1. Introduction
The increasing prevalence of maternal obesity is emerging as a significant public health concern due to its potential impact on offspring health. Recent studies have demonstrated that maternal obesity and excess weight are associated with substantial alterations in infant intestinal microbiome composition, particularly among vaginally delivered children from families with higher socioeconomic status [
1,
2]. Basak et al. (2022), in their review, demonstrated that maternal obesity and gut microbiota are closely associated with neurodevelopment, whereby alterations in the a mother’s microbiota composition influence the development and function of the gastrointestinal tract of her offspring [
3].
The human gut microbiome plays a crucial role in maintaining metabolic health. During pregnancy, maternal obesity alters the structure and composition of the gut microbiota, characterized by an increase in the relative abundance of
Firmicutes and depletion of
Proteobacteria [
4]. These microbial changes may be transmitted to the infant, eventually influencing colonization and increasing the risk of metabolic disorders. Multiple factors may influence this process, including maternal pre-pregnancy body mass index, gestational weight gain, mode of delivery, and breastfeeding practices [
5,
6].
Furthermore, according to Guzzardi et al. (2022), maternal pre-pregnancy overweight and neonatal gut bacterial colonization are significantly associated with cognitive development in pre-school-age offspring. Their longitudinal study revealed that maternal overweight leads to the depletion of beneficial bacteria such as
Bifidobacterium,
Blautia, and
Ruminococcus, and correlates with reduced practical reasoning scores in offspring at 36 months of age. These findings indicate the influence of maternal metabolic status on both the gut microbiota composition and cognitive function in early childhood [
7].
Research over the past decade has significantly expanded our understanding of microbial colonization processes in the infant gut. There are suggestions that microbial colonization of the infant intestine begins in utero through amniotic fluid [
8]. This shifts the traditional paradigm of a microbe-free intrauterine environment and emphasizes the importance of maternal microbiota at the prenatal stage. Furthermore, children born to mothers with obesity exhibit significant differences in gut microbiome composition, including changes in specific bacterial taxa such as
Faecalibacterium, Eubacterium, and
Blautia. These microbial modifications may influence the development of the infant’s immune system, metabolism, and appetite regulation [
9]. As noted by Gohir et al. (2015), the gut microbiome of obese mothers is associated with inflammatory processes and increased risk of obesity in offspring [
10].
In recent years, interest in the relationship between maternal obesity and the infant microbiome has increased significantly. Buffington et al. (2016) demonstrated that maternal obesity induced by a high-fat diet impacts the microbiota structure and may contribute to behavioral disorders associated with alterations in oxytocin transmission in the ventral tegmental area [
11]. The authors emphasize the potential significance of the microbiome in the development of neurological and behavioral aspects related to maternal obesity. Meanwhile, Denizli et al. (2022) showed in their review that maternal obesity may program long-term changes in inflammatory and immune responses in offspring. These changes are potentially associated with an increased risk of developing chronic diseases, including metabolic syndrome [
12].
Chu et al. (2016) discovered that the composition and structure of the neonatal gut microbiome varied significantly depending on maternal pre-pregnancy weight, with infants born to overweight mothers exhibiting reduced
Bacteroides diversity and increased levels of pro-inflammatory taxa [
13]. These differences in early-life microbial communities may have long-term implications for metabolic programming and disease susceptibility.
Recent investigations have unveiled potential mechanisms underlying the influence of maternal obesity on infant microbiome. Soderborg and colleagues (2018), conducting microbiota transplantation from the offspring of obese mothers, found that maternal obesity is associated with alterations in the gut microbiota metabolic pathways in offspring, potentially contributing to increased intestinal permeability and metabolic dysfunction [
14], suggesting a causal role of the altered microbiome in programming metabolic health. Ferretti et al. (2018) identified, through studying the longitudinal dynamics of the infant microbiome during the first year of life, that BMI and other maternal factors have a prolonged influence on infant microbiome development, which is particularly noticeable in the first months of life [
15].
In the context of metabolic programming, Hasan et al. (2018) identified specific bacterial taxa that were differentially represented in the offspring of mothers with gestational diabetes 5 years after birth. Specifically, elevated levels of the genus
Anaerotruncus, associated with glucose intolerance, indicate a potential link between maternal metabolic disturbances and long-term changes in the offspring microbiome [
16]. Furthermore, maternal obesity may significantly affect lipid biosynthesis pathways in infants, with studies indicating changes in gene expression related to mitochondrial and lipid metabolism in the umbilical vein endothelial cells of infants [
17]. These changes, in conjunction with restructuring in the intestinal microbiome, suggest a complex interaction between maternal metabolic status and infant development, potentially contributing to the intergenerational transmission of obesity risk [
18].
Given the growing body of information indicating the importance of early gut colonization for future child health and development, there is an urgent need for a deeper understanding of the impact of maternal obesity on infant microbiome formation. The aim of this research is to investigate age-specific alterations in infant gut microbiota composition associated with maternal obesity, with a particular focus on the dynamics of taxonomic composition, metabolic pathways, and antibiotic resistance genes during early postnatal development.
3. Results
The recruitment of volunteers for the study and control groups was conducted at Perinatal Center No. 2 in Astana from February 2023 to February 2024. The samples for shotgun sequencing and metagenomic analysis were collected at the following time points: 3 DB—first 72 h of life, 1 MB—after 30 days of life, 3 MB—after 3 months of life, and 6 MB—after 6 months.
