


To Predict the Prognosis and Immunological Characteristics of Pancreatic Cancer Based on Disulfide-Death Gene Death-Related lncRNA



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Present Study
Synopsis
3. Materials and Methods
3.1. TCGA Data Collection
3.2. Expression Extraction of Disulfide-Death Genes and Analysis of Their Co-Expressed lncRNA
3.3. Prognostic Model Construction and Validation
3.4. Functional Enrichment Analysis
3.5. Immune Cell Infiltration Analysis and Mutation Load Analysis
3.6. Immunotherapy Analysis and Drug Sensitivity Analysis
4. Results
4.1. Extract Disulfide Death-Related LncRNAs with Prognostic Value for PAAD Patients
4.2. Construction and Validation of Disulfide Death-Related lncRNA Prognostic Model
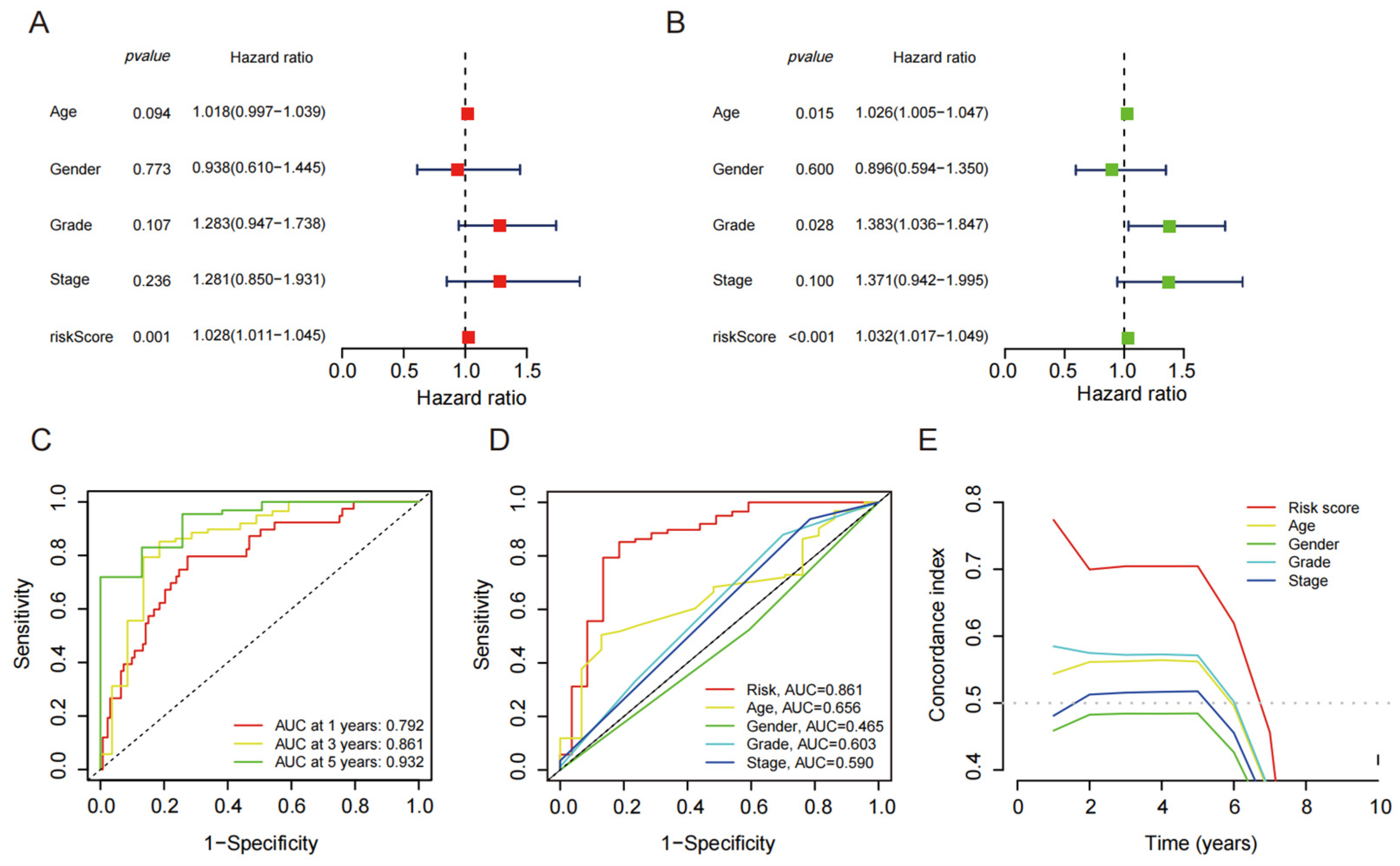
4.3. Independent Prognostic Value of Disulfide Death-Related lncRNAs
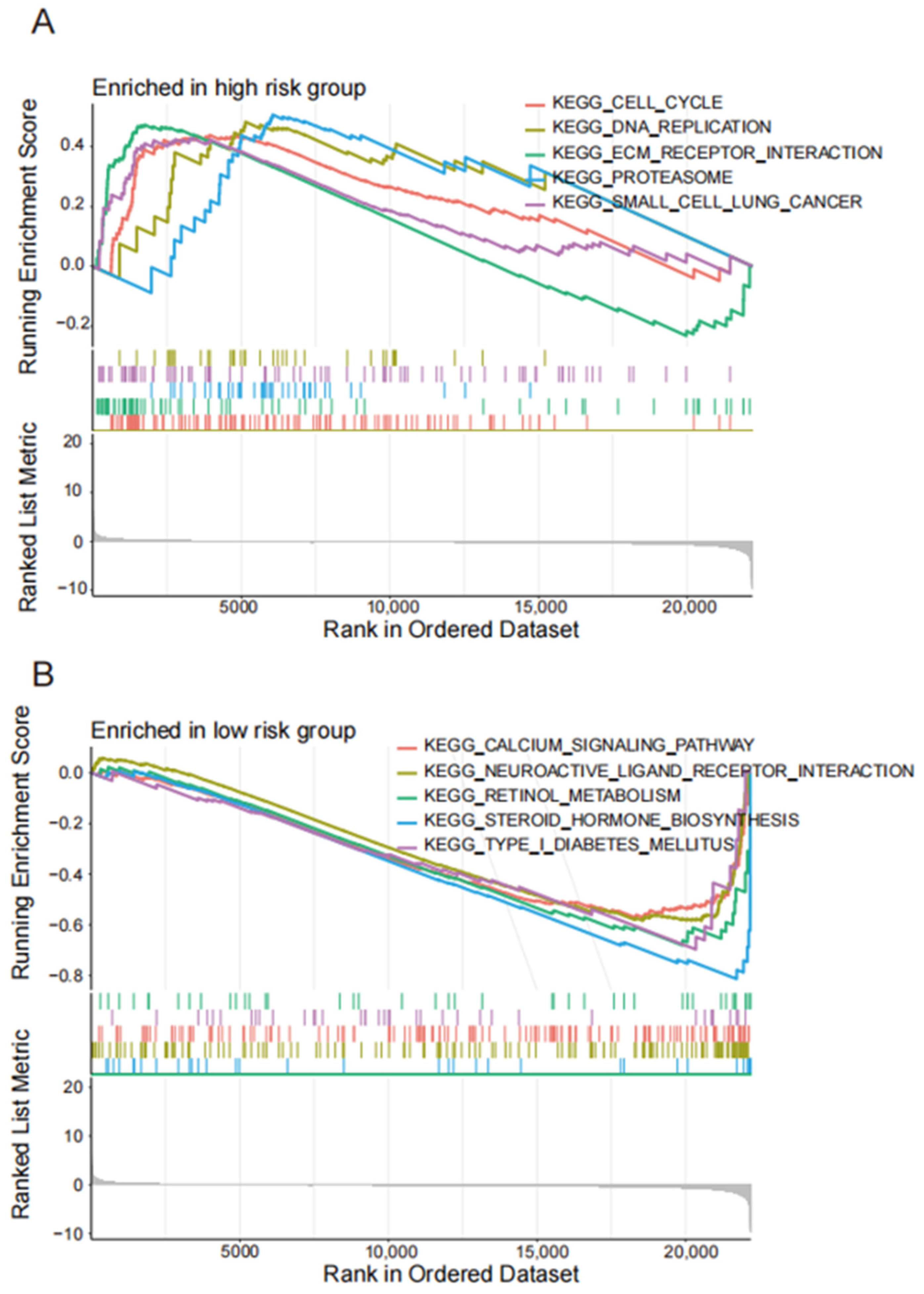
4.4. Enrichment Analysis of lncRNA Related Pathways Related to Disulfide Mortality
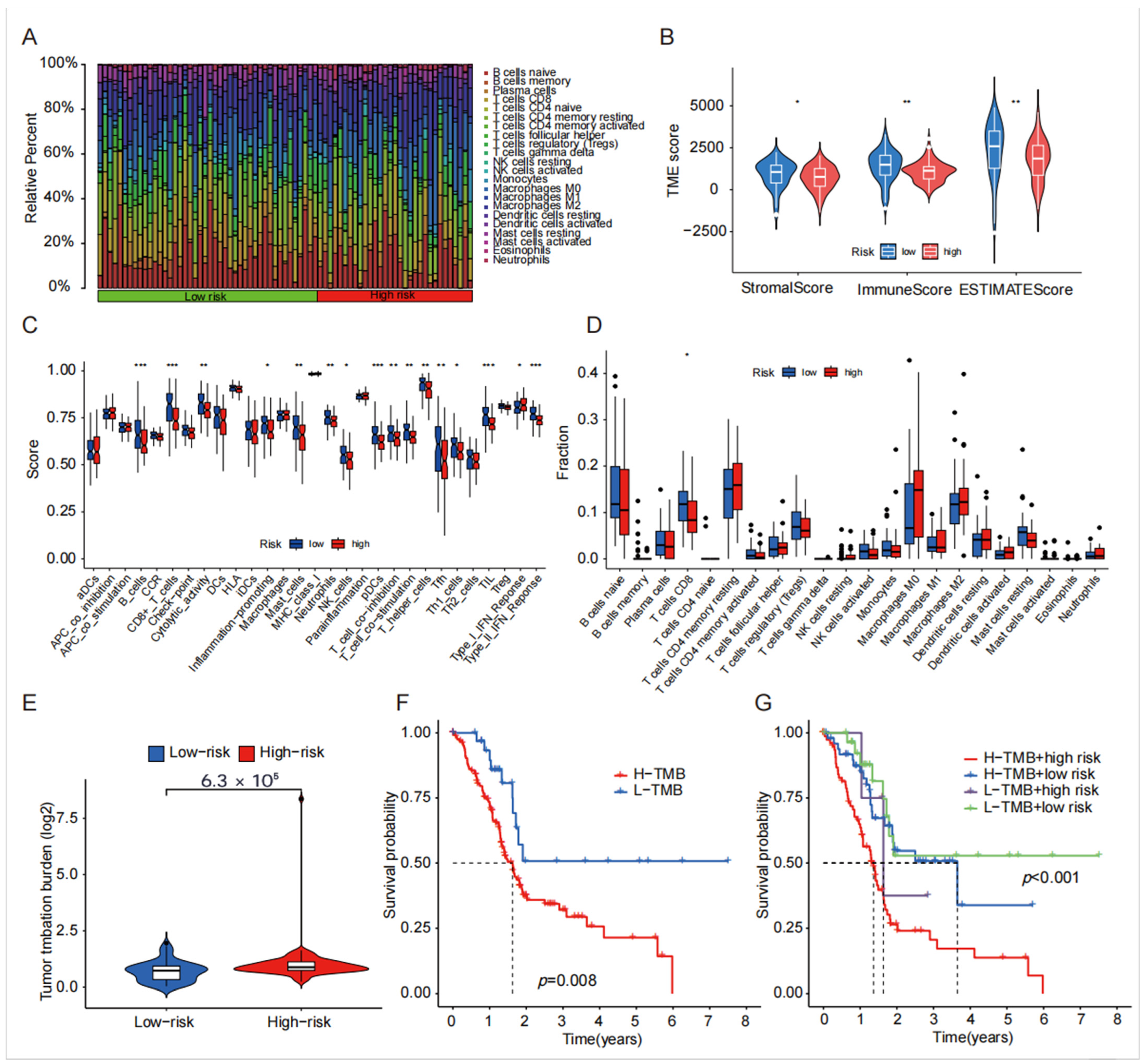
4.5. TME, Immune Cell Infiltration and Tumor Mutational Burden Analysis of Disulfide-Related lncRNA
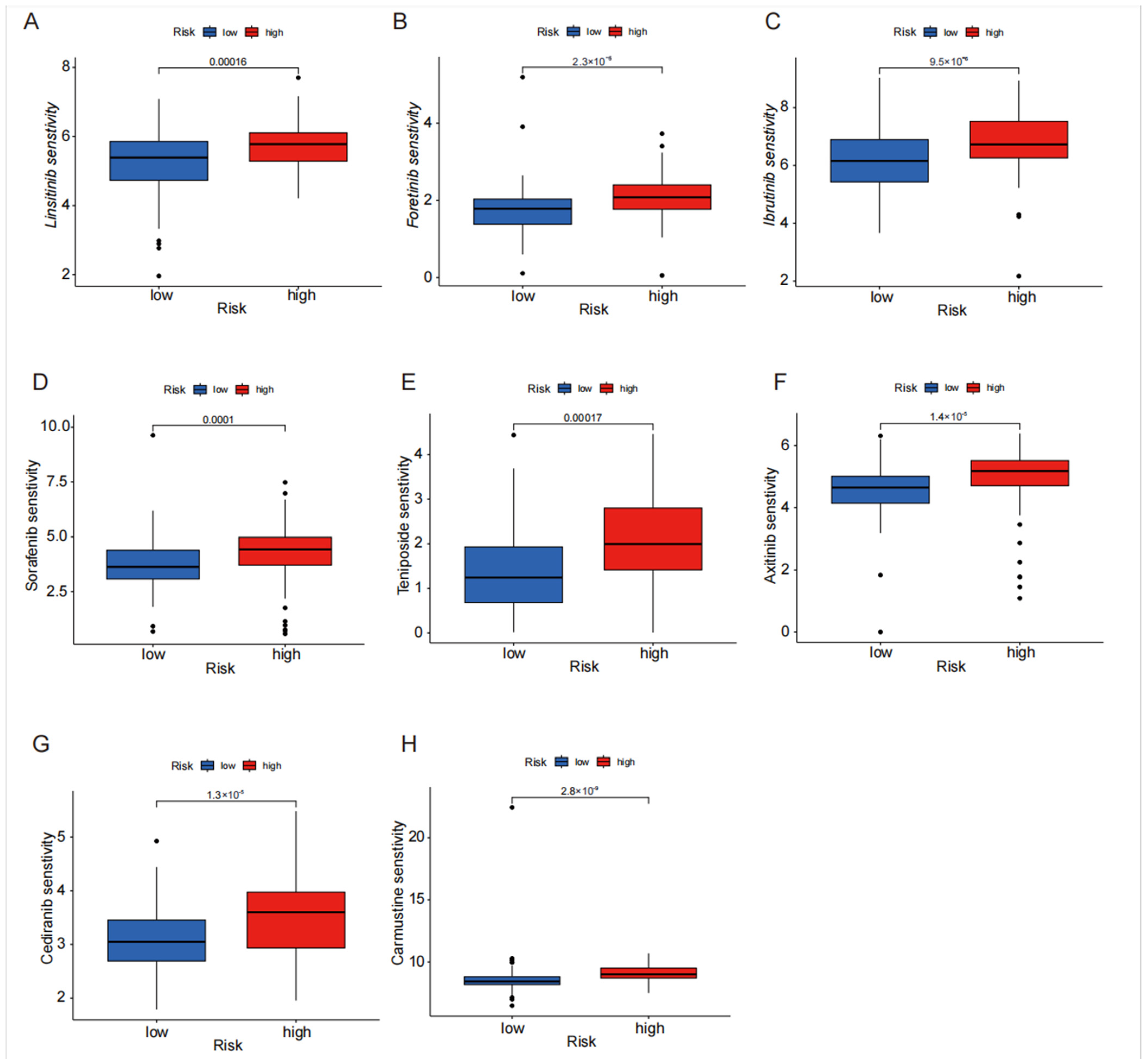
4.6. Drug Sensitivity Analysis of Disulfide Death-Related lncRNA in High- and Low-Risk Groups
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Klein, A.P. Pancreatic cancer epidemiology: Understanding the role of lifestyle and inherited risk factors. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 493–502. [Google Scholar] [CrossRef]
