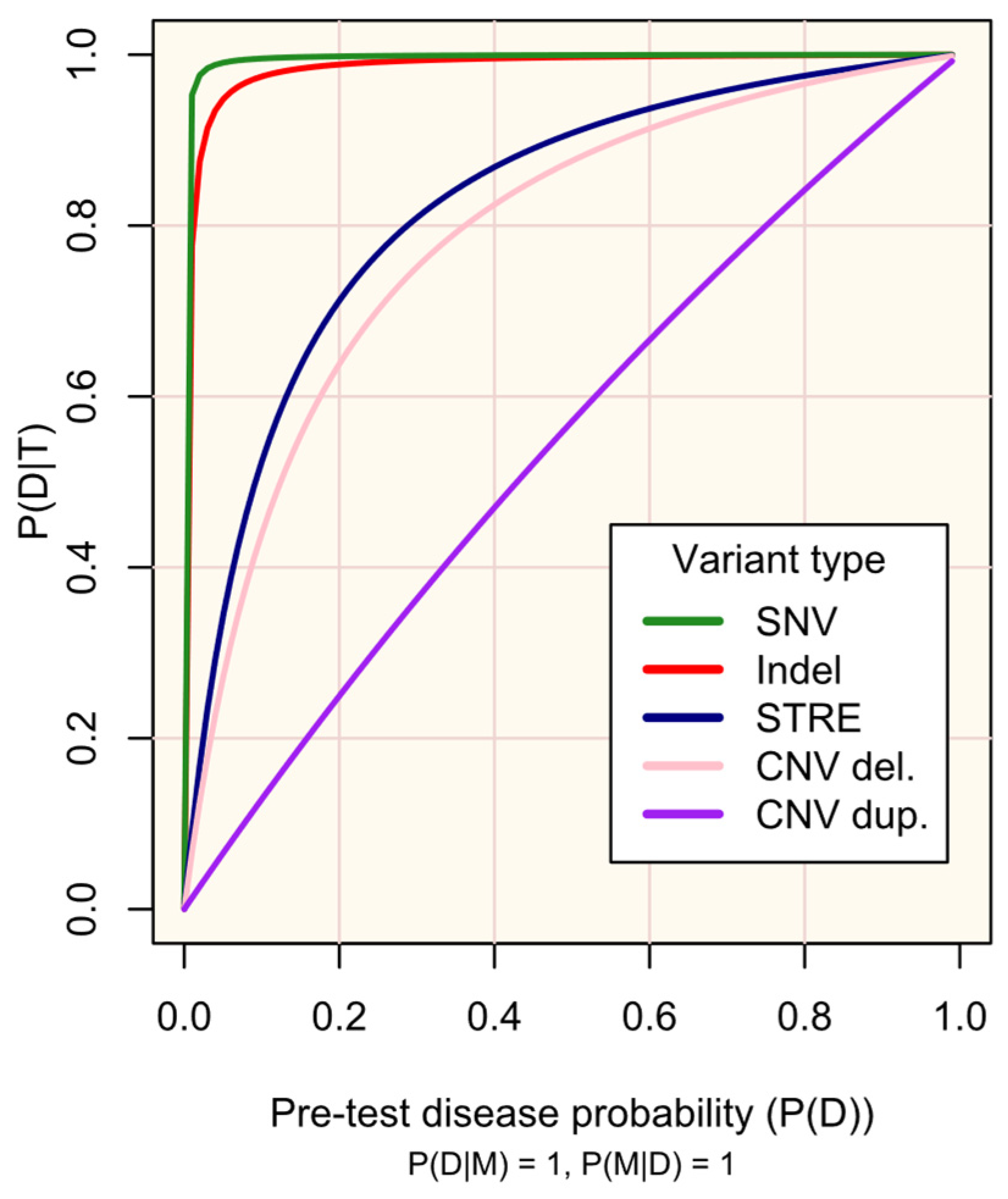
3.1. Case 1—Huntington’s Disease
3.2. Case 2—Amyotrophic Lateral Sclerosis
- SOD1 (all)—M includes any rare variant reported in people with ALS of European ancestry contained within the meta-analysis sample set from which the variant frequencies were derived (see Supplementary Materials S3.2) [38];
- FUS (all)—M includes any rare variant reported in people with ALS of European ancestry contained within the meta-analysis sample set from which the variant frequencies were derived (see Supplementary Materials S3.2) [38];
- FUS (ClinVar)—M includes any of 21 FUS variants reported as pathogenic or likely pathogenic for ALS within ClinVar and present within databases of familial and sporadic ALS (see Tables S3, S5 and S6) [31,32,33];
- C9orf72—M represents a pathogenic C9orf72 STRE of 30≤ repeat GGGGCC units within the first intron of the C9orf72 gene.