The study cohort comprised a total of 48 participants divided into two groups: 12 mothers with class 2 and 3 obesity (BMI 37.9 ± 1.58 kg/m2 [95.0% CI: 36.05–40.0]) and their 12 infants. The control group consisted of 12 women with normal weight (BMI 27.7 ± 2.3 kg/m2 [95.0% CI: 24.3–31.9]) and their infants. The mean age of the participants was 33.3 ± 5.33 years (range 19–42 years), with no statistically significant intergroup differences (p = 0.43). The median parity was comparable between the groups: 3.0 ± 1.3 and 3.0 ± 1.15, respectively (p = 0.52). Analysis of the obstetric parameters revealed that gestational age at delivery in the obese mothers group was 278.8 ± 2.6 days, compared to 282.0 ± 3.4 days in the control group (p = 0.12). The proportions of vaginal deliveries were 33.0% (4/12) and 83.0% (10/12), respectively (p = 0.34). The assessment of the neonatal anthropometric parameters showed that the mean birth weight was 3843.0 ± 554.2 g in the maternal obesity group and 4167.5 ± 282.4 g in the control group (p = 0.24). Neonatal body length was 55.8 ± 1.5 cm and 57.0 ± 1.79 cm, respectively (p = 0.26).
During the follow-up period, by six months of age, comparable weight gain was observed in both groups (
Table 1), with a similar trend noted for body length.
Gender distribution was characterized by a predominance of males in both groups: the female/male ratio was 3/9 in the study group and 5/7 in the control group (p = 0.67). At each analysis stage, some samples were excluded due to quality control failure or insufficient read counts, resulting in varying final sample numbers across analytical phases.
At each stage of the analysis, some samples were excluded due to non-compliance with quality control requirements or insufficient number of reads, resulting in different results in the final quantities.
For the analysis of gut microbiota at each temporal stage, we applied shotgun sequencing to a subset of fecal samples (N = 77, 64.1% of the initial cohort). The shotgun sequencing analysis yielded a total of 6,670,000,000 paired reads, with a mean of 35,000,000 sequences per sample.
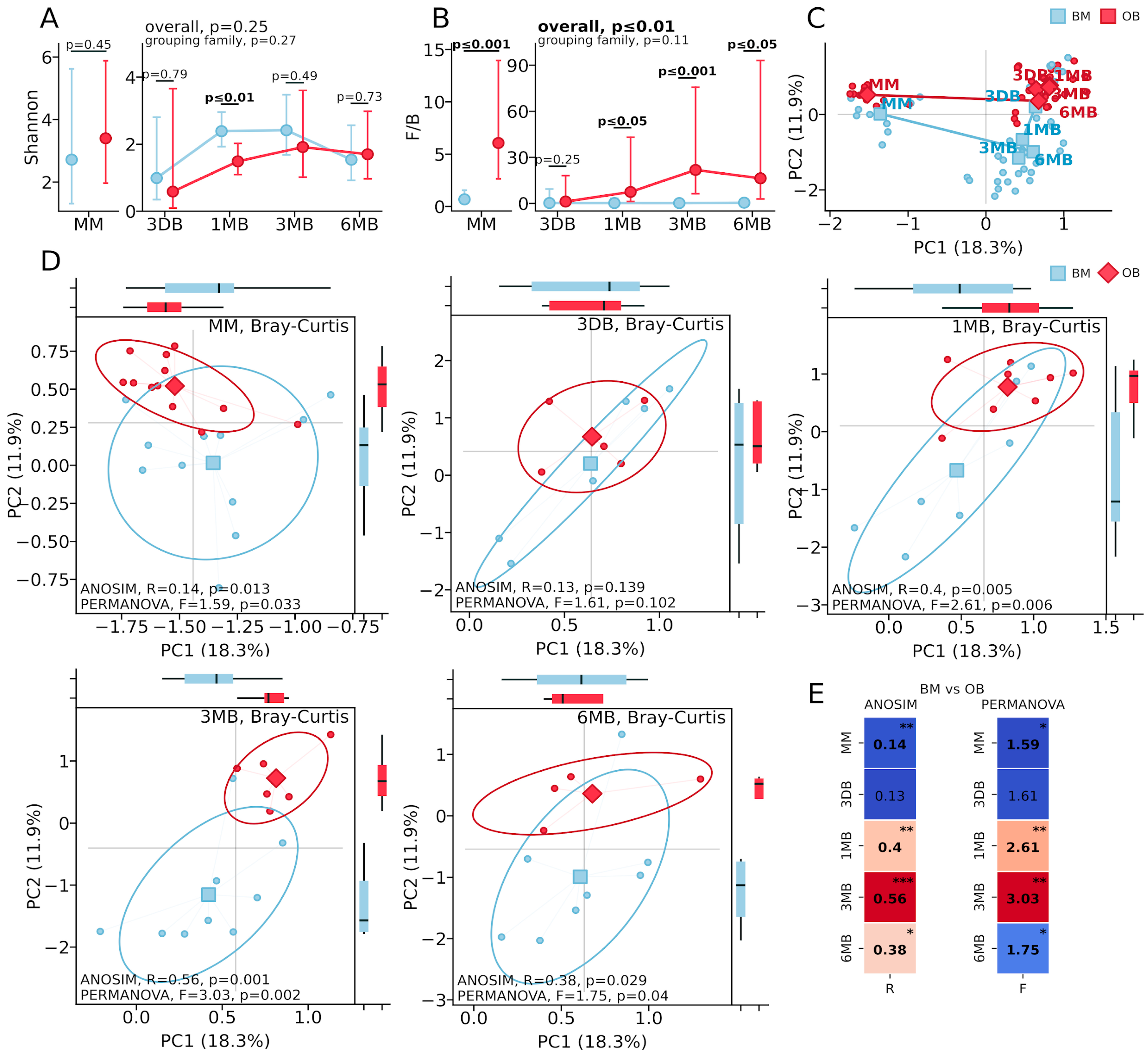
The microbiota diversity analysis revealed distinct patterns between the OB and BM groups. Alpha diversity, assessed using the Shannon index (
Figure 1A), showed a significant reduction in the OB group only at one month of age (
p ≤ 0.01), while differences at other time points were not statistically significant (MM:
p = 0.45; 3 DB:
p = 0.79; 3 MB:
p = 0.49; 6 MB:
p = 0.73). The Firmicutes/Bacteroidetes ratio (
Figure 1B) was significantly elevated in the OB group, beginning with maternal samples (
p ≤ 0.001), reaching maximum differences at 3 months (
p ≤ 0.0001), and maintaining differences at 6 months (
p ≤ 0.01). Principal Coordinates Analysis (PCoA) based on the Bray–Curtis distance (
Figure 1C), demonstrated the sequential formation of microbial communities from the maternal samples through the meconium to the mature infant microbiota, with a clear separation of samples both across time points and between the OB and BM groups.