- Sugawara, T.; Rodriguez Franco, S.; Sherman, S.; Kirsch, M.J.; Colborn, K.; Ishida, J.; Grandi, S.; Al-Musawi, M.H.; Gleisner, A.; Schulick, R.D.; et al. Association of Adjuvant Chemotherapy in Patients with Resected Pancreatic Adenocarcinoma After Multiagent Neoadjuvant Chemotherapy. JAMA Oncol. 2023, 9, 316–323. [Google Scholar] [CrossRef]
- Wu, H.; Guo, S.; Liu, X.; Li, Y.; Su, Z.; He, Q.; Liu, X.; Zhang, Z.; Yu, L.; Shi, X.; et al. Noninvasive detection of pancreatic ductal adenocarcinoma using the methylation signature of circulating Tumour DNA. BMC Med. 2022, 20, 458. [Google Scholar] [CrossRef]
- Wu, S.; Zhuang, L.; Poyurovsky, M.V.; James You, M.; Hart, T.; Billadeau, D.D.; Chen, J.; Gan, B. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat. Cell Biol. 2023, 25, 404–414. [Google Scholar] [CrossRef]
- Zheng, P.; Zhou, C.; Ding, Y.; Duan, S. Disulfidptosis: A new target for metabolic cancer therapy. J. Exp. Clin. Cancer Res. 2023, 42, 103. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Wu, F.; Zhao, M.; Yang, M. Bibliometric Evaluation of 2012–2020 Publications on Ferroptosis in Cancer Treatment. Front. Cell Dev. Biol. 2022, 9, 793347. [Google Scholar] [CrossRef] [PubMed]
- Nair, L.; Chung, H.; Basu, U. Regulation of long non-coding RNAs and genome dynamics by the RNA surveillance machinery. Nat. Rev. Mol. Cell Biol. 2020, 21, 123–136. [Google Scholar] [CrossRef]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef]
- Van Heesch, S.; van Iterson, M.; Jacobi, J.; Boymans, S.; Essers, P.B.; de Bruijn, E.; Hao, W.; MacInnes, A.W.; Cuppen, E.; Simonis, M. Extensive localization of long noncoding RNAs to the cytosol and mono- and polyribosomal complexes. Genome Biol. 2014, 15, R6. [Google Scholar] [CrossRef]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Akhade, V.S.; Dighe, S.N.; Kataruka, S.; Rao, M.R. Mechanism of Wnt signaling induced down regulation of mrhl long non-coding RNA in mouse spermatogonial Cells. Nucleic Acids Res. 2016, 44, 387–401. [Google Scholar] [CrossRef]