{kind=link}
{kind=link}
{kind=link}
| Case Study | Gene Containing Marker (Case Study Scenario) | Variant Type | Pre-Test Disease Probability § | Marker Frequency in People Affected § | Penetrance § | Test Sensitivity ¶ | Test Specificity ¶ | Disease Risk After Positive Test | Disease Risk After Negative Test | Relative Disease Risk After Positive Rather than Negative Test |
|---|---|---|---|---|---|---|---|---|---|---|
| - | - | - | P(D) | P(M|D) | P(D|M) | P(T|M) | P(T’|M’) | P(D|T) | P(D|T′) | - |
| 1: HD | HTT (screening) | STRE | 0.000410 | 1.000 | 1.000 | 0.990 | 0.900 | 0.00404 | 0.00000456 | 887 |
| HTT (targeted) | STRE | 0.500 | 1.000 | 1.000 | 0.990 | 0.900 | 0.908 | 0.011 | 82.7 | |
| 2: ALS | SOD1 (all) | SNV | 0.00333 | 0.0188 (0.0138, 0.0238) | 0.701 (0.491, 0.926) | 0.9996 | 0.9995 | 0.109 | 0.00327 | 33.3 |
| SOD1 (A5V) | SNV | 0.00333 | 0.000529 (4.43 × 10−5, 0.00101) | 0.91 | 0.9996 | 0.9995 | 0.00683 | 0.00333 | 2.05 | |
| FUS (all) | SNV | 0.00333 | 0.00425 (0.0023, 0.0062) | 0.579 (0.291, 0.884) | 0.9996 | 0.9995 | 0.0302 | 0.00332 | 9.09 | |
| FUS (ClinVar *) | SNV | 0.00333 | 0.00251 (0.00125, 0.00377) | 0.536 (0.211, 0.877) | 0.9996 | 0.9995 | 0.0194 | 0.00333 | 5.84 | |
| C9orf72 | STRE | 0.00333 | 0.0635 (0.0538, 0.0732) | 0.439 (0.358, 0.520) | 0.990 | 0.900 | 0.00519 | 0.00313 | 1.66 | |
| C9orf72 (positive sequencing screening confirmation) | STRE | 0.0052 | 0.0635 (0.0538, 0.0732) | 0.439 (0.358, 0.520) | 0.95 † | 0.98 † | 0.0198 | 0.00489 | 4.06 (6.35 Ω) | |
| 3: PKU | PAH (screening) | SNV | 0.000100 | 0.743 | 0.892 | 0.9996 | 0.9995 | 0.127 | 0.0000257 | 4.961 |
| PAH (positive metabolic screening confirmation) | SNV | 0.167 | 0.743 | 0.892 | 0.9996 | 0.9995 | 0.889 | 0.0497 | 17.9 (889,000 Ω) |
3.3. Case 3—Phenylketonuria
4. Results and Discussion
4.1. Post-Test Disease Probability
4.1.1. Screening Versus Diagnostic Testing
4.1.2. Relative Risk and Secondary Testing
4.1.3. Constraints upon Post-Test Disease Probability
4.2. Practical Implementation of Genetic Screening
4.2.1. Marker Selection
4.2.2. Utility over Time and Actionability
4.3. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iacoangeli, A.; Al Khleifat, A.; Sproviero, W.; Shatunov, A.; Jones, A.R.; Morgan, S.L.; Pittman, A.; Dobson, R.J.; Newhouse, S.J.; Al-Chalabi, A. DNAscan: Personal computer compatible NGS analysis, annotation and visualisation. BMC Bioinform. 2019, 20, 213. [Google Scholar] [CrossRef] [PubMed]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.-C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Illumina. Accuracy Improvements in Germline Small Variant Calling with the DRAGEN Platform2019 07/09/2019. Available online: https://science-docs.illumina.com/documents/Informatics/dragen-v3-accuracy-appnote-html-970-2019-006/Content/Source/Informatics/Dragen/dragen-v3-accuracy-appnote-970-2019-006/dragen-v3-accuracy-appnote-970-2019-006.html (accessed on 13 March 2023).
