

Identification of KIFC3 as a Colorectal Cancer Biomarker and Its Regulatory Mechanism in the Immune Microenvironment Based on Integrated Analysis of Multi-Omics Databases

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection
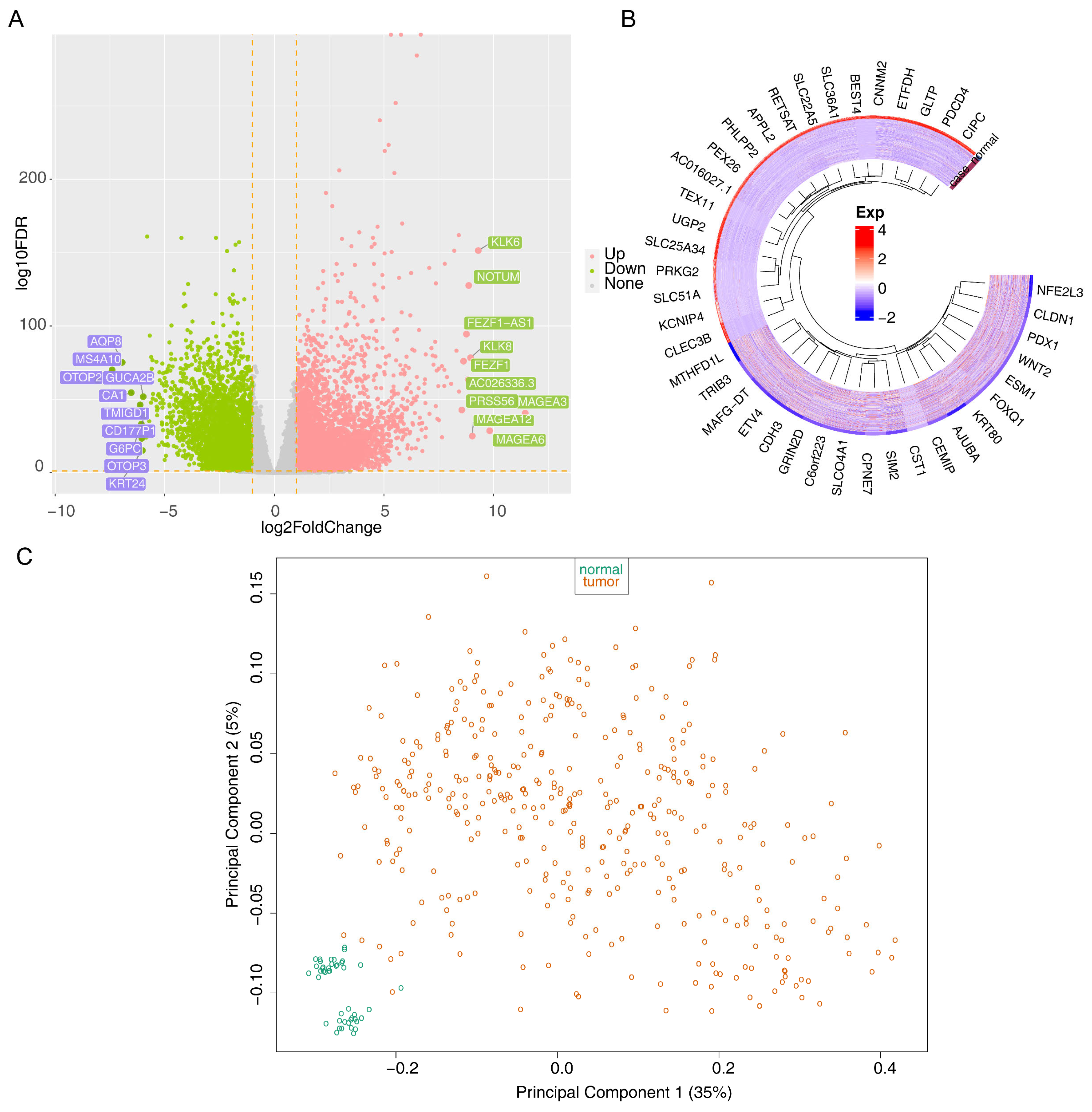
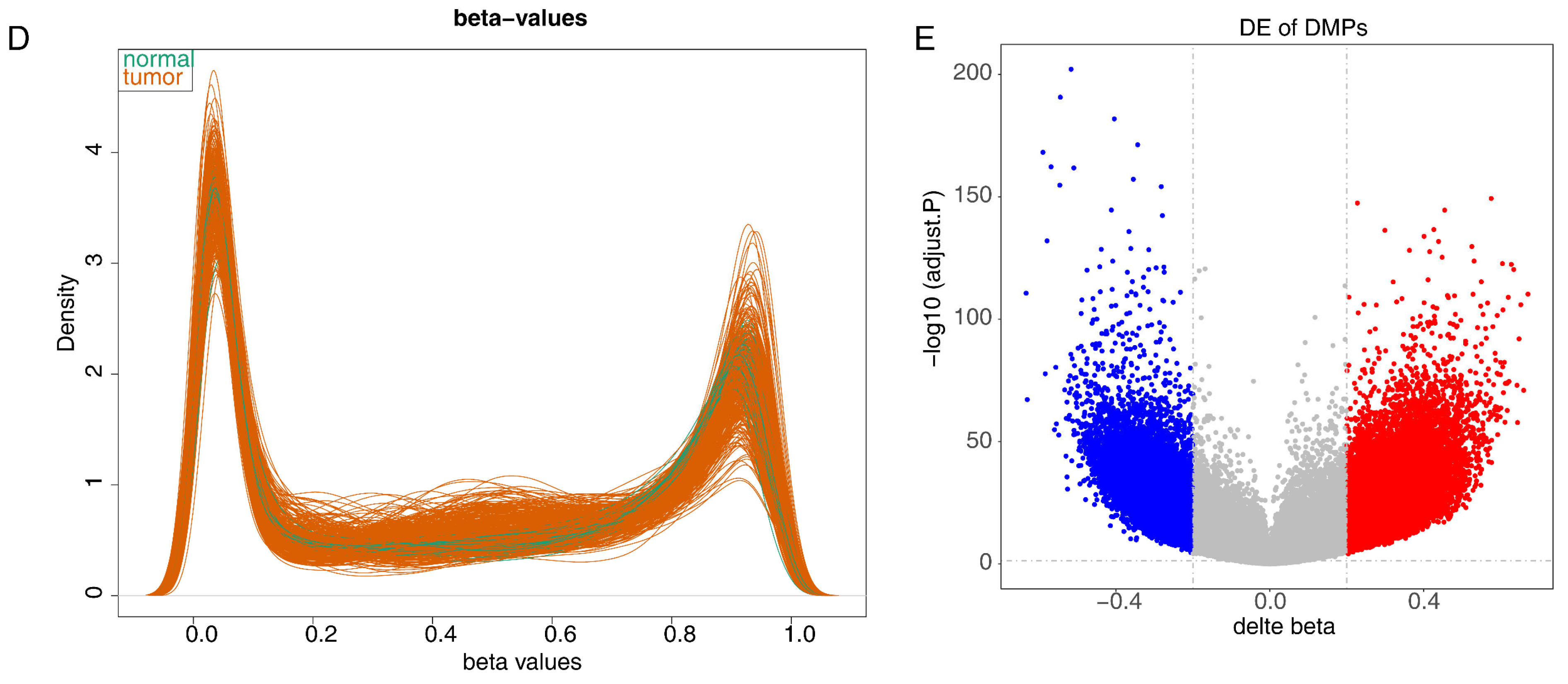
2.2. Differential Expression Analysis of the TCGA-CRC Dataset
2.3. Functional Analysis of the Intersecting Genes in the TCGA-CRC Dataset
2.4. Survival Analysis of Intersecting Genes in the TCGA-CRC Dataset
2.5. Identification of Biomarkers in Multiple Datasets
2.6. Gene Set Enrichment Analysis (GSEA) in the TCGA-CRC Dataset
2.7. Immune Infiltration and Immune Checkpoint Analysis in the TCGA-CRC Dataset
2.8. Regulation Network Analysis
2.9. Drug Sensitivity Analysis in the TCGA-CRC Dataset
2.10. Single-Cell RNA Sequencing (scRNA-Seq) Analysis
2.11. Differential Expression Analysis, GSEA, and Biomarkers Expression Analysis in Annotated Cells
2.12. Pseudo-Temporal Analysis and Single-Cell Regulatory Network Inference and Clustering (SCENIC)
2.13. Experimental Validation Analysis of Target Genes
2.14. Statistical Analysis
3. Results
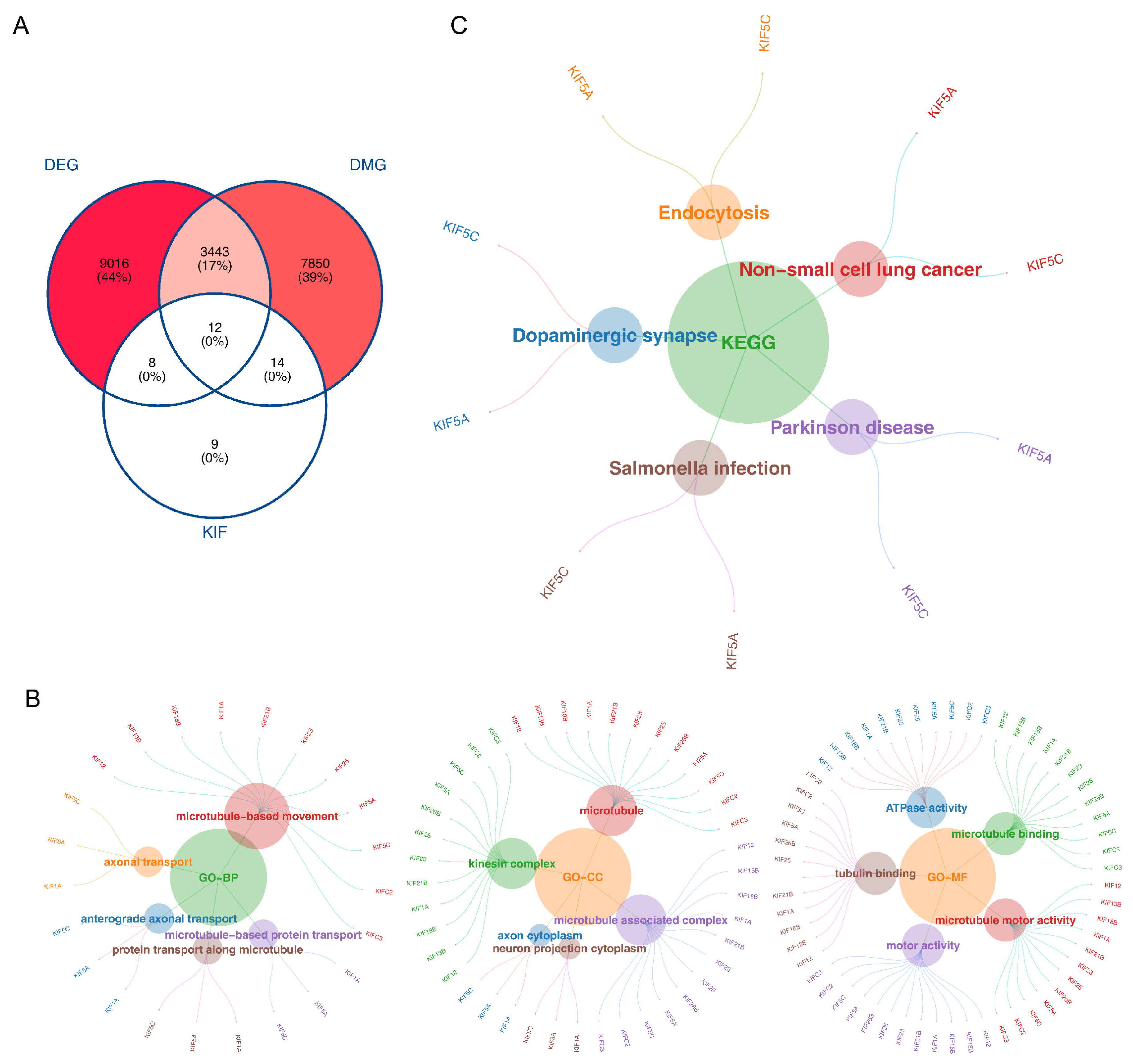
3.1. The 12 Intersecting Genes Were Mainly Enriched in Microtubule Related Function
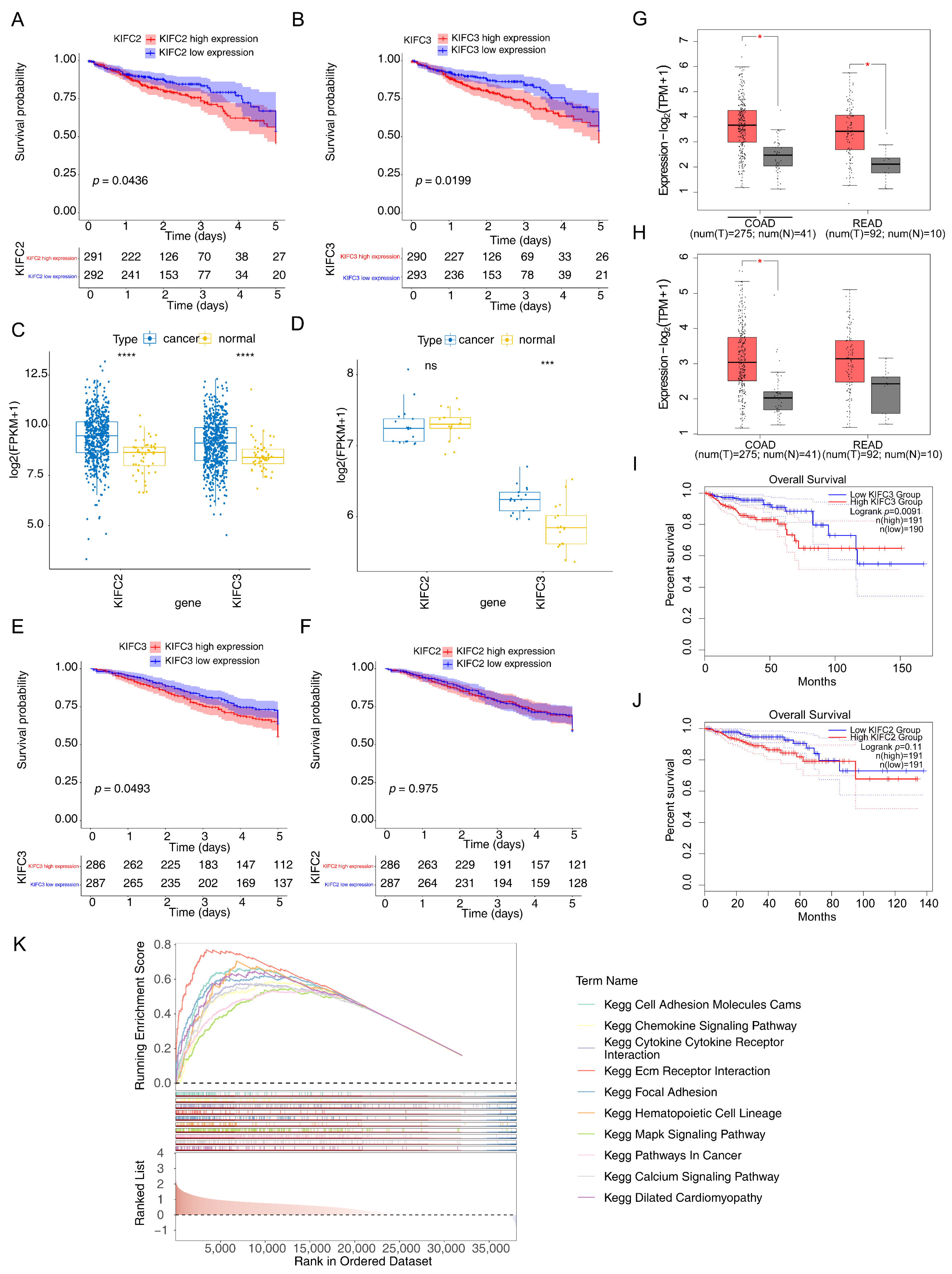
3.2. KIFC3 Were Identified as Biomarkers
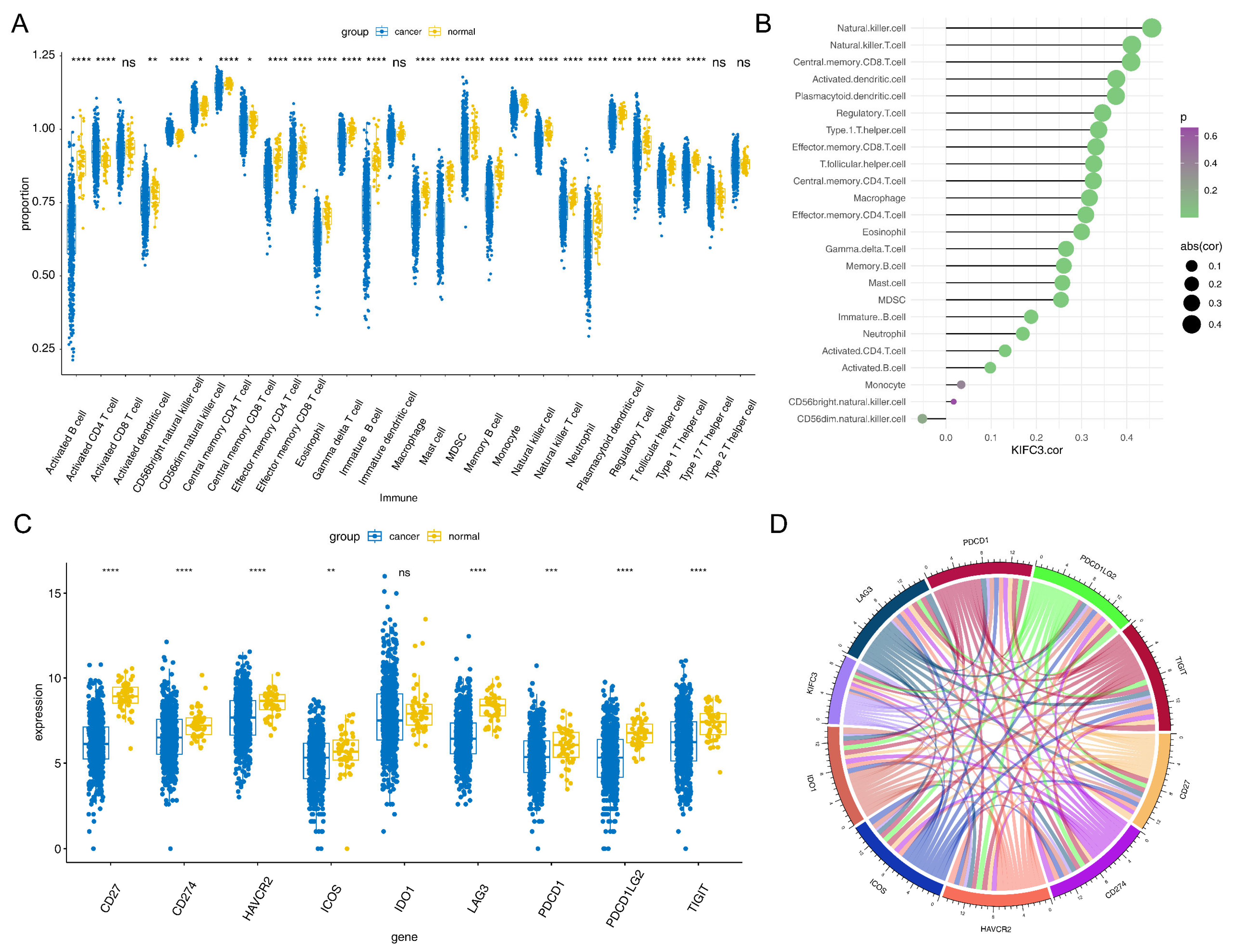
3.3. KIFC3 Had a Positive Correlation with Differential Immune Cells and Common Checkpoints
3.4. Regulatory Network of KIFC3 in CRC
3.5. AZD.2281, Nilotinib, PD.173074, and Shikonin Were Helpful for Treating CRC