Analysis of the microbiota beta diversity based on the Bray–Curtis distance revealed dynamic differences in the gut microbiota structure among infants born to mothers with obesity (
Figure 1). Significant differences between the groups were detected in the maternal gut microbiota samples (
Figure 1D, MM: ANOSIM: R = 0.14,
p = 0.013; PERMANOVA: F = 1.59,
p = 0.033). No significant differences in the microbial community structure were observed in the first-pass meconium samples (
Figure 1D, 3 DB: ANOSIM: R = 0.13,
p = 0.139; PERMANOVA: F = 1.61,
p = 0.102). By one month of age (
Figure 1D, 1 MB), substantial differences in the microbiota composition between the groups had developed (ANOSIM: R = 0.4,
p = 0.005; PERMANOVA: F = 2.61,
p = 0.006), reaching maximum divergence at three months of age (
Figure 1D, 3 MB: ANOSIM: R = 0.56,
p = 0.001; PERMANOVA: F = 3.03,
p = 0.002). By six months (
Figure 1D, 6 MB), differences between the groups persisted but were less pronounced, though remaining statistically significant (ANOSIM: R = 0.38,
p = 0.029; PERMANOVA: F = 1.75,
p = 0.04).
Principal Coordinates Analysis (PCoA) based on the Bray–Curtis distance (
Figure 1C) and beta diversity visualization at individual time points (
Figure 1D) demonstrated clear a separation of the microbial communities between the OB and BM groups, which was particularly pronounced at 1 and 3 months of age. The first two principal components explain 18.3% and 11.9% of the total variance, respectively. The statistical metrics heat map (
Figure 1E) further corroborates the pronounced differences between groups at the 1 MB and 3 MB time points, with a slight attenuation at 6 MB while maintaining statistical significance.
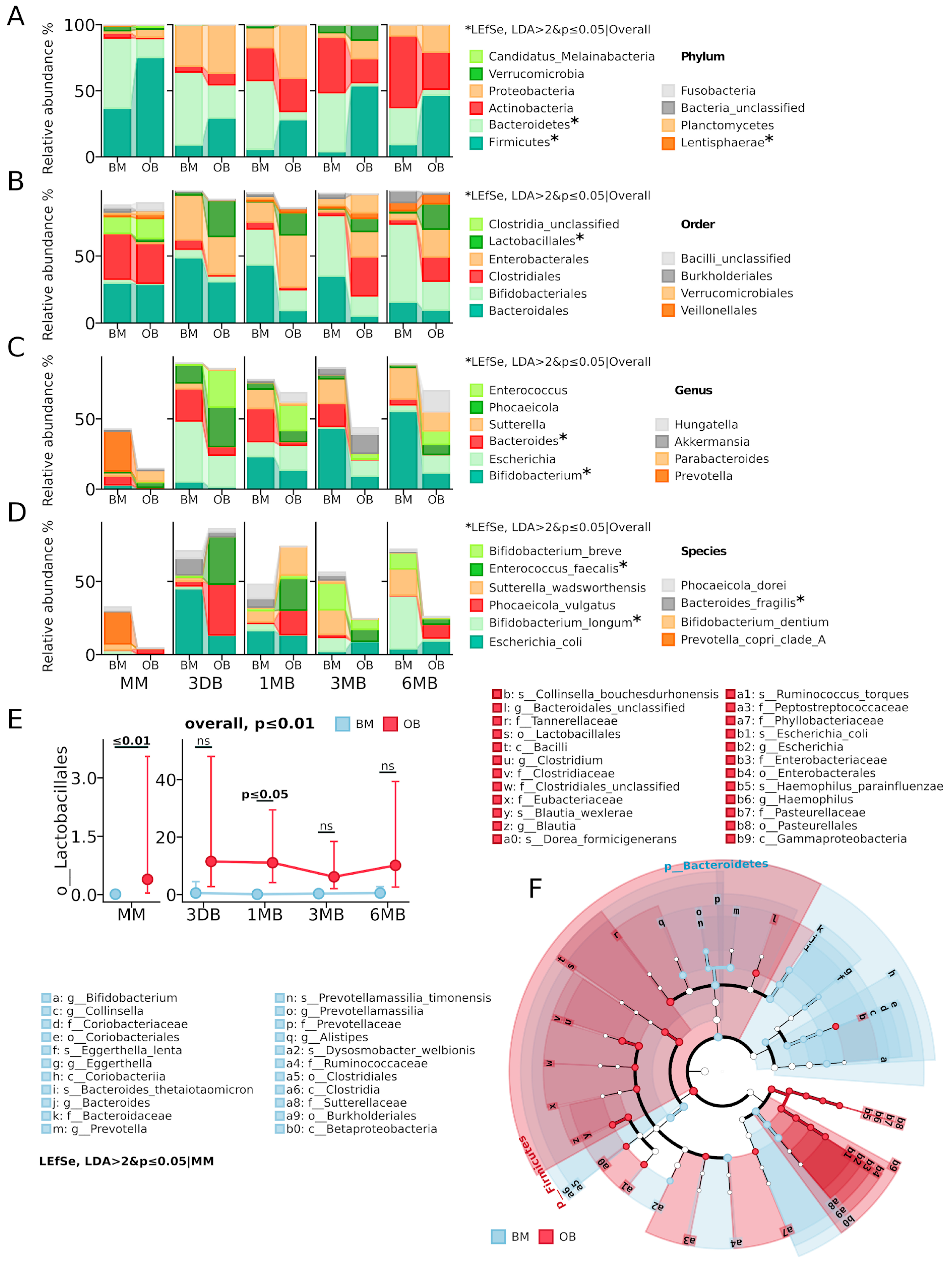
Taxonomic analysis revealed substantial differences in the microbiota composition between the OB and BM groups (
Figure 2). At the phylum level (
Figure 2A), the maternal microbiota showed significant differences in the abundance of
Firmicutes (current name
Bacillota) on average (OB: 75.2% vs. BM: 36.9%) and
Bacteroidetes (current name
Bacteroidota) (OB: 14.3% vs. BM: 53.1%). The OB group also showed elevated levels of
Proteobacteria (current name
