

Glycolysis-Driven Prognostic Model for Acute Myeloid Leukemia: Insights into the Immune Landscape and Drug Sensitivity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
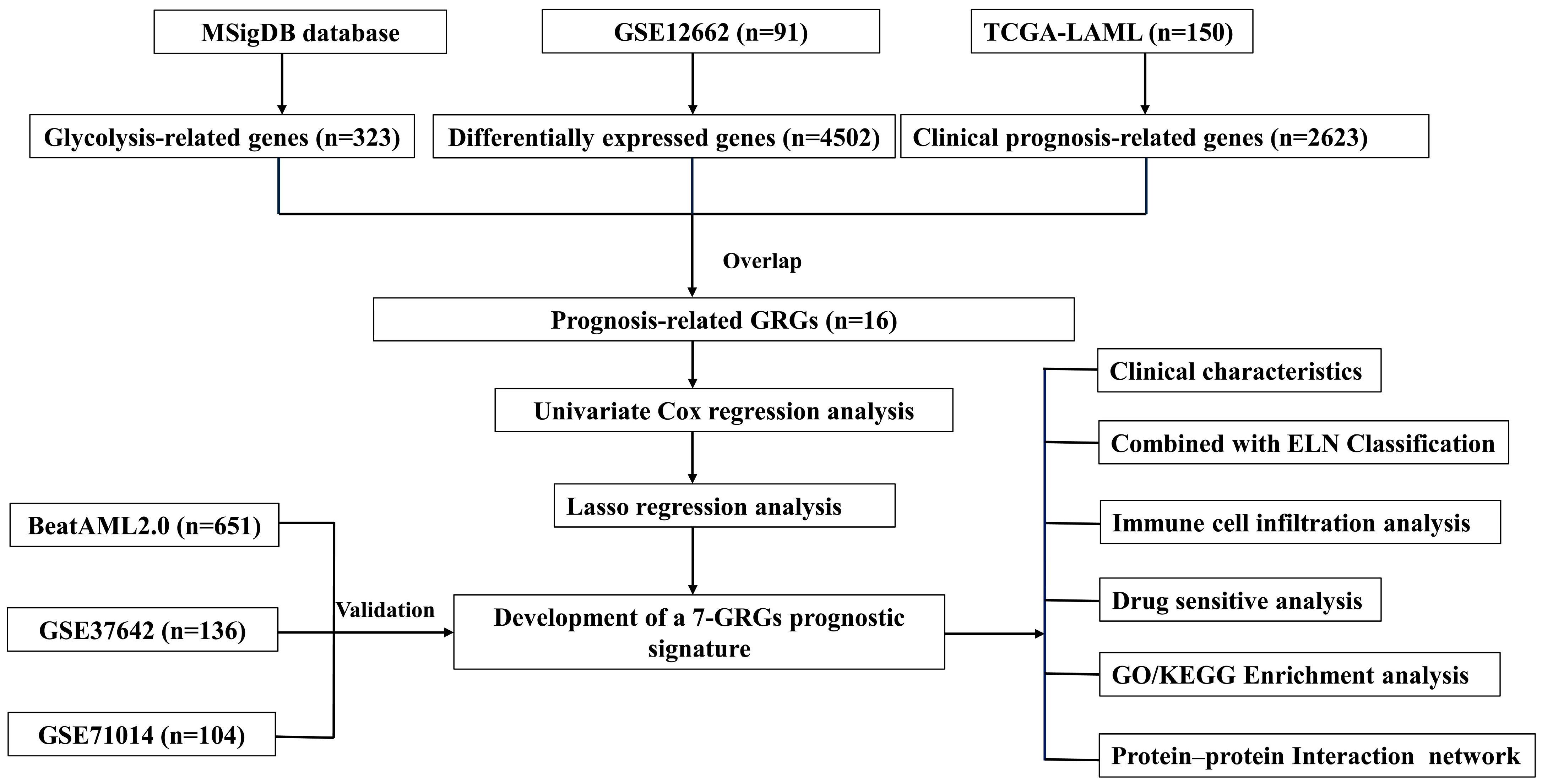
2.1. Data Acquisition and Processing
2.2. Analysis of Differential Expression and Prognosis
2.3. Construction of a Novel AML Prognostic Risk Model Based on GRGs
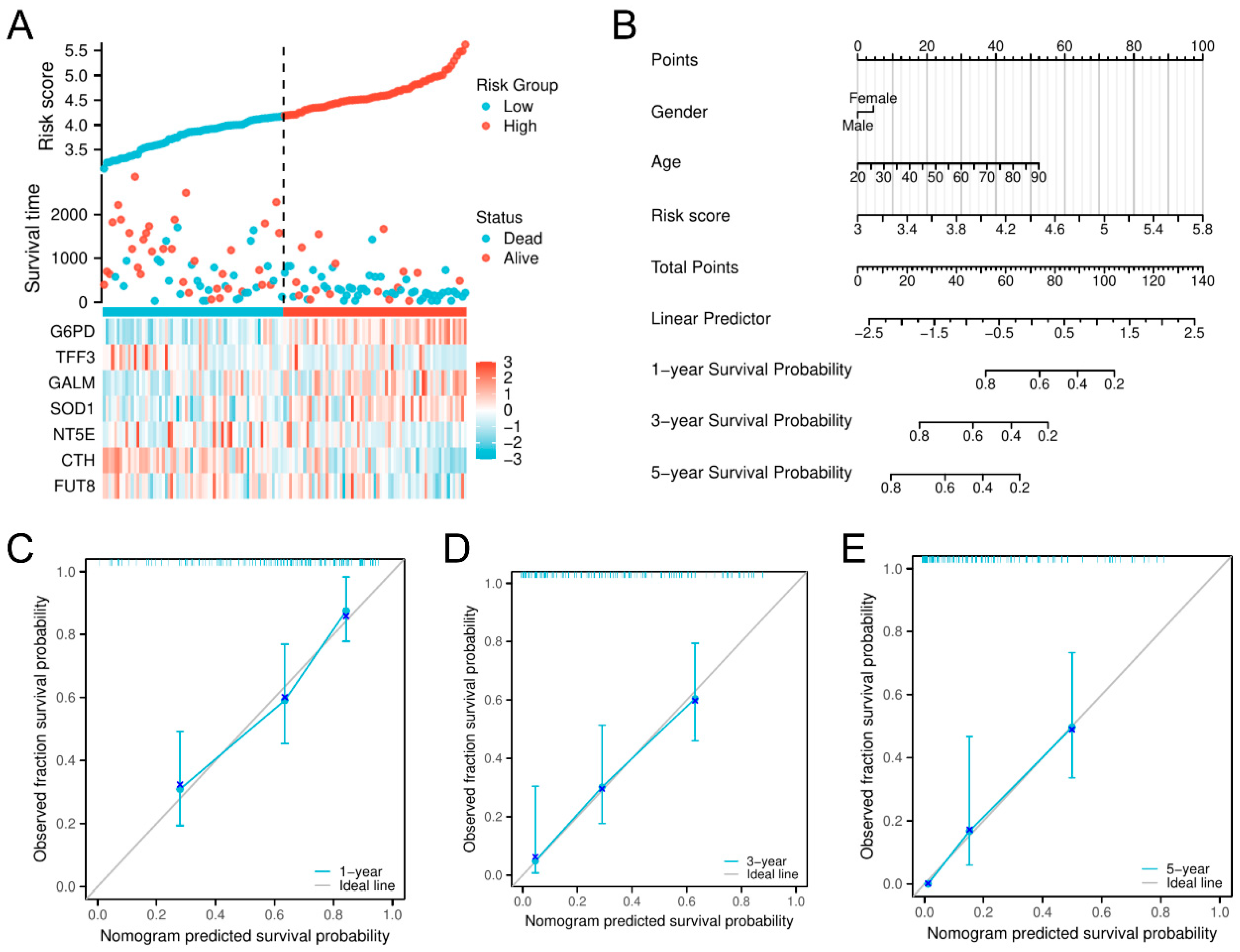
2.4. Development of a Nomogram and Calibration Curve
2.5. Immune Infiltration Analysis
2.6. Drug Sensitivity Analysis
2.7. Differentially Enriched Genes and PPI Network Interaction
2.8. Statistical Analyses
3. Results