- Huang, M.D.; Qi, W.M.F.Z.; Sun, M.; Xu, T.P.; Ma, P.; Shu, Y.Q. Long non-coding RNA TUG1 is up-regulated in hepatocellular carcinoma and promotes cell growth and Apoptosis by epigenetically silencing of KLF2. Mol. Cancer 2015, 14, 165. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, X.; Liu, N.; Shi, Y.; Liu, Y.; Ouyang, L.; Tam, S.; Xiao, D.; Liu, S.; Wen, F.; et al. A Nuclear Long Non-Coding RNA LINC00618 Accelerates Ferroptosis in a Manner Dependent upon Apoptosis. Mol. Ther. J. Am. Soc. Gene Ther. 2021, 29, 263–274. [Google Scholar] [CrossRef]
- Liu, L.; Liu, J.; Lyu, Q.; Huang, J.; Chen, Y.; Feng, C.; Liu, Y.; Chen, F.; Wang, Z. Disulfidptosis-associated lncRNAs index predicts prognosis and chemotherapy drugs sensitivity in cervical Cancer. Sci. Rep. 2023, 13, 12470. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Cao, S.; Wang, F.; Du, K.; Hu, F. Characterization and Prognosis of Biological Microenvironment in Lung Adenocarcinoma through a Disulfidptosis-Related lncRNAs Signature. Genet. Res. 2023, 2023, 6670514. [Google Scholar] [CrossRef]
- Dong, X.; Liao, P.; Liu, X.; Yang, Z.; Wang, Y.; Zhong, W.; Wang, B. Construction and Validation of a Reliable Disulfidptosis-Related lncRNAs Signature of the Subtype, Prognostic, and Immune Landscape in Colon Cancer. Int. J. Mol. Sci. 2023, 24, 12915. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.; Li, M.; Liu, Z.; Li, T.; Zhao, L.; Fu, W. Glycolysis-Related LINC02432/Hsa-miR-98-5p/HK2 Axis Inhibits Ferroptosis and Predicts Immune Infiltration, Tumor Mutation Burden, and Drug Sensitivity in Pancreatic Adenocarcinoma. Front. Pharmacol. 2022, 13, 937413. [Google Scholar] [CrossRef]
- Xie, W.; Chu, M.; Song, G.; Zuo, Z.; Han, Z.; Chen, C.; Li, Y.; Wang, Z.W. Emerging roles of long noncoding RNAs in chemoresistance of pancreatic cancer. Semin. Cancer Biol. 2022, 83, 303–318. [Google Scholar] [CrossRef]
- Gao, J.; Kwan, P.W.; Shi, D. Sparse kernel learning with LASSO and Bayesian inference algorithm. Neural Netw. Off. J. Int. Neural Netw. Soc. 2010, 23, 257–264. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar] [CrossRef] [PubMed]
- Halbrook, C.J.; Lyssiotis, C.A.; Pasca di Magliano, M.; Maitra, A. Advances and challenges. Cell 2023, 186, 1729–1754. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Gillen, S.; Schuster, T.; Meyer Zum Buschenfelde, C.; Friess, H.; Kleeff, J. Preoperative/neoadjuvant therapy in pancreatic cancer: A systematic review and meta-analysis of response and resection percentages. PLoS Med. 2010, 7, e1000267. [Google Scholar] [CrossRef]
- Nie, W.; Ge, H.J.; Yang, X.Q.; Sun, X.; Huang, H.; Tao, X.; Chen, W.S.; Li, B. lncRNA-UCA1 exerts oncogenic functions in non-small cell lung cancer by targeting miR-193a-3p. Cancer Lett. 2016, 371, 99–106. [Google Scholar] [CrossRef]
- Spizzo, R.; Almeida, M.I.; Colombatti, A.; Calin, G.A. Long non-coding RNAs and cancer: A new frontier of translational research? Oncogene 2012, 31, 4577–4587. [Google Scholar] [CrossRef]