Pseudomonadota) (6.0% vs. 1.8% in BM) and
Candidatus_Melainabacteria (1.7% vs. 0.3% in BM). In the meconium samples (3 DB,
Figure 2D), Proteobacteria dominated in both groups (OB: 36.5% vs. BM: 30.9%), primarily due to
E. coli, with the OB group characterized by increased
Firmicutes (29.5% vs. 9.1% in BM) and decreased
Bacteroidetes (25.0% vs. 55.0% in BM). By one month of age (1 MB), a significant increase in
Actinobacteria (current name
Actinomycetota) was observed in both groups (OB: 25.0% vs. BM: 24.9%), with the OB group maintaining elevated levels of
Proteobacteria (40.6% vs. 14.9%) and
Firmicutes (28.1% vs. 5.9%), while
Bacteroidetes predominated in the BM group (51.9% vs. 6.2%). The most pronounced intergroup differences were recorded at three months of age (3 MB), when
Firmicutes dominated in the OB group (53.9% vs. 4.1%) and
Verrucomicrobia (current name
Verrucomicrobiota) proportion increased (11.6% vs. 5.8%), while the BM group was characterized by high levels of
Bacteroidetes (44.5% vs. 2.3%) and
Actinobacteria (41.7% vs. 18.2%). By six months (6 MB), the difference profile persisted: a predominance of
Firmicutes (46.7% vs. 9.3%) and elevated
Proteobacteria (20.8% vs. 8.3%) in the OB group, with
Actinobacteria dominating in the BM group (54.4% vs. 27.9%).
At the order level (
Figure 2B,E), a significant increase in
Lactobacillales abundance was observed in the OB group (
p ≤ 0.01), with maximum differences in the maternal samples (
p ≤ 0.01) and after 1 month of early infant life (
p ≤ 0.05). A consistent decrease in the relative abundance of
Bacteroidales and increase in
Clostridiales (current name
Eubacteriales) was also noted in the OB group throughout the observation period. At the genus and species levels (
Figure 2C,D), the OB group showed elevated levels of
Enterococcus faecalis (
p ≤ 0.05), while the BM group was characterized by a predominance of
Bifidobacterium (
p ≤ 0.05) representatives, especially
B. longum (
p ≤ 0.05) and
Bacteroides (
p ≤ 0.01). These results indicate persistent differences in taxonomic composition between the groups at all taxonomic levels, starting from the maternal microbiota and persisting throughout the first six months of life.
