
Multiplex Ligation Probe Amplification and Sanger Sequencing: Light and Shade in the Diagnosis of Lysosomal Storage Disorders

 , and
, and
Abstract

1. Introduction
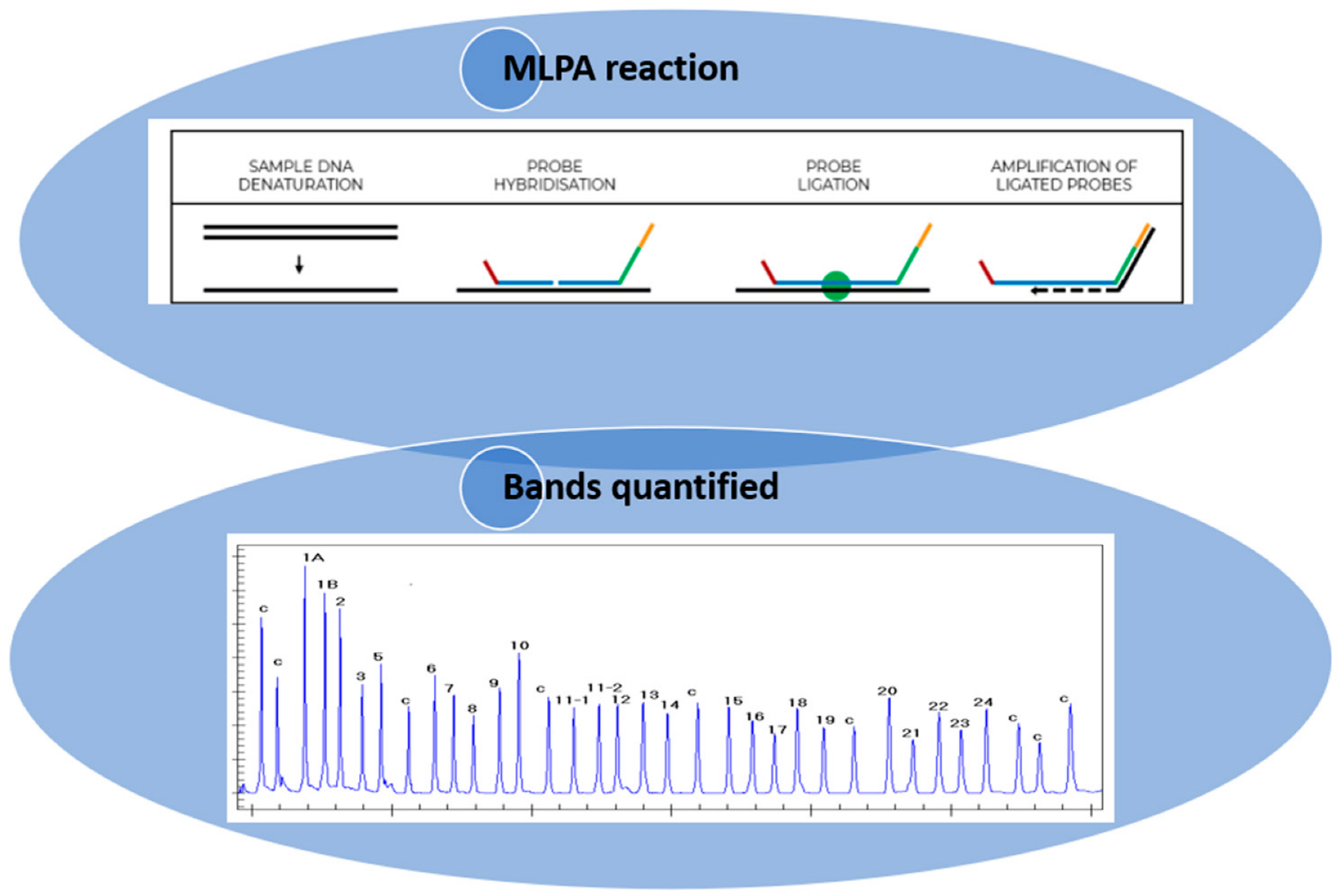
1.1. Principles of the MLPA Technique
1.2. MLPA in the Diagnosis of Fabry, Gaucher, and Pompe Diseases
2. Materials and Methods
2.1. Patients
2.2. Enzyme Activity Assays
2.3. Genetic Analysis
2.4. Biomarkers Assay
2.5. MLPA Analyses
2.6. Data Analysis
3. Results
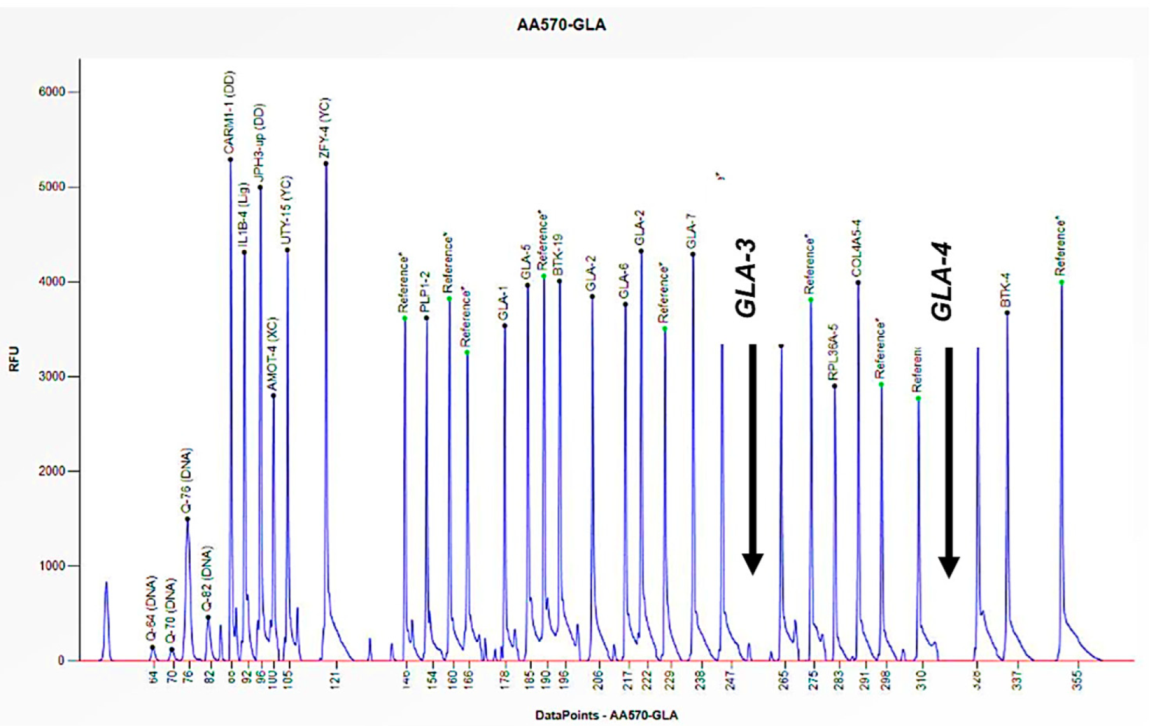
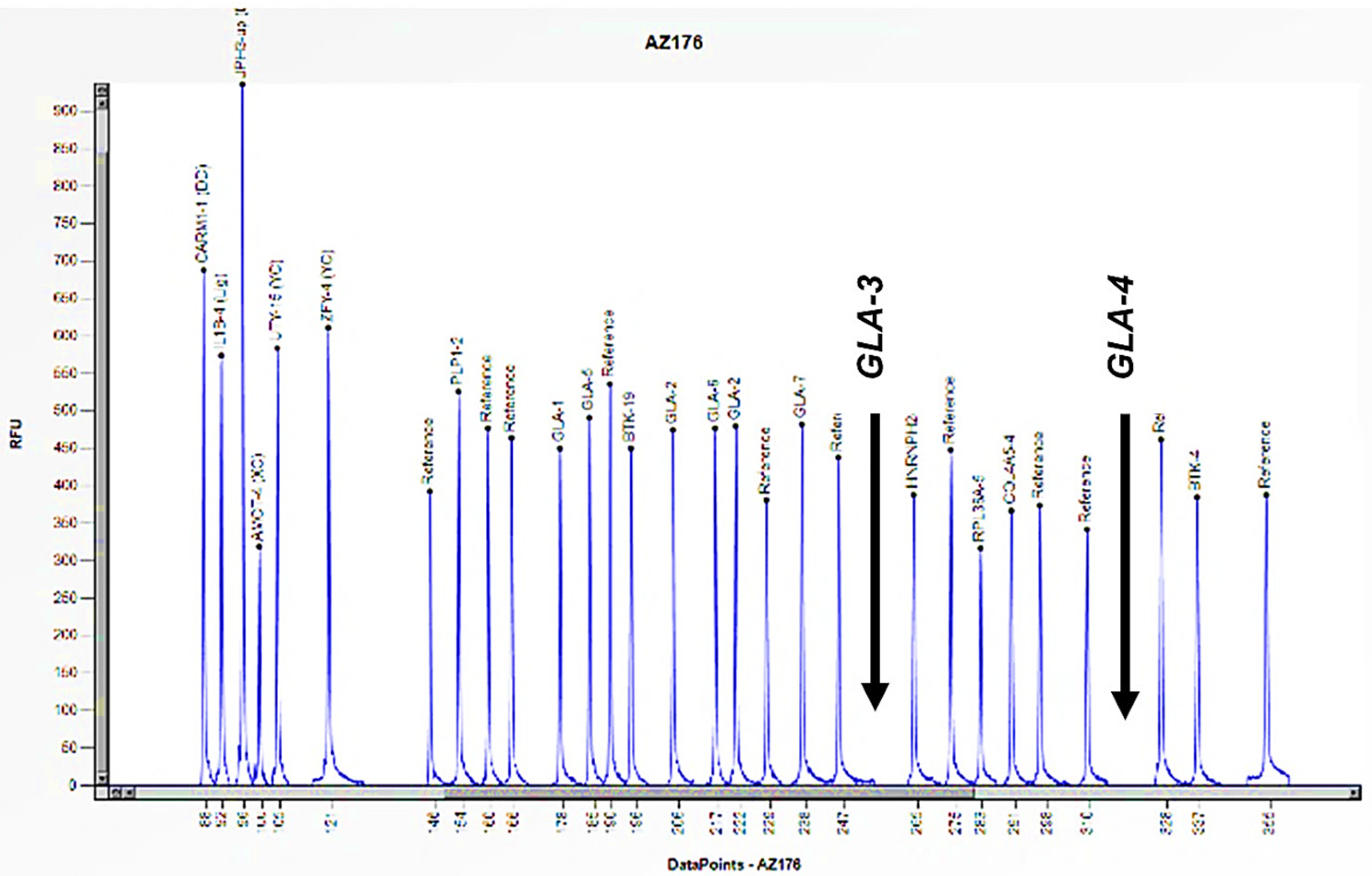
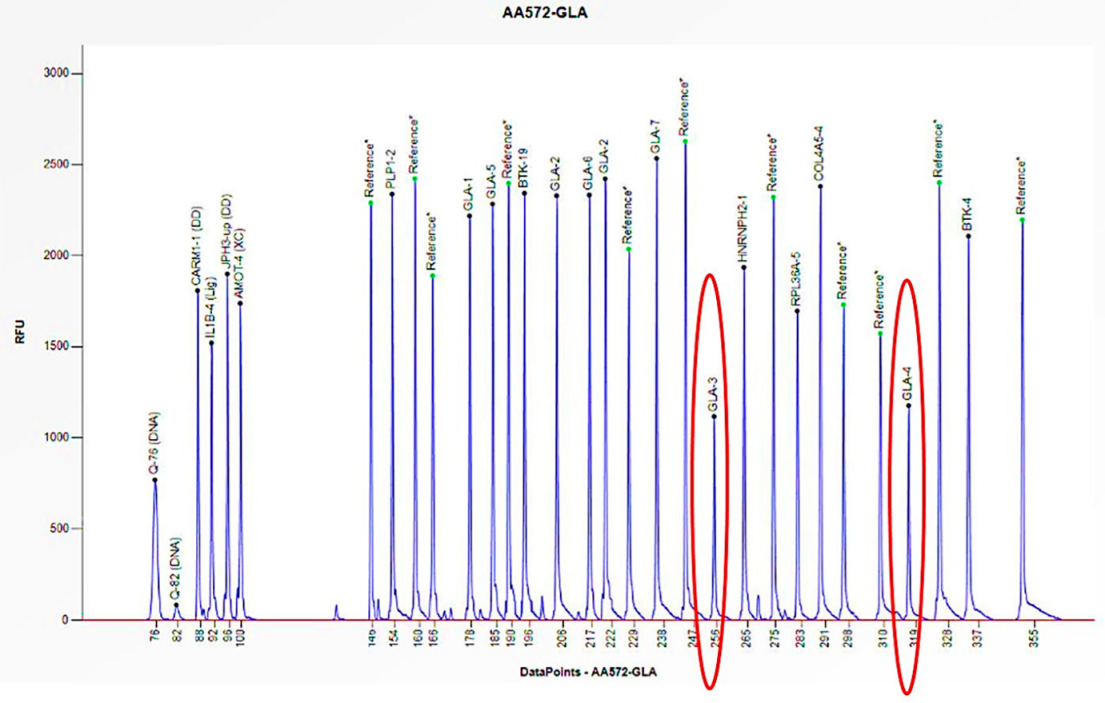
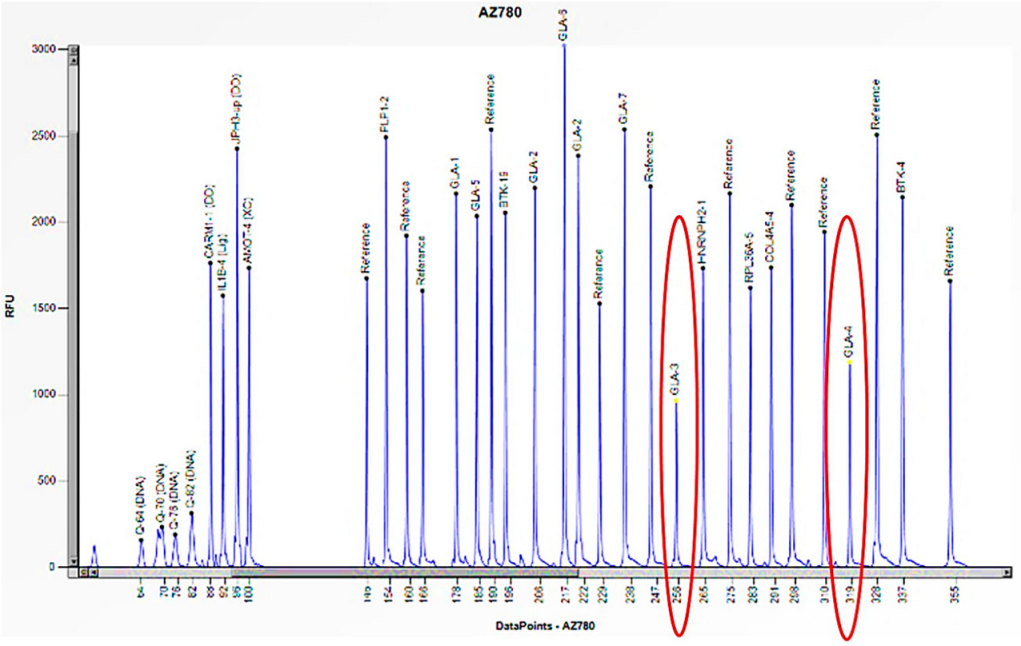
3.1. Patients with Clinical Suspicion of Fabry Disease
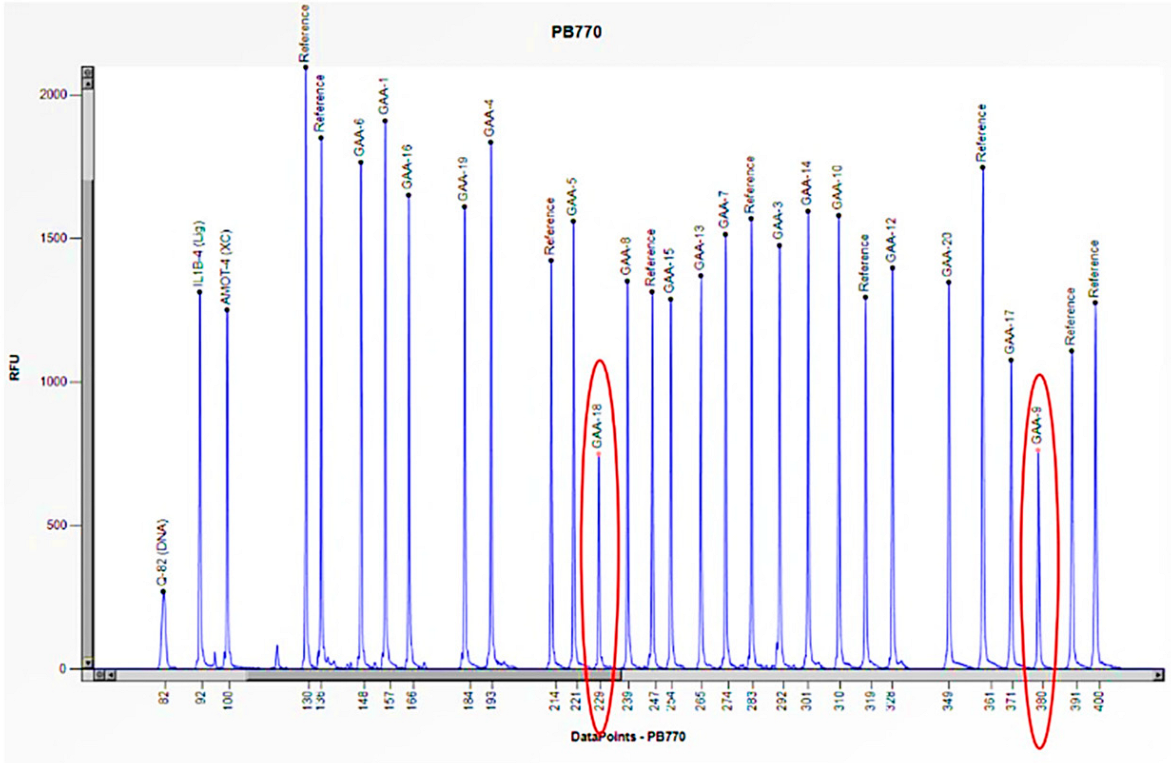
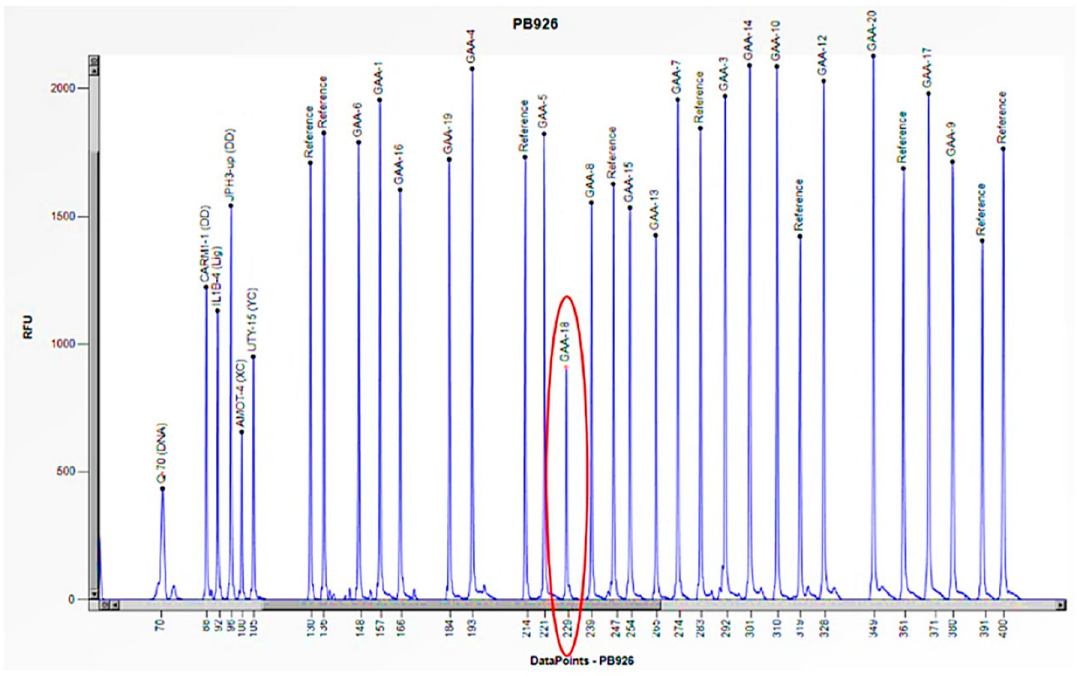
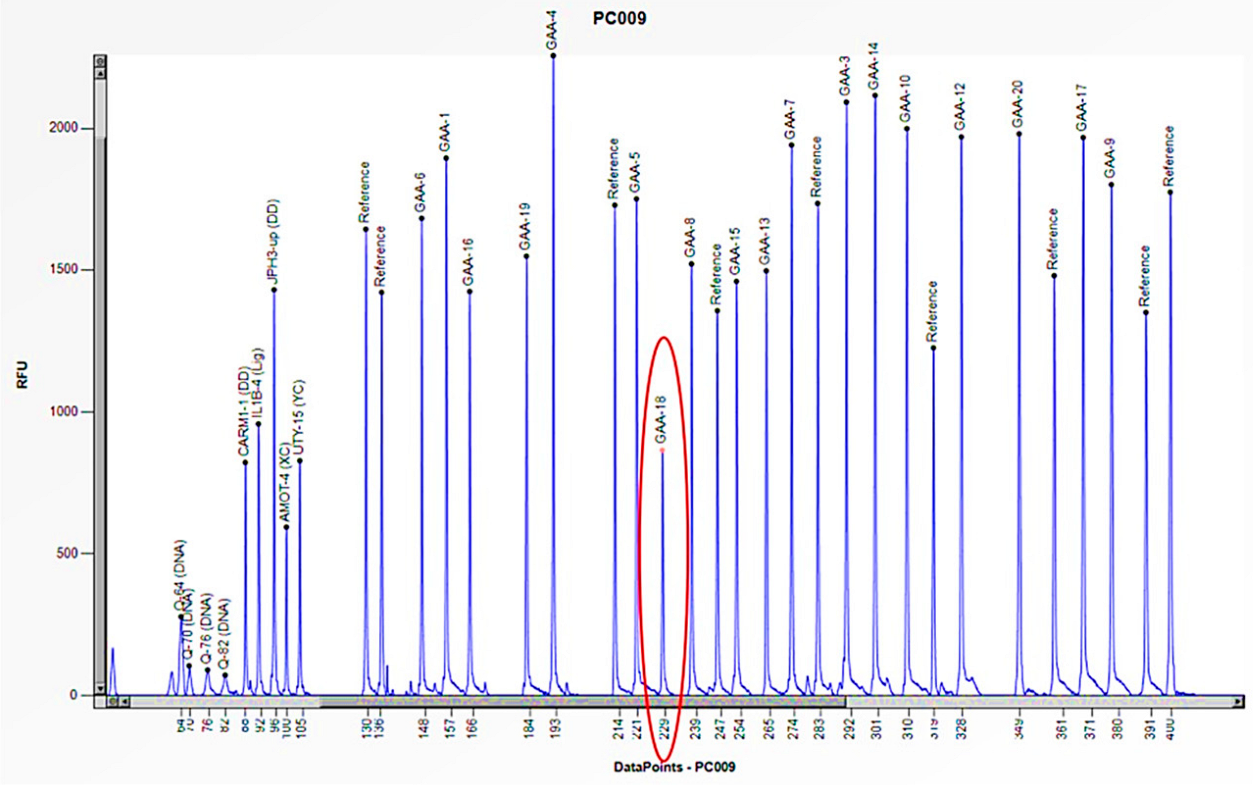
3.2. Patients with Clinical Suspicion of Pompe Disease
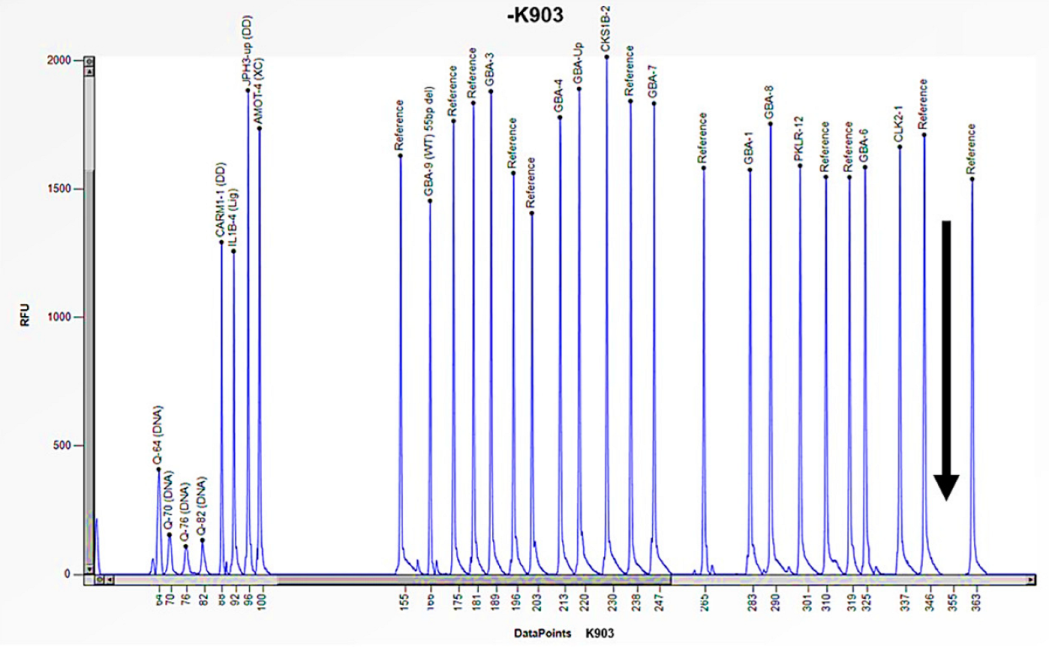
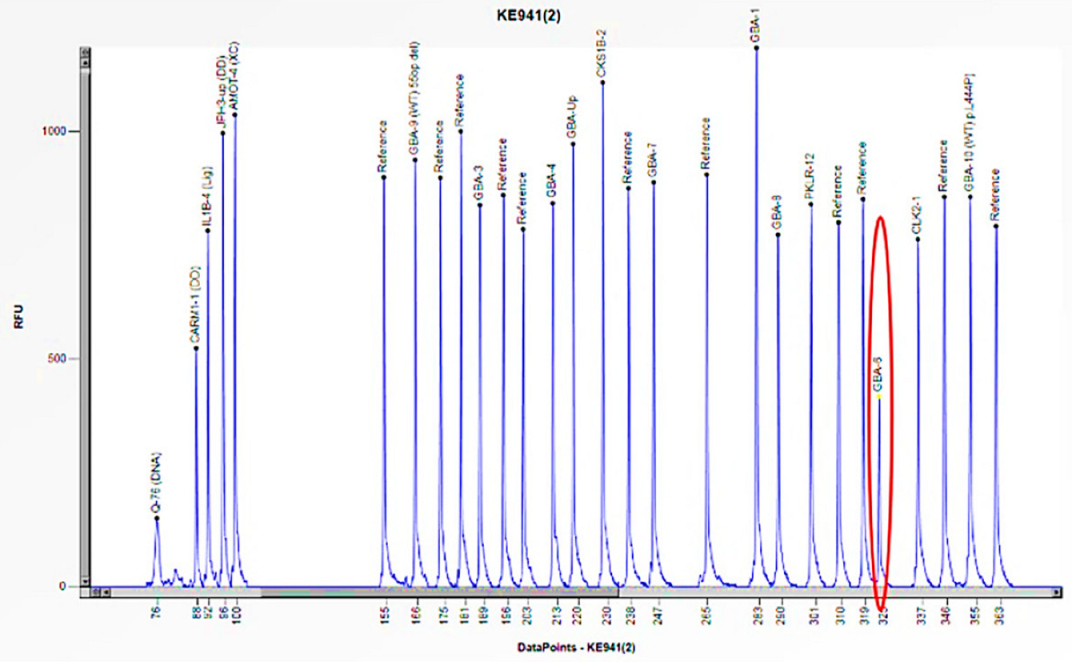
3.3. Patients with Clinical Suspicion of Gaucher Disease
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Armour, J.A.; Barton, D.E.; Cockburn, D.J.; Taylor, G.R. The detection of large deletions or duplications in genomic DNA. Hum. Mutat. 2002, 20, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Iafrate, A.J.; Brothman, A.R. Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat. Genet. 2007, 39 (Suppl. S7), S48–S54. [Google Scholar] [CrossRef] [PubMed]