- Kosugi, S.; Momozawa, Y.; Liu, X.; Terao, C.; Kubo, M.; Kamatani, Y. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 2019, 20, 117. [Google Scholar] [CrossRef] [PubMed]
- Rhoades, R.; Jackson, F.; Teng, S. Discovery of rare variants implicated in schizophrenia using next-generation sequencing. J. Transl. Genet. Genom. 2019, 3, 1. [Google Scholar] [CrossRef]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef]
- Lee, J.J.; Wedow, R.; Okbay, A.; Kong, E.; Maghzian, O.; Zacher, M.; Nguyen-Viet, T.A.; Bowers, P.; Sidorenko, J.; Linnér, R.K.; et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet. 2018, 50, 1112–1121. [Google Scholar] [CrossRef]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J.; et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018, 562, 203–209. [Google Scholar] [CrossRef]
- Hörster, F.; Kölker, S.; Loeber, J.G.; Cornel, M.C.; Hoffmann, G.F.; Burgard, P. Newborn Screening Programmes in Europe, Arguments and Efforts Regarding Harmonisation: Focus on Organic Acidurias. JIMD Rep. 2017, 32, 105–115. [Google Scholar]
- Perrone, F.; Cacace, R.; Van Mossevelde, S.; Van den Bossche, T.; De Deyn, P.P.; Cras, P.; Engelborghs, S.; van der Zee, J.; Van Broeckhoven, C. Genetic screening in early-onset dementia patients with unclear phenotype: Relevance for clinical diagnosis. Neurobiol. Aging. 2018, 69, 292.e7–292.e14. [Google Scholar] [CrossRef]
- Majumder, M.A.; Guerrini, C.J.; McGuire, A.L. Direct-to-Consumer Genetic Testing: Value and Risk. Annu. Rev. Med. 2021, 72, 151–166. [Google Scholar] [CrossRef]
- Murray, M.F.; Evans, J.P.; Khoury, M.J. DNA-Based Population Screening: Potential Suitability and Important Knowledge Gaps. JAMA 2020, 323, 307–308. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, J.A.; Pimlott, N.; Grad, R.; Singh, H.; Szafran, O.; Wilson, B.J.; Groulx, S.; Thériault, G.; Bell, N.R. Screening: When things go wrong. Can. Fam. Physician 2018, 64, 502–508. [Google Scholar] [PubMed]
- Jansen, M.E.; Lister, K.J.; van Kranen, H.J.; Cornel, M.C. Policy Making in Newborn Screening Needs a Structured and Transparent Approach. Front. Public Health 2017, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Genome U.K.: The Future of Healthcare. Available online: https://www.gov.uk/government/publications/genome-uk-the-future-of-healthcare (accessed on 13 March 2023).