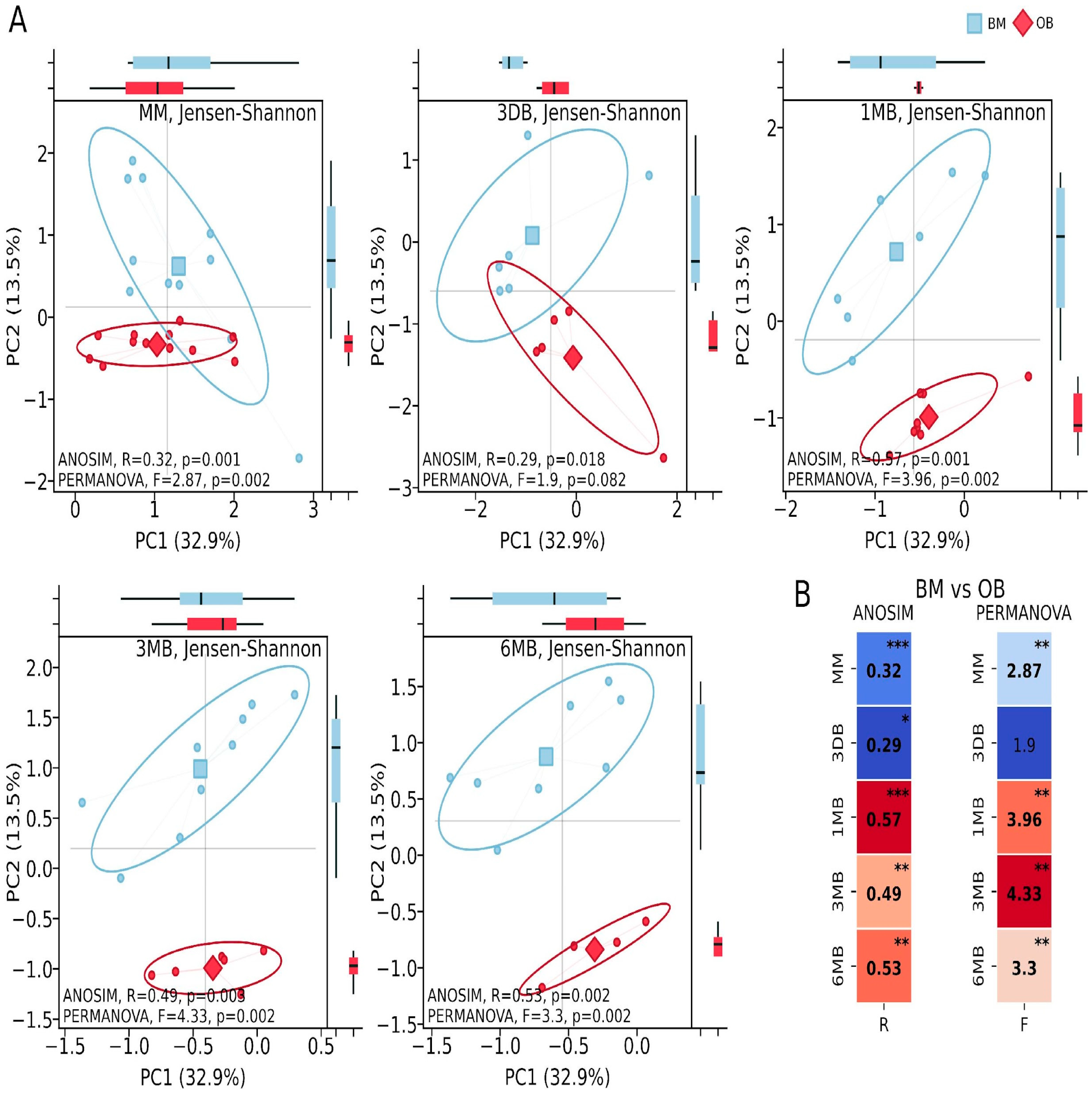
Metabolic activity analysis identified 415 metabolic pathways, of which 43 showed statistically significant differences between the study groups (
p ≤ 0.05 and non-overlapping 95.0% CI and effect difference between means > 0.3). Pathway identification was conducted using the curated MetaCyc database (Version 28.5). Principal Coordinates Analysis (PCoA) of the metabolic pathways showed substantial intergroup differences in the maternal samples between the OB and BM groups (ANOSIM: R = 0.32,
p = 0.001; PERMANOVA: F = 2.87,
p = 0.002). Considerable differences were found in the first-pass meconium samples (ANOSIM: R = 0.29,
p = 0.018; PERMANOVA: F = 1.9,
p = 0.082). In the infants, significant differences were observed during periods of intensive growth and increasing microbial diversity: after one month of life (ANOSIM: R = 0.57,
p = 0.001; PERMANOVA: F = 3.96,
p = 0.002), three months (ANOSIM: R = 0.49,
p = 0.003; PERMANOVA: F = 4.33,
p = 0.002) and six months (ANOSIM: R = 0.53,
p = 0.002; PERMANOVA: F = 3.33,
p = 0.002) (
Figure 3).