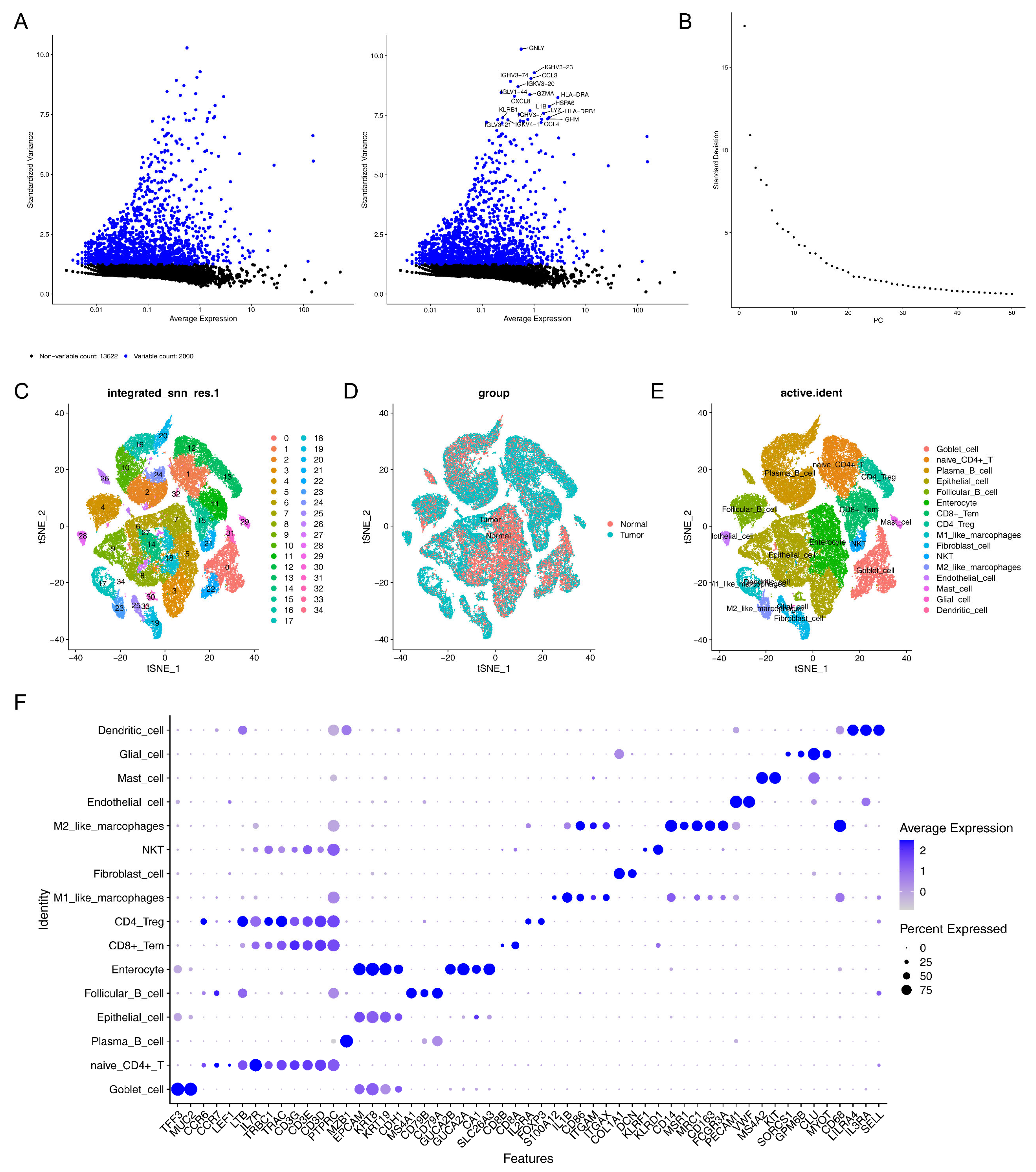
3.6. A Total of 16 Cell Clusters Were Annotated
3.7. GSEA Revealed 16 Cell Clusters Involved in Pathways, Including Antigen Processing and Presentation, Among Others
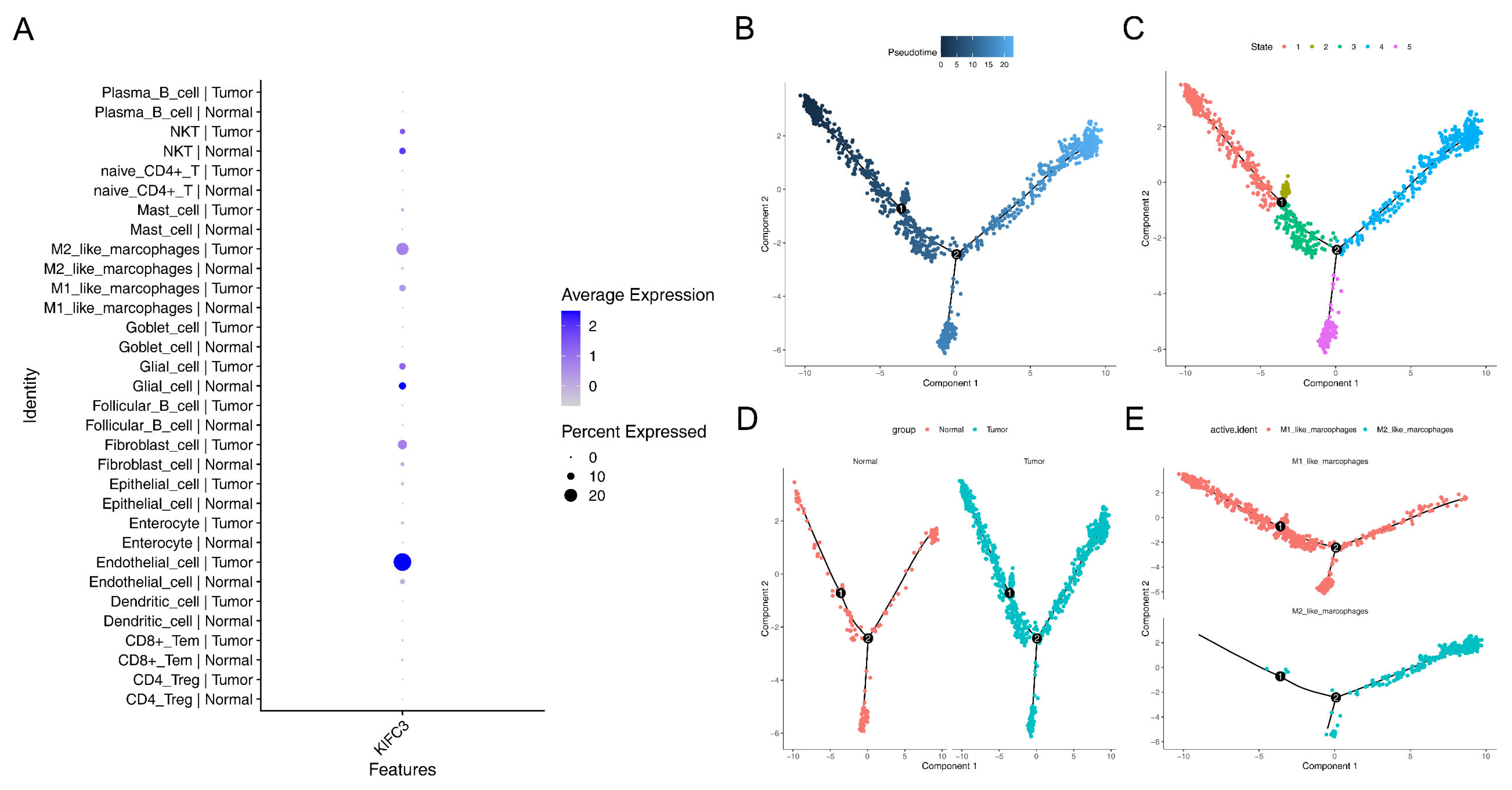
3.8. Macrophages Play a Critical Role in the Development of CRC
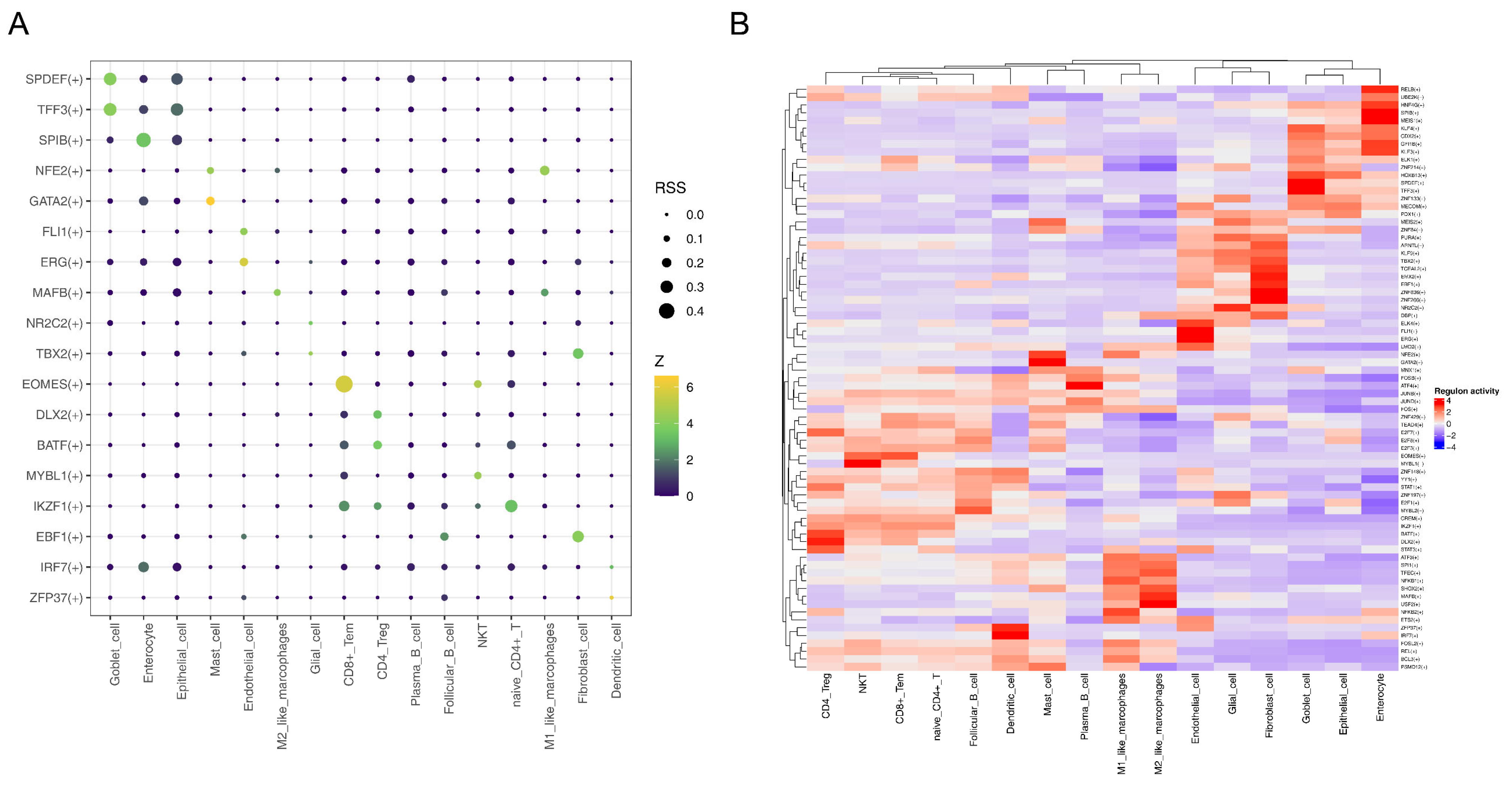
3.9. MAFB Was Key TF for M2-like Macrophages
3.10. Expression of KIFC3in CRC Tissues and Normal Tissues in qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef] [PubMed]
- Vacante, M.; Ciuni, R.; Basile, F.; Biondi, A. The Liquid Biopsy in the Management of Colorectal Cancer: An Overview. Biomedicines 2020, 8, 308. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.K.; Kim, H.C.; Lee, W.Y.; Yun, S.H.; Cho, Y.B.; Huh, J.W.; Park, Y.A.; Chun, H.K. High preoperative serum CA 19-9 levels can predict poor oncologic outcomes in colorectal cancer patients on propensity score analysis. Ann. Surg. Treat. Res. 2019, 96, 107–115. [Google Scholar] [CrossRef]
- Hirokawa, N.; Noda, Y.; Tanaka, Y.; Niwa, S. Kinesin superfamily motor proteins and intracellular transport. Nature reviews. Mol. Cell Biol. 2009, 10, 682–696. [Google Scholar] [CrossRef]
- Novikov, N.M.; Zolotaryova, S.Y.; Gautreau, A.M.; Denisov, E.V. Mutational drivers of cancer cell migration and invasion. Br. J. Cancer 2021, 124, 102–114. [Google Scholar] [CrossRef]