- Moorthie, S.; Hall, A.; Janus, J.; Brigden, T.; de Villiers, C.B.; Blackburn, L.; Johnson, E.; Kroese, M. Polygenic Scores and Clinical Utility; University of Cambridge: Cambridge, UK, 2021. [Google Scholar]
- Adhikari, A.N.; Gallagher, R.C.; Wang, Y.; Currier, R.J.; Amatuni, G.; Bassaganyas, L.; Chen, F.; Kundu, K.; Kvale, M.; Mooney, S.D.; et al. The role of exome sequencing in newborn screening for inborn errors of metabolism. Nat. Med. 2020, 26, 1392–1397. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Use of Genomics in Newborn Screening Programs: The Promise and Challenges 2021. Available online: https://archive.cdc.gov/www_cdc_gov/genomics/events/newborn_screening_2021.htm (accessed on 16 April 2025).
- Pain, O.; Gillett, A.C.; Austin, J.C.; Folkersen, L.; Lewis, C.M. A tool for translating polygenic scores onto the absolute scale using summary statistics. Eur. J. Hum. Genet. 2022, 30, 339–348. [Google Scholar] [CrossRef]
- Loeber, J.G.; Burgard, P.; Cornel, M.C.; Rigter, T.; Weinreich, S.S.; Rupp, K.; Hoffmann, G.F.; Vittozzi, L. Newborn screening programmes in Europe; arguments and efforts regarding harmonization. Part 1—From blood spot to screening result. J. Inherit. Metab. Dis. 2012, 35, 603–611. [Google Scholar] [CrossRef]
- Southern, K.W.; Munck, A.; Pollitt, R.; Travert, G.; Zanolla, L.; Dankert-Roelse, J.; Castellani, C.; ECFS CF Neonatal Screening Working Group. A survey of newborn screening for cystic fibrosis in Europe. J. Cyst. Fibros. 2007, 6, 57–65. [Google Scholar] [CrossRef]
- Rinaldo, P.; Zafari, S.; Tortorelli, S.; Matern, D. Making the case for objective performance metrics in newborn screening by tandem mass spectrometry. Ment. Retard. Dev. Disabil. Res. Rev. 2006, 12, 255–261. [Google Scholar] [CrossRef]
- Rehm, H.L.; Berg, J.S.; Brooks, L.D.; Bustamante, C.D.; Evans, J.P.; Landrum, M.J.; Ledbetter, D.H.; Maglott, D.R.; Martin, C.L.; Nussbaum, R.L.; et al. ClinGen—The Clinical Genome Resource. N. Engl. J. Med. 2015, 372, 2235–2242. [Google Scholar] [CrossRef]
- Hunter, J.E.; Irving, S.A.; Biesecker, L.G.; Buchanan, A.; Jensen, B.; Lee, K.; Martin, C.L.; Milko, L.; Muessig, K.; Niehaus, A.D.; et al. A standardized, evidence-based protocol to assess clinical actionability of genetic disorders associated with genomic variation. Genet. Med. 2016, 18, 1258–1268. [Google Scholar] [CrossRef]
- Biesecker, L.G. Genomic screening and genomic diagnostic testing-two very different kettles of fish. Genome Med. 2019, 11, 75. [Google Scholar] [CrossRef] [PubMed]
- Hunink, M.G.M.; Weinstein, M.C.; Wittenberg, E.; Drummond, M.F.; Pliskin, J.S.; Wong, J.B.; Glasziou, P.P. Decision Making in Health and Medicine: Integrating Evidence and Values, 2nd ed.; Cambridge University Press: Cambridge, UK, 2014. [Google Scholar]
- Bertram, L.; Tanzi, R.E. The genetic epidemiology of neurodegenerative disease. J. Clin. Invest. 2005, 115, 1449–1457. [Google Scholar] [CrossRef] [PubMed]
- Langbehn, D.R.; Brinkman, R.R.; Falush, D.; Paulsen, J.S.; Hayden, M.R. A new model for prediction of the age of onset and penetrance for Huntington’s disease based on CAG length. Clin. Genet. 2004, 65, 267–277. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef]
- Hillert, A.; Anikster, Y.; Belanger-Quintana, A.; Burlina, A.; Burton, B.K.; Carducci, C.; Chiesa, A.E.; Christodoulou, J.; Đorđević, M.; Desviat, L.R.; et al. The Genetic Landscape and Epidemiology of Phenylketonuria. Am. J. Hum. Genet. 2020, 107, 234–250. [Google Scholar] [CrossRef]
- van der Spek, R.A.A.; van Rheenen, W.; Pulit, S.L.; Kenna, K.P.; van den Berg, L.H.; Veldink, J.H. The project MinE databrowser: Bringing large-scale whole-genome sequencing in ALS to researchers and the public. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 432–440. [Google Scholar] [CrossRef]
- ALS Variant Server. Available online: https://www.umassmed.edu/als-clinic-research-center/ (accessed on 12 February 2021).