- Schouten, J.P.; McElgunn, C.J.; Waaijer, R.; Zwijnenburg, D.; Diepvens, F.; Pals, G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002, 30, e57. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ferri, L.; Cavicchi, C.; Fiumara, A.; Parini, R.; Guerrini, R.; Morrone, A. Pitfalls in the detection of gross gene rearrangements using MLPA in Fabry disease. Clin. Chim. Acta 2016, 452, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, P.; Jasinska, A.J.; Kwiatkowski, D.J. New applications and developments in the use of multiplex ligation-dependent probe amplification. Electrophoresis 2008, 29, 4627–4636. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, S.; Currie-Fraser, E.; Xu, L.; Coffa, J. Multiplex ligation-dependent probe amplification analysis on capillary electrophoresis instruments for a rapid gene copy number study. J. Biomol. Tech. 2008, 19, 238–243. [Google Scholar] [PubMed] [PubMed Central]
- Coffa, J.; van de Wiel, M.A.; Diosdado, B.; Carvalho, B.; Schouten, J.; Meijer, G.A. MLPAnalyzer: Data analysis tool for reliable automated normalization of MLPA fragment data. Cell. Oncol. 2008, 30, 323–335. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cáceres, A.; Armengol, L.; Villatoro, S.; González, J.R. MLPAstats: An R GUI package for the integrated analysis of copy number alterations using MLPA data. BMC Bioinform. 2011, 12, 147. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schirinzi, A.; Centra, M.; Prattichizzo, C.; Gigante, M.; De Fabritiis, M.; Giancaspro, V.; Petrarulo, F.; Santacroce, R.; Margaglione, M.; Gesualdo, L.; et al. Identification of GLA gene deletions in Fabry patients by Multiplex Ligation-dependent Probe Amplification (MLPA). Mol. Genet. Metab. 