- Wang, J.; Cui, F.; Wang, X.; Xue, Y.; Chen, J.; Yu, Y.; Lu, H.; Zhang, M.; Tang, H.; Peng, Z. Elevated kinesin family member 26B is a prognostic biomarker and a potential therapeutic target for colorectal cancer. J. Exp. Clin. Cancer Res. 2015, 34, 13. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, L.; Xiong, L.; Wang, K.; Hou, X.; Li, Q.; Kong, F.; Liu, X.; He, J. KIF11 is upregulated in colorectal cancer and silencing of it impairs tumor growth and sensitizes colorectal cancer cells to oxaliplatin via p53/GSK3β signaling. J. Cancer 2021, 12, 3741–3753. [Google Scholar] [CrossRef]
- Akabane, S.; Oue, N.; Sekino, Y.; Asai, R.; Thang, P.Q.; Taniyama, D.; Sentani, K.; Yukawa, M.; Toda, T.; Kimura, K.I.; et al. KIFC1 regulates ZWINT to promote tumor progression and spheroid formation in colorectal cancer. Pathol. Int. 2021, 71, 441–452. [Google Scholar] [CrossRef]
- Huo, D.; Yang, H.; Huang, J.D.; Cai, J.P.; Cui, J. Roles of kinesin superfamily proteins in colorectal cancer carcinogenesis (Review). Oncol. Rep. 2021, 46, 121. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, M.; Zhang, B.; Zhao, W. KIF18A improves migration and invasion of colorectal cancer (CRC) cells through inhibiting PTEN signaling. Aging 2023, 15, 9182–9192. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Saito, M.; Saito, K.; Kanke, Y.; Watanabe, Y.; Onozawa, H.; Hayase, S.; Sakamoto, W.; Ishigame, T.; Momma, T.; et al. Enhanced expression of KIF4A in colorectal cancer is associated with lymph node metastasis. Oncol. Lett. 2018, 15, 2188–2194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhu, G.; Wang, X.; Liao, X.; Huang, R.; Huang, C.; Huang, P.; Zhang, J.; Wang, P. Genome-wide investigation of the clinical significance and prospective molecular mechanisms of kinesin family member genes in patients with lung adenocarcinoma. Oncol. Rep. 2019, 42, 1017–1034. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Gustavsson, E.K.; Zhang, D.; Reynolds, R.H.; Garcia-Ruiz, S.; Ryten, M. ggtranscript: An R package for the visualization and interpretation of transcript isoforms using ggplot2. Bioinformatics 2022, 38, 3844–3846. [Google Scholar] [CrossRef]
- Zheng, X.; Ma, Y.; Bai, Y.; Huang, T.; Lv, X.; Deng, J.; Wang, Z.; Lian, W.; Tong, Y.; Zhang, X.; et al. Identification and validation of immunotherapy for four novel clusters of colorectal cancer based on the tumor microenvironment. Front. Immunol. 2022, 13, 984480. [Google Scholar] [CrossRef]
- Wei, T.; Nie, J.; Larson, N.B.; Ye, Z.; Eckel-Passow, J.E.; Robertson, K.D.; Kocher, J.A.; Wang, L. CpGtools: A python package for DNA methylation analysis. Bioinformatics 2021, 37, 1598–1599. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Wang, L.; Wang, D.; Yang, L.; Zeng, X.; Zhang, Q.; Liu, G.; Pan, Y. Cuproptosis related genes associated with Jab1 shapes tumor microenvironment and pharmacological profile in nasopharyngeal carcinoma. Front. Immunol. 2022, 13, 989286. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Germain, P.L.; Lun, A.; Garcia Meixide, C.; Macnair, W.; Robinson, M.D. Doublet identification in single-cell sequencing data using scDblFinder. F1000Research 2021, 10, 979. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, B.; Zhao, Y. Inflammation in Preeclampsia: Genetic Biomarkers, Mechanisms, and Therapeutic Strategies. Front. Immunol. 2022, 13, 883404. [Google Scholar] [CrossRef]