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Santos, R.; Lee, H.; Williams, A.; Baffour-Kyei, A.; Breen, G.; Iacoangeli, A. Investigating the performance of Oxford Nanopore long-read sequencing with respect to Illumina microarrays and short-read sequencing. bioRxiv 2024. [Google Scholar] [CrossRef]
- Mitsuhashi, S.; Frith, M.C.; Mizuguchi, T.; Miyatake, S.; Toyota, T.; Adachi, H.; Oma, Y.; Kino, Y.; Mitsuhashi, H.; Matsumoto, N. Tandem-genotypes: Robust detection of tandem repeat expansions from long DNA reads. Genome biology 2019, 20, 1–17. [Google Scholar] [CrossRef]
- Paulson, H. Repeat expansion diseases. Handb. Clin. Neurol. 2018, 147, 105–123. [Google Scholar]
- Iacoangeli, A.; Al Khleifat, A.; Sproviero, W.; Shatunov, A.; Jones, A.R.; Opie-Martin, S.; Naselli, E.; Topp, S.D.; Fogh, I.; Hodges, A.; et al. ALSgeneScanner: A pipeline for the analysis and interpretation of DNA sequencing data of ALS patients. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.-Y.; Zhou, Z.-R.; Che, C.-H.; Liu, C.-Y.; He, R.-L.; Huang, H.-P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.R.; Iacoangeli, A.; Opie-Martin, S.; van Vugt, J.; Al Khleifat, A.; Bredin, A.; Ossher, L.; Andersen, P.M.; Hardiman, O.; Mehta, A.R.; et al. The impact of age on genetic testing decisions in amyotrophic lateral sclerosis. Brain 2022, 145, 4440–4447. [Google Scholar] [CrossRef]
- Abel, O.; Powell, J.F.; Andersen, P.M.; Al-Chalabi, A. ALSoD: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum. Mutat. 2012, 33, 1345–1351. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Cudkowicz, M.E.; McKenna-Yasek, D.; Sapp, P.E.; Chin, W.; Geller, B.; Hayden, D.L.; Schoenfeld, D.A.; Hosler, B.A.; Horvitz, H.R.; Brown, R.H. Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis. Ann. Neurol. 1997, 41, 210–221. [Google Scholar] [CrossRef]
- Spargo, T.P.; Opie-Martin, S.; Bowles, H.; Lewis, C.M.; Iacoangeli, A.; Al-Chalabi, A. Calculating variant penetrance from family history of disease and average family size in population-scale data. Genome Med. 2022, 14, 141. [Google Scholar] [CrossRef]
- Saeed, M.; Yang, Y.; Deng, H.X.; Hung, W.Y.; Siddique, N.; Dellefave, L.; Gellera, C.; Andersen, P.M.; Siddique, T. Age and founder effect of SOD1 A4V mutation causing ALS. Neurol. 2009, 72, 1634–1639. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Akimoto, C.; Volk, A.E.; van Blitterswijk, M.; Van den Broeck, M.; Leblond, C.S.; Lumbroso, S.; Camu, W.; Neitzel, B.; Onodera, O.; van Rheenen, W.; et al. A blinded international study on the reliability of genetic testing for GGGGCC-repeat expansions in C9orf72 reveals marked differences in results among 14 laboratories. J. Med. Genet. 2014, 51, 419. [Google Scholar] [CrossRef]