- Tan, T.Y.; Lin, J.F.; Li, T.; Li, J.J.; Xu, R.H.; Ju, H.Q. lncRNA-mediated posttranslational modifications and reprogramming of energy metabolism in cancer. Cancer Commun. 2021, 41, 109–120. [Google Scholar] [CrossRef]
- Stoffel, E.M.; Brand, R.E.; Goggins, M. Pancreatic Cancer: Changing Epidemiology and New Approaches to Risk Assessment, Early Detection, and Prevention. Gastroenterology 2023, 164, 752–765. [Google Scholar] [CrossRef]
- Wolfgang, C.L.; Herman, J.M.; Laheru, D.A.; Klein, A.P.; Erdek, M.A.; Fishman, E.K.; Hruban, R.H. Recent progress in pancreatic cancer. CA Cancer J. Clin. 2013, 63, 318–348. [Google Scholar] [CrossRef]
- Ducreux, M.; Cuhna, A.S.; Caramella, C.; Hollebecque, A.; Burtin, P.; Goéré, D.; Seufferlein, T.; Haustermans, K.; Van Laethem, J.L.; Conroy, T.; et al. Cancer of the pancreas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2015, 26 (Suppl. S5), v56–v68. [Google Scholar] [CrossRef]
- Khorana, A.A.; Mangu, P.B.; Berlin, J.; Engebretson, A.; Hong, T.S.; Maitra, A.; Mohile, S.G.; Mumber, M.; Schulick, R.; Shapiro, M.; et al. Potentially Curable Pancreatic Cancer: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 2541–2556. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhu, C.; He, Z.; Chen, S.; Li, L.; Sun, C. lncRNA PSMB8-AS1 contributes to pancreatic cancer progression via modulating miR-382-3p/STAT1/PD-L1 Axis. J. Exp. Clin. Cancer Res. 2020, 39, 179. [Google Scholar] [CrossRef]
- Yang, A.; Liu, X.; Liu, P.; Feng, Y.; Liu, H.; Gao, S.; Huo, L.; Han, X.; Wang, J.; Kong, W. lncRNA UCA1 promotes development of gastric cancer via the miR-145/MYO6 axis. Cell. Mol. Biol. Lett. 2021, 26, 33. [Google Scholar] [CrossRef]