2008, 94, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Duro, G.; Anania, M.; Zizzo, C.; Francofonte, D.; Giacalone, I.; D’Errico, A.; Marsana, E.M.; Colomba, P. Diagnosis of Fabry Disease Using Alpha-Galactosidase A Activity or LysoGb3 in Blood Fails to Identify Up to Two Thirds of Female Patients. Int. J. Mol. Sci. 2024, 25, 5158. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schäfer, E.; Baron, K.; Widmer, U.; Deegan, P.; Neumann, H.P.; Sunder-Plassmann, G.; Johansson, J.O.; Whybra, C.; Ries, M.; Pastores, G.M.; et al. Thirty-four novel mutations of the GLA gene in 121 patients with Fabry disease. Hum. Mutat. 2005, 25, 412. [Google Scholar] [CrossRef] [PubMed]
- Shabbeer, J.; Yasuda, M.; Benson, S.D.; Desnick, R.J. Fabry disease: Identification of 50 novel alpha-galactosidase A mutations causing the classic phenotype and three-dimensional structural analysis of 29 missense mutations. Hum. Genom. 2006, 2, 297–309. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Marziliano, N.; Sapere, N.; Orsini, F.; Motta, V.; Veronese, S.; Gambacorta, M.; Merlini, P.A.; Intrieri, M. A quantitative-PCR protocolrapidlydetectsαGAL deletions/duplications in patients with Anderson-Fabrydisease. Mol. Genet. Metab. 2012, 105, 687–689. [Google Scholar] [CrossRef] [PubMed]
- Feldt-Rasmussen, U.; Dobrovolny, R.; Nazarenko, I.; Ballegaard, M.; Hasholt, L.; Rasmussen, A.K.; Christensen, E.I.; Sorensen, S.S.; Wibrand, F.; Desnick, R.J. Diagnostic dilemma: A young woman with Fabry disease symptoms, no family history, and a “sequencing cryptic” α-galactosidase a large deletion. Mol. Genet. Metab. 