- Nachbar, J.; Lázaro-Diéguez, F.; Prekeris, R.; Cohen, D.; Müsch, A. KIFC3 promotes mitotic progression and integrity of the central spindle in cytokinesis. Cell Cycle 2014, 13, 426–433. [Google Scholar] [CrossRef]
- Lu, S.; Liu, Y.; Tian, S.; He, Y.; Dong, W. KIFC3 regulates progression of hepatocellular carcinoma via EMT and the AKT/mTOR pathway. Exp. Cell Res. 2023, 426, 113564. [Google Scholar] [CrossRef]
- He, Y.; He, P.; Lu, S.; Dong, W. KIFC3 Regulates the progression and metastasis of gastric cancer via Notch1 pathway. Dig. Liver Dis. 2023, 55, 1270–1279. [Google Scholar] [CrossRef]
- Liao, H.; Zhang, L.; Lu, S.; Li, W.; Dong, W. KIFC3 Promotes Proliferation, Migration, and Invasion in Colorectal Cancer via PI3K/AKT/mTOR Signaling Pathway. Front. Genet. 2022, 13, 848926. [Google Scholar] [CrossRef]
- Machackova, T.; Vychytilova-Faltejskova, P.; Souckova, K.; Trachtova, K.; Brchnelova, D.; Svoboda, M.; Kiss, I.; Prochazka, V.; Kala, Z.; Slaby, O. MiR-215-5p Reduces Liver Metastasis in an Experimental Model of Colorectal Cancer through Regulation of ECM-Receptor Interactions and Focal Adhesion. Cancers 2020, 12, 3518. [Google Scholar] [CrossRef]
- Mellado, M.; Rodríguez-Frade, J.M.; Mañes, S.; Martínez, A.C. Chemokine signaling and functional responses: The role of receptor dimerization and TK pathway activation. Annu. Rev. Immunol. 2001, 19, 397–421. [Google Scholar] [CrossRef]
- Ma, T.; Peng, C.; Wu, D.; Yang, S.; Ji, L.; Cheng, Z.; Gao, C. Immune-based prognostic biomarkers associated with metastasis of osteosarcoma. Gen. Physiol. Biophys. 2023, 42, 1–12. [Google Scholar] [CrossRef]