- Ye, P.; Lv, X.; Aizemaiti, R.; Cheng, J.; Xia, P.; Di, M. H3K27ac-activated LINC00519 promotes lung squamous cell carcinoma progression by targeting MiR-450-b-5 p/p/YAP1 miR-515-5 axis. Cell Effort 2020, 53, e12797. [Google Scholar] [CrossRef]
- Cao, P.W.; Liu, L.; Li, Z.H.; Cao, F.; Liu, F.B. Prognostic Value of Drug Targets Predicted Using Deep Bioinformatic Analysis of m6A-Associated lncRNA-Based Pancreatic Cancer Model Characteristics and Its Tumour Microenvironment. Front. Genet. 2022, 13, 853471. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Zhao, Q.; Wu, Y.; Wang, G.; Huang, Y.; Sun, W.; Zhang, M. Construction of a cancer-associated fibroblasts-related long non-coding RNA signature to predict prognosis and immune landscape in pancreatic adenocarcinoma. Front. Genet. 2022, 13, 989719. [Google Scholar] [CrossRef]
- Kalantari, S.; Kazemi, B.; Roudi, R.; Zali, H.; D’Angelo, A.; Mohamadkhani, A.; Madjd, Z.; Pourshams, A. RNA-sequencing for transcriptional profiling of whole blood in early stage and metastatic pancreatic cancer patients. Cell Biol. Int. 2023, 47, 238–249. [Google Scholar] [CrossRef]
- Guenther, M.G.; Levine, S.S.; Boyer, L.A.; Jaenisch, R.; Young, R.A. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 2007, 130, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, A.; Bertolotti, A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 697–712. [Google Scholar] [CrossRef]
- Toska, A.; Modi, N.; Chen, L.F. RUNX3 Meets the Ubiquitin-Proteasome System in Cancer. Cells 2023, 12, 717. [Google Scholar] [CrossRef]
- Chen, F.; Zhuang, X.; Lin, L.; Yu, P.; Wang, Y.; Shi, Y.; Hu, G.; Sun, Y. New horizons in tumor microenvironment biology: Challenges and opportunities. BMC Med. 2015, 13, 45. [Google Scholar] [CrossRef] [PubMed]