2011, 104, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Torras, J.; Navarro, I.; Giese, A.K.; Böttcher, T.; Mascher, H.; Lackner, K.J.; Fauler, G.; Paschke, E.; Cruzado, J.M.; et al. Broad spectrum of Fabry disease manifestation in an extended Spanish family with a new deletion in the GLA gene. Clin. Kidney J. 2012, 5, 395–400. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Horowitz, M.; Wilder, S.; Horowitz, Z.; Reiner, O.; Gelbart, T.; Beutler, E. The human glucocerebrosidase gene and pseudogene: Structure and evolution. Genomics 1989, 4, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Amico, G.; Grossi, S.; Vijzelaar, R.; Lanza, F.; Mazzotti, R.; Corsolini, F.; Ketema, M.; Filocamo, M. MLPA-based approach for initial and simultaneous detection of GBA deletions and recombinant alleles in patients affected by Gaucher Disease. Mol. Genet. Metab. 2016, 119, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Taverna, S.; Cammarata, G.; Colomba, P.; Sciarrino, S.; Zizzo, C.; Francofonte, D.; Zora, M.; Scalia, S.; Brando, C.; Curto, A.L.; et al. Pompe disease: Pathogenesis, moleculargenetics and diagnosis. Aging 2020, 12, 15856–15874. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Moschetti, M.; Lo Curto, A.; Giacomarra, M.; Francofonte, D.; Zizzo, C.; Messina, E.; Duro, G.; Colomba, P. Mutation Spectrum of GAA Gene in Pompe Disease: Current Knowledge and Results of an Italian Study. Int. J. Mol. Sci. 2024, 25, 9139. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chamoles, N.A.; Blanco, M.; Gaggioli, D. Fabry disease: Enzymatic diagnosis in dried blood spots on filter paper. Clin. Chim. Acta 2001, 308, 195–196. [Google Scholar] [CrossRef] [PubMed]