- Krijgsman, D.; de Vries, N.L.; Skovbo, A.; Andersen, M.N.; Swets, M.; Bastiaannet, E.; Vahrmeijer, A.L.; van de Velde, C.J.H.; Heemskerk, M.H.M.; Hokland, M.; et al. Characterization of circulating T-, NK-, and NKT cell subsets in patients with colorectal cancer: The peripheral blood immune cell profile. Cancer Immunol. Immunother. 2019, 68, 1011–1024. [Google Scholar] [CrossRef] [PubMed]
- Fionda, C.; Scarno, G.; Stabile, H.; Molfetta, R.; Di Censo, C.; Gismondi, A.; Paolini, R.; Sozzani, S.; Santoni, A.; Sciumè, G. NK Cells and Other Cytotoxic Innate Lymphocytes in Colorectal Cancer Progression and Metastasis. Int. J. Mol. Sci. 2022, 23, 7859. [Google Scholar] [CrossRef]
- Prizment, A.E.; Anderson, K.E.; Visvanathan, K.; Folsom, A.R. Inverse association of eosinophil count with colorectal cancer incidence: Atherosclerosis risk in communities study. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1861–1864. [Google Scholar] [CrossRef]
- Bochum, S.; Berger, S.; Martens, U.M. Olaparib. In Small Molecules in Oncology; Recent Results in Cancer Research (RECENTCANCER); Springer: Berlin/Heidelberg, Germany, 2018; Volume 211, pp. 217–233. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Gresse, M.; Kim, T.D.; le Coutre, P. Nilotinib. In Small Molecules in Hematology; Recent Results in Cancer Research (RECENTCANCER); Springer: Berlin/Heidelberg, Germany, 2018; Volume 212, pp. 69–85. [Google Scholar] [CrossRef]
- Jeitany, M.; Leroy, C.; Tosti, P.; Lafitte, M.; Le Guet, J.; Simon, V.; Bonenfant, D.; Robert, B.; Grillet, F.; Mollevi, C.; et al. Inhibition of DDR1-BCR signalling by nilotinib as a new therapeutic strategy for metastatic colorectal cancer. EMBO Mol. Med. 2018, 10, e7918. [Google Scholar] [CrossRef]