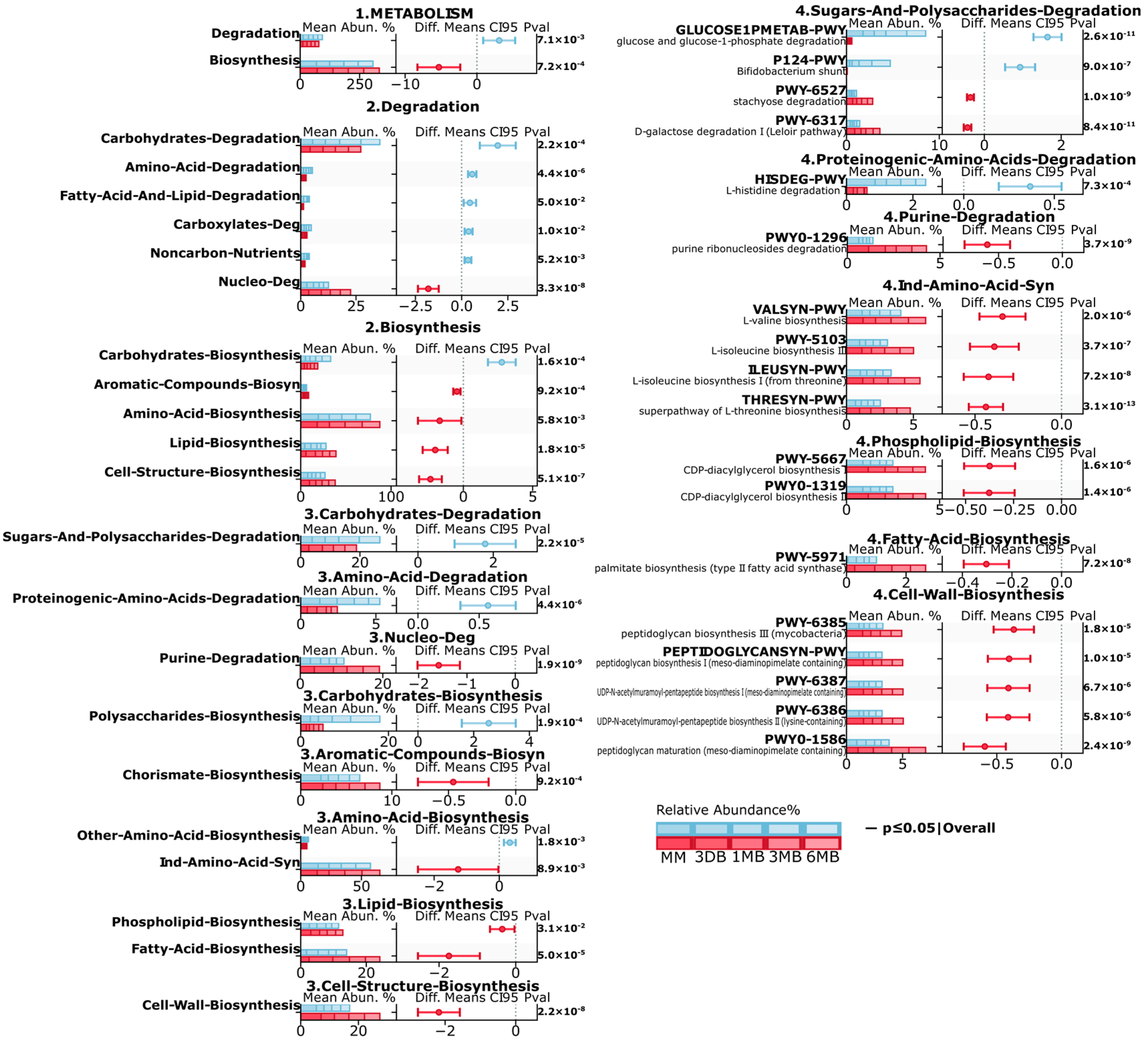
Metagenomic analysis revealed substantial differences in the microbiota metabolic pathways between the infants of mothers with obesity (OB) and normal weight (BM) (
Figure 4). At the first level (1. METABOLISM), a significant decrease in the degradation (
p ≤ 0.01) and increase in the biosynthesis (
p ≤ 0.001) pathways were observed in the OB group. Second-level analysis revealed a significant decrease in the carbohydrate (
p ≤ 0.001, amino acid (
p ≤ 0.001), carboxylate (
p ≤ 0.01), degradation pathways in the OB group, increases in lipid biosynthesis (
p ≤ 0.001), cellular structure biosynthesis (
p ≤ 0.001), and amino acid biosynthesis (
p ≤ 0.01), and elevated activity in the aromatic compound biosynthesis pathways (
p ≤ 0.001). At the third level, the BM group showed increases in carbohydrate and polysaccharide degradation (
p ≤ 0.001) and proteinogenic amino acid degradation (
p ≤ 0.001), while purine degradation (
p ≤ 0.001), as well as phospholipid biosynthesis (
p ≤ 0.05), fatty acid biosynthesis (
p ≤ 0.001), and cell wall component biosynthesis (
p ≤ 0.001), showed increases in the OB group.
Detailed fourth-level analysis revealed significant increases in the specific glucose degradation (GLUCOSE1PMETAB-PWY, p ≤ 0.001), Bifidobacterium shunt (P124-PWY, p ≤ 0.001), and histidine degradation (HISDEG-PWY, p ≤ 0.001) pathways in the BM group. The OB group also showed increased branched-chain amino acid biosynthesis pathways (VALSYN-PWY, ILEUSYN-PWY, p ≤ 0.001), CDP-diacylglycerol biosynthesis (PWY-5667, PWY0-1319, p ≤ 0.001), and peptidoglycan biosynthesis (PWY-6385, PEPTIDOGLYCANSYN-PWY, p ≤ 0.001). These differences indicate substantial restructuring of the microbiota metabolic potential in the OB group, characterized by increased activity in anabolic processes, particularly in amino acid, and the lipid metabolism pathways.