- Polo, G.; Burlina, A.P.; Kolamunnage, T.B.; Zampieri, M.; Dionisi-Vici, C.; Strisciuglio, P.; Zaninotto, M.; Plebani, M.; Burlina, A.B. Diagnosis of sphingolipidoses: A new simultaneous measurement of glycosphingolipids by LC-MS/MS. Clin. Chem. Lab. Med. 2017, 55, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Guggenbuhl, P.; Grosbois, B.; Chalès, G. Gaucher disease. Jt. Bone Spine 2008, 75, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Torralba, M.A.; Alfonso, P.; Pérez-Calvo, J.I.; Cenarro, A.; Pastores, G.M.; Giraldo, P.; Civeira, F.; Pocoví, M. High prevalence of the 55-bp deletion (c.1263del55) in exon 9 of the glucocerebrosidase gene causing misdiagnosis (for homozygous N370S (c.1226A > G) mutation) in Spanish Gaucher disease patients. Blood Cells Mol. Dis. 2002, 29, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Mokhtariye, A.; Hagh-Nazari, L.; Varasteh, A.R.; Keyfi, F. Diagnostic methods for Lysosomal Storage Disease. Rep. Biochem. Mol. Biol. 2019, 7, 119–128. [Google Scholar] [PubMed] [PubMed Central]
- Chin, E.L.; da Silva, C.; Hegde, M. Assessment of clinical analytical sensitivity and specificity of next-generation sequencing for detection of simple and complex mutations. BMC Genet. 2013, 14, 6. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Giacomarra, M.; Colomba, P.; Francofonte, D.; Zora, M.; Caocci, G.; Diomede, D.; Giuffrida, G.; Fiori, L.; Montanari, C.; Sapuppo, A.; et al. Gaucher Disease or Acid Sphingomyelinase Deficiency? The Importance of Differential Diagnosis. J. Clin. Med. 2024, 13, 1487. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zampieri, S.; Cattarossi, S.; Pavan, E.; Barbato, A.; Fiumara, A.; Peruzzo, P.; Scarpa, M.; Ciana, G.; Dardis, A. Accurate Molecular Diagnosis of Gaucher Disease Using Clinical Exome Sequencing as a First-Tier Test. Int. J. Mol. Sci. 2021, 22, 5538. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yoshimitsu, M.; Higuchi, K.; Miyata, M.; Devine, S.; Mattman, A.; Sirrs, S.; Medin, J.A.; Tei, C.; Takenaka, T. Identification of novel mutations in the α-galactosidase A gene in patients with Fabry disease: Pitfalls of mutation analyses in patients with low α-galactosidase A activity. J. Cardiol. 2011, 57, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Gal, A.; Grosz, Z.; Borsos, B.; Szatmari, I.; Sebők, A.; Jávor, L.; Harmath, V.; Szakszon, K.; Dezsi, L.; Balku, E.; et al. Correlation of GAA Genotype and Acid-α-Glucosidase Enzyme Activity in Hungarian Patients with Pompe Disease. Life 2021, 11, 507. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family Member | Sex (F: Female, M: Male) | Age | Enzyme Activity (≥3 nmol/mL/h) | Lyso-Gb3 (0.08–1.13 nmol/L) | GLA Gene | Status |
|---|---|---|---|---|---|---|
| Case 1 Family Study | ||||||
| 1 | M | 32 | 0 | 36.64 | Hemizygous deletion of exons 3 and 4 | Acroparesthesias, cornea verticillata, angiokeratomas. Ischemic cardiomyopathy. |
| Mother | F | 64 | 3.5 | 5.47 | Heterozygous deletion of exons 3 and 4 | Rheumatoid Arthritis |
| Case 2 Family Study | ||||||
| 2 | M | 46 | 0.2 | 35.37 | Hemizygous deletion of exons 3 and 4 | Left Ventricular Hypertrophy (LVH) |
| Mother | F | 70 | 9.2 | 9.67 | Heterozygous deletion of exons 3 and 4 | Left Ventricular Hypertrophy (LVH) |
| Family Member | Sex (F: Female, M: Male) | Age | Enzyme Activity (≥6 nmol/mL/h) | GAA Gene | Status |
|---|---|---|---|---|---|
| 3 | F | 1 month | 0.9 | E262K/Heterozygous deletion of exons 9 and 18 | Severe cardiac involvement, generalized hypotonia, muscle weakness, proteinuria, and poor growth |
| Mother | F | 37 | 5.9 | Heterozygous deletion of exons 9 and 18 | Asymptomatic |
| Father | M | 42 | 5.0 | E262K heterozygote | Asymptomatic |
| 4 | M | 9 | 1.5 | c.-32-13T > G/Heterozygous deletion of exon 18 | Increased CPK, increased AST/ALT, increased LDH, slight neck weakness |
| 5 | M | 24 | 1.2 | c.-32-13T > G/Heterozygous deletion of exon 18 | Hyperkemia, increased AST/ALT, muscle weakening, hepatomegaly. Muscle biopsy analysis (vacuolar myopathy with glycogen accumulation) |
| Family Member | Sex (F: female, M: male) | Age | Enzyme Activity (≥2.5 nmol/mL/h) | Lyso-Gb1 (≤6.8 ng/mL) | GBA1 Gene | Status |
|---|---|---|---|---|---|---|
| Case 6 Family Study | ||||||
| 6 | M | 2 | 0.4 | -- | Homozygous deletion of exon 10 | Hepatosplenomegaly, anaemia, thrombocytopenia, and gastroesophageal reflux |
| Mother | F | 38 | 3.5 | -- | Heterozygous deletion of exon 10 | Asymptomatic |
| Father | M | 43 | 4.5 | -- | Heterozygous deletion of exon 10 | Asymptomatic |
| Case 7 Family Study | ||||||
| 7 | F | 22 | 0.5 | 498.0 | N409S/Heterozygous deletion of exon 6 | Hepatomegaly, splenomegaly, and thrombocytopenia |
| Mother | F | 53 | 3.0 | -- | Heterozygous deletion of exon 6 | Asymptomatic |
| Father | M | 58 | 4.2 | -- | N409S heterozygote | Asymptomatic |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vinci, M.; Zizzo, C.; Moschetti, M.; Giacomarra, M.; Anania, M.; Duro, G.; Di Chiara, T.; Russo, M.; Messina, E.; Colomba, P.; et al. Multiplex Ligation Probe Amplification and Sanger Sequencing: Light and Shade in the Diagnosis of Lysosomal Storage Disorders. Biomedicines 2025, 13, 973. https://doi.org/10.3390/biomedicines13040973
Vinci M, Zizzo C, Moschetti M, Giacomarra M, Anania M, Duro G, Di Chiara T, Russo M, Messina E, Colomba P, et al. Multiplex Ligation Probe Amplification and Sanger Sequencing: Light and Shade in the Diagnosis of Lysosomal Storage Disorders. Biomedicines. 2025; 13(4):973. https://doi.org/10.3390/biomedicines13040973
Chicago/Turabian StyleVinci, Martina, Carmela Zizzo, Marta Moschetti, Miriam Giacomarra, Monia Anania, Giulia Duro, Tiziana Di Chiara, Maria Russo, Elisa Messina, Paolo Colomba, and et al. 2025. "Multiplex Ligation Probe Amplification and Sanger Sequencing: Light and Shade in the Diagnosis of Lysosomal Storage Disorders" Biomedicines 13, no. 4: 973. https://doi.org/10.3390/biomedicines13040973
APA StyleVinci, M., Zizzo, C., Moschetti, M., Giacomarra, M., Anania, M., Duro, G., Di Chiara, T., Russo, M., Messina, E., Colomba, P., & Duro, G. (2025). Multiplex Ligation Probe Amplification and Sanger Sequencing: Light and Shade in the Diagnosis of Lysosomal Storage Disorders. Biomedicines, 13(4), 973. https://doi.org/10.3390/biomedicines13040973