- De Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Xiao, Y.; Yu, D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol. Ther. 2021, 221, 107753. [Google Scholar] [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef] [PubMed]
- Van der Leun, A.M.; Thommen, D.S.; Schumacher, T.N. CD8+ T cell states in human cancer: Insights from single-cell analysis. Nat. Rev. Cancer 2020, 20, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Addeo, A.; Friedlaender, A.; Banna, G.L.; Weiss, G.J. TMB or not TMB as a biomarker: That is the question. Crit. Rev. Oncol./Hematol. 2021, 163, 103374. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Lim, L.J.; Wong, S.Y.S.; Huang, F.; Lim, S.; Chong, S.S.; Ooi, L.L.; Kon, O.L.; Lee, C.G. Roles and Regulation of Long Noncoding RNAs in Hepatocellular Carcinoma. Cancer Res. 2019, 79, 5131–5139. [Google Scholar] [CrossRef]
- Chen, Y.; Zitello, E.; Guo, R.; Deng, Y. The function of lncRNAs and their role in the prediction, diagnosis, and prognosis of lung cancer. Clin. Transl. Med. 2021, 11, E367. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.Y.; Yuan, X.R.; Tu, C.X.; Shen, J.L.; Li, Y.W.; Yan, A.H.; Ru, Y.; Han, H.Y.; Yang, Y.M.; Liu, Y.; et al. Long Noncoding RNA 00472: A Novel Biomarker in Human Diseases. Front. Pharmacol. 2021, 12, 726908. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Assaraf, Y.G.; Gacche, R.N. Long non-coding RNA mediated drug resistance in breast cancer. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2022, 63, 100851. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sun, M.; Lu, K.; Liu, J.; Zhang, M.; Wu, W.; De, W.; Wang, Z.; Wang, R. The long noncoding RNA HOTAIR contributes to cisplatin resistance of human lung adenocarcinoma cells via Downregualtion of p21 [WAF1/CIP1) expression. PLoS ONE 2013, 8, e77293. [Google Scholar] [CrossRef]
- Luo, Y.; Zheng, S.; Wu, Q.; Wu, J.; Zhou, R.; Wang, C.; Wu, Z.; Rong, X.; Huang, N.; Sun, L.; et al. Long noncoding RNA (lncRNA) EIF3J-DT induces chemoresistance of gastric cancer via autophagy Activation. Autophagy 2021, 17, 4083–4101. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, Z.; Dai, T.; Yuan, F.; Li, K.; Wang, G. To Predict the Prognosis and Immunological Characteristics of Pancreatic Cancer Based on Disulfide-Death Gene Death-Related lncRNA. Biomedicines 2025, 13, 924. https://doi.org/10.3390/biomedicines13040924
Liao Z, Dai T, Yuan F, Li K, Wang G. To Predict the Prognosis and Immunological Characteristics of Pancreatic Cancer Based on Disulfide-Death Gene Death-Related lncRNA. Biomedicines. 2025; 13(4):924. https://doi.org/10.3390/biomedicines13040924
Chicago/Turabian StyleLiao, Zhihong, Tianxing Dai, Feng Yuan, Kai Li, and Guoying Wang. 2025. "To Predict the Prognosis and Immunological Characteristics of Pancreatic Cancer Based on Disulfide-Death Gene Death-Related lncRNA" Biomedicines 13, no. 4: 924. https://doi.org/10.3390/biomedicines13040924
APA StyleLiao, Z., Dai, T., Yuan, F., Li, K., & Wang, G. (2025). To Predict the Prognosis and Immunological Characteristics of Pancreatic Cancer Based on Disulfide-Death Gene Death-Related lncRNA. Biomedicines, 13(4), 924. https://doi.org/10.3390/biomedicines13040924