- Schulze, A.; Lindner, M.; Kohlmüller, D.; Olgemöller, K.; Mayatepek, E.; Hoffmann, G.F. Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: Results, outcome, and implications. Pediatrics 2003, 111, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Ferini-Strambi, L.; Marelli, S.; Galbiati, A.; Rinaldi, F.; Giora, E. REM Sleep Behavior Disorder (RBD) as a marker of neurodegenerative disorders. Arch. Ital. Biol. 2014, 152, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Amado, D.A.; Davidson, B.L. Gene therapy for ALS: A review. Mol. Ther. 2021, 29, 3345–3358. [Google Scholar] [CrossRef] [PubMed]
- Paquin, R.S.; Mittendorf, K.F.; Lewis, M.A.; Hunter, J.E.; Lee, K.; Berg, J.S.; Williams, M.S.; Goddard, K.A.B. Expert and lay perspectives on burden, risk, tolerability, and acceptability of clinical interventions for genetic disorders. Genet. Med. 2019, 21, 2561–2568. [Google Scholar] [CrossRef]
- Batish, S.D.; Meservey, M.; Haji-Sheikhi, F.; Nedzweckas, E.; Wang, Z.; Datta, V.; Mullen, T.; Radcliff, J. HTT CAG Repeat Length Variation in Huntington Disease (HD) Patients: Experience from a US Reference Laboratory (2376). Neurology 2021, 96 (Suppl. S15), 2376. [Google Scholar] [CrossRef]
- Iacoangeli, A.; Al Khleifat, A.; Jones, A.R.; Sproviero, W.; Shatunov, A.; Opie-Martin, S.; Alzheimer’s Disease Neuroimaging Initiative; Morrison, K.E.; Shaw, P.J.; Shaw, C.E.; et al. C9orf72 intermediate expansions of 24–30 repeats are associated with ALS. Acta Neuropathol. Commun. 2019, 7, 115. [Google Scholar] [CrossRef]
- Iacoangeli, A.; Dilliott, A.A.; Al Khleifat, A.; Andersen, P.M.; Başak, N.A.; Cooper-Knock, J.; Corcia, P.; Couratier, P.; de Carvalho, M.; Drory, V.; et al. Oligogenic structure of amyotrophic lateral sclerosis has genetic testing, counselling and therapeutic implications. J. Neurol. Neurosurg. Psychiatry 2025. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spargo, T.P.; Iacoangeli, A.; Ryten, M.; Forzano, F.; Pearce, N.; Al-Chalabi, A. Modelling Population Genetic Screening in Rare Neurodegenerative Diseases. Biomedicines 2025, 13, 1018. https://doi.org/10.3390/biomedicines13051018
Spargo TP, Iacoangeli A, Ryten M, Forzano F, Pearce N, Al-Chalabi A. Modelling Population Genetic Screening in Rare Neurodegenerative Diseases. Biomedicines. 2025; 13(5):1018. https://doi.org/10.3390/biomedicines13051018
Chicago/Turabian StyleSpargo, Thomas P., Alfredo Iacoangeli, Mina Ryten, Francesca Forzano, Neil Pearce, and Ammar Al-Chalabi. 2025. "Modelling Population Genetic Screening in Rare Neurodegenerative Diseases" Biomedicines 13, no. 5: 1018. https://doi.org/10.3390/biomedicines13051018
APA StyleSpargo, T. P., Iacoangeli, A., Ryten, M., Forzano, F., Pearce, N., & Al-Chalabi, A. (2025). Modelling Population Genetic Screening in Rare Neurodegenerative Diseases. Biomedicines, 13(5), 1018. https://doi.org/10.3390/biomedicines13051018