- Salih, J.; Hilpert, J.; Placke, T.; Grünebach, F.; Steinle, A.; Salih, H.R.; Krusch, M. The BCR/ABL-inhibitors imatinib, nilotinib and dasatinib differentially affect NK cell reactivity. Int. J. Cancer 2010, 127, 2119–2128. [Google Scholar] [CrossRef]
- Chen, J.; Schmitt, A.; Chen, B.; Rojewski, M.; Rübeler, V.; Fei, F.; Yu, Y.; Yu, X.; Ringhoffer, M.; von Harsdorf, S.; et al. Nilotinib hampers the proliferation and function of CD8+ T lymphocytes through inhibition of T cell receptor signalling. J. Cell. Mol. Med. 2008, 12, 2107–2118. [Google Scholar] [CrossRef]
- Pei, J.; Gao, Y.; Wu, A. An inflammation-related subtype classification for analyzing tumor microenvironment and clinical prognosis in colorectal cancer. Front. Immunol. 2024, 15, 1369726. [Google Scholar] [CrossRef]
- Massimino, M.; Martorana, F.; Stella, S.; Vitale, S.R.; Tomarchio, C.; Manzella, L.; Vigneri, P. Single-Cell Analysis in the Omics Era: Technologies and Applications in Cancer. Genes 2023, 14, 1330. [Google Scholar] [CrossRef]
- Lin, B.; Yuejiao, X.; Dingyu, D.; Yi, X. Advances in macrophage function and its anti-inflammatory and proresolving activity and role in periodontitis development. West China J. Stomatol. 2017, 35, 427–432. [Google Scholar] [CrossRef]
- Ge, W.; Wu, W. Influencing Factors and Significance of Tumor-associated Macrophage Polarization in Tumor Microenvironment. Chin. J. Lung Cancer 2023, 26, 228–237. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | p-Value |
|---|---|
| KIF12 | 0.796061026 |
| KIF13B | 0.277954542 |
| KIF18B | 0.505774554 |
| KIF1A | 0.842258936 |
| KIF21B | 0.778932753 |
| KIF23 | 0.85609048 |
| KIF25 | 0.805087841 |
| KIF26B | 0.112269083 |
| KIF5A | 0.377814398 |
| KIF5C | 0.340239373 |
| KIFC2 | 0.043597233 |
| KIFC3 | 0.019886808 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Zeng, X.; Wen, J.; Xian, K.; Jin, F.; Jiang, S.; Sun, L. Identification of KIFC3 as a Colorectal Cancer Biomarker and Its Regulatory Mechanism in the Immune Microenvironment Based on Integrated Analysis of Multi-Omics Databases. Biomedicines 2025, 13, 859. https://doi.org/10.3390/biomedicines13040859
Wang F, Zeng X, Wen J, Xian K, Jin F, Jiang S, Sun L. Identification of KIFC3 as a Colorectal Cancer Biomarker and Its Regulatory Mechanism in the Immune Microenvironment Based on Integrated Analysis of Multi-Omics Databases. Biomedicines. 2025; 13(4):859. https://doi.org/10.3390/biomedicines13040859
Chicago/Turabian StyleWang, Fen, Xinxin Zeng, Jielun Wen, Kexin Xian, Feng Jin, Sunfang Jiang, and Liyue Sun. 2025. "Identification of KIFC3 as a Colorectal Cancer Biomarker and Its Regulatory Mechanism in the Immune Microenvironment Based on Integrated Analysis of Multi-Omics Databases" Biomedicines 13, no. 4: 859. https://doi.org/10.3390/biomedicines13040859
APA StyleWang, F., Zeng, X., Wen, J., Xian, K., Jin, F., Jiang, S., & Sun, L. (2025). Identification of KIFC3 as a Colorectal Cancer Biomarker and Its Regulatory Mechanism in the Immune Microenvironment Based on Integrated Analysis of Multi-Omics Databases. Biomedicines, 13(4), 859. https://doi.org/10.3390/biomedicines13040859