Metabolomic analysis of gut microbiota revealed significant differences between infants of mothers with obesity (OB) and normal weight (BM). Fecal pH analysis showed dynamic differences between groups (
Supplementary Table S1). Mothers in the OB group showed significantly lower pH (6.2 ± 0.23 vs. 6.88 ± 0.24,
p < 0.001). No significant differences were found in meconium samples (3 DB,
p = 0.2) and at one month (1 MB,
p = 0.39). By three months, the OB group showed decreased pH (5.61 ± 0.35 vs. 6.35 ± 0.36,
p ≤ 0.01), however, by six months, a significant increase in pH compared to the BM group was observed (6.31 ± 0.97 vs. 5.54 ± 0.11,
p ≤ 0.05). Correlation analysis between metabolic pathways and pH reduction (
Supplementary Table S1) showed strong negative correlations with fatty acid biosynthesis (r = −0.497,
p < 0.001) and polyprenol biosynthesis (r = −0.480,
p < 0.001), moderate positive correlateions with coenzyme A biosynthesis (r = 0.353,
p < 0.01) and chorismate biosynthesis (r = 0.327,
p < 0.01), and a weak negative correlation with other amino acid biosynthesis (other than those used for protein synthesis) (r = −0.279,
p < 0.05).
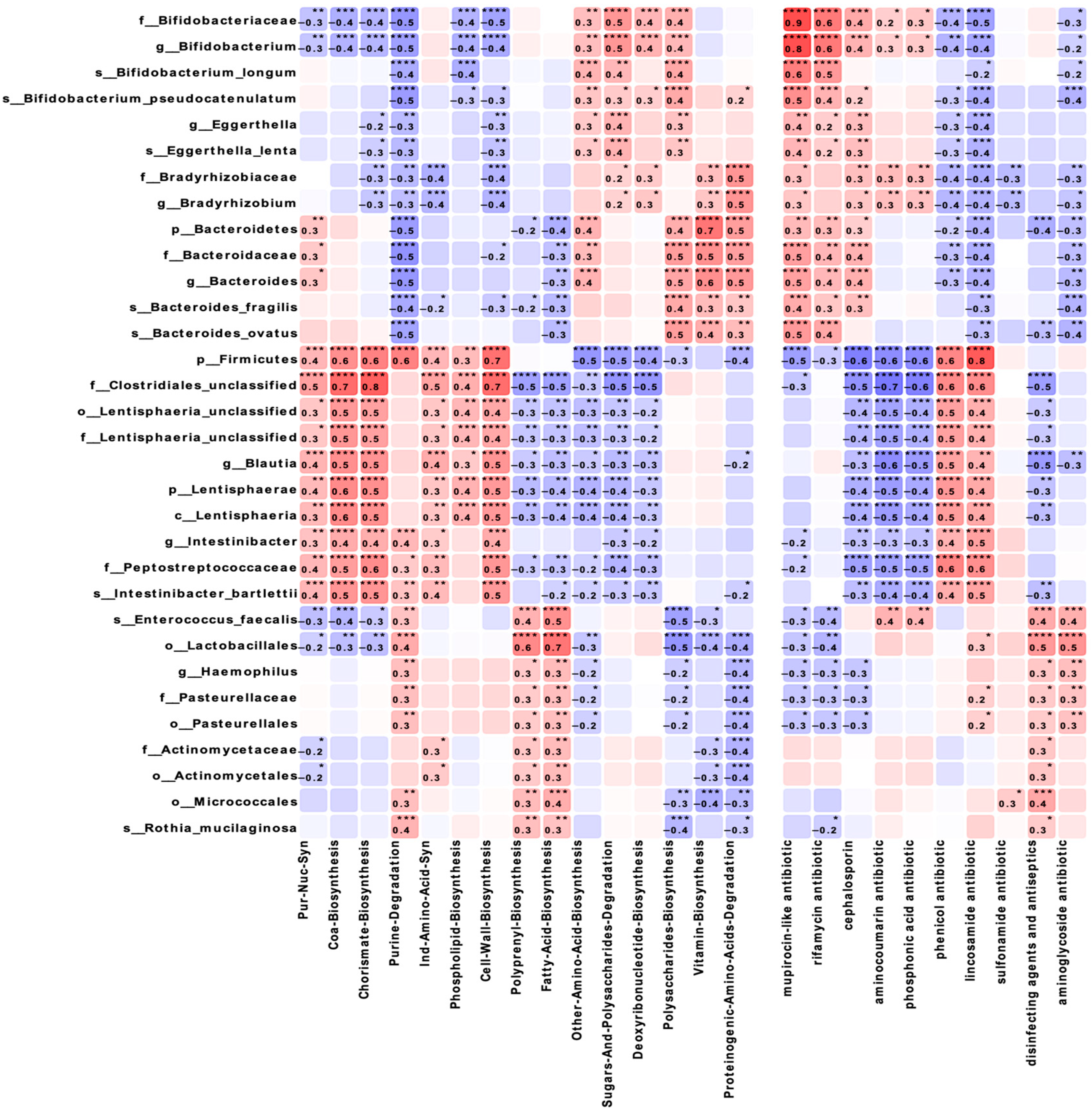
Correlation analysis of the taxonomic composition and functional characteristics of the microbiota (
Figure 5) revealed that
Lactobacillales showed a strong positive correlation with fatty acid biosynthesis (r = 0.7,
p ≤ 0.0001) and a negative correlation with polysaccharide biosynthesis (r = −0.5,
p ≤ 0.001). Firmicutes demonstrated significant correlations with coenzyme A biosynthesis (r = 0.6,
p ≤ 0.0001) and chorismate (r = 0.6,
p ≤ 0.0001), as well as with lincosamide resistance genes (r = 0.8,
p ≤ 0.0001).
Bifidobacteriaceae showed negative correlations with purine degradation and cell wall biosynthesis (r = −0.5,
p ≤ 0.0001), while
Bacteroidetes demonstrated moderate positive correlations with amino acid, polysaccharide biosynthesis, and proteinogenic amino acids degradation (r = 0.4–0.5,
p ≤ 0.01) and a strong positive correlation with vitamin biosynthesis (r = 0.7,
p ≤ 0.01).
4. Discussion
This study presents a comprehensive analysis of how maternal obesity affects the development of the infant gut microbiome during the critical first 6 months of life. Our shotgun metagenomic approach revealed taxonomic alterations and important functional changes in metabolic pathways that may have long-term consequences for infant health. The dynamic patterns observed across multiple time points highlight critical windows of microbiome development where maternal obesity exerts its most potent influence.
In this study, we present a comprehensive dynamic picture of intestinal microbiome formation in infants born to obese mothers during the first 6 months of life. While previous studies have demonstrated general changes in infant microbiota associated with maternal obesity [
10,
13,
20], our work extends these observations by demonstrating not only taxonomic changes but also their functional metabolic consequences using whole metagenomic sequencing. The significant decrease in alpha diversity in infants from the OB group at one month of age indicates a critical window in microbiome establishment, which is consistent with the concept of early metabolic programming [
21]. A comparison with longitudinal studies of the infant microbiome, such as TEDDY [
22], shows that the temporal patterns of the microbiota we identified represent distinct developmental trajectories. The observed elevated
Firmicutes/Bacteroidetes ratio in the OB group, which is particularly pronounced at 3 months (
p ≤ 0.001), is of particular interest. Unlike previous studies [
23,
24], we observe that this imbalance forms dynamically, beginning with meconium microbiota and reaching a maximum at 3 months of infant life. This fact may indicate vertical transmission of the microbiome from mother to child, which is supported by studies by Ferretti et al. [
15]. The depletion of the relative abundance of
Bacteroides and
Bifidobacterium longum in the OB group has potentially serious implications for future metabolic health. Vatanen et al. [
25] showed that
Bacteroides play a key role in degrading complex polysaccharides and maintaining the intestinal barrier, and their deficiency may contribute to future metabolic disorders.
An important aspect is the identification of the substantial reprogramming of the metabolic potential of the microbiome in the OB group. Unlike other studies focused predominantly on taxonomic changes [
13,
26], we found complex changes in metabolic pathways that may explain the mechanisms of maternal obesity’s influence on children’s health. The decreased presence of carbohydrate degradation pathways, amino acids, and carboxylates, with a simultaneous increase in lipid biosynthesis pathways and cellular structures in our study cohort, suggests early metabolic programming. These changes are consistent with a study by Costa et al. [
17], which showed the effect of maternal obesity on the expression of genes associated with the mitochondrial and lipid metabolism in the umbilical vein endothelial cells of infants, but we demonstrate for the first time that similar changes are reflected in the metabolic potential of the intestinal microbiota. These changes can potentially contribute to programming the child’s metabolic status through the production of bioactive lipids and modulation of host signaling pathways.
The identified strong correlations between the taxonomic groups and metabolic pathways reveal new potential aspects in understanding the functional ecology of the microbiome in maternal obesity. The positive correlation of
Lactobacillales with fatty acid biosynthesis and negative correlation with polysaccharide biosynthesis may reflect compensatory mechanisms aimed at maintaining metabolic homeostasis [
27,
28]. The correlations of
Firmicutes with genes for resistance to lincosamides and the phenicol resistance gene are of particular interest in the context of the growing problem of antibiotic resistance. Compared to data on the resistome of the intestinal microbiota (Gasparrini et al., 2019) [
29], our study shows that maternal obesity may influence the formation of the resistome in infants, which has potentially important clinical implications. The dynamics of fecal pH, reflecting the metabolic activity of the microbiota, in our opinion, deserve special attention. Increased fecal acidity in mothers in the OB group and infants at 3 months may be associated with the increased production of short-chain fatty acids. This is consistent with data on the influence of maternal obesity on the metabolic activity of newborn microbiota [
20], but our study demonstrates dynamic changes in this parameter for the first time during the first 6 months of life.
The identified specific time windows (1 and 3 months) may represent the optimal periods for therapeutic intervention. Unlike previous works proposing general strategies for microbiome modulation [
30], our data on specific metabolic pathways allow the development of more targeted approaches. The strong correlations between specific taxa and metabolic functions provide new targets for probiotic and prebiotic therapy. For example, stimulation of
Bacteroidetes and
Bifidobacterium representatives may help restore the polysaccharide degradation pathways, while the modulation of
Lactobacillales activity could potentially normalize lipid metabolism. Our study has several limitations to consider when interpreting results. The relatively small sample size (24 mother–infant pairs) and single-center study design may limit result generalizability. The 6-month observation period, while covering a critical microbiome establishment period, does not allow assessment of the long-term consequences of the observed changes. Additionally, the lack of detailed maternal diet composition data during pregnancy and lactation, along with sample loss due to quality control failure, may have affected result completeness. Despite these limitations, the obtained data on taxonomic composition and the metabolic potential differences between the OB and BM groups remain statistically significant and align with other research findings [
20], confirming their value and contribution to understanding maternal obesity’s influence on infant microbiome formation. Future studies with larger cohorts, longer observation periods, and the integration of host metabolomic data will allow a more comprehensive characterization of the interaction between maternal obesity, the infant microbiome, and long-term health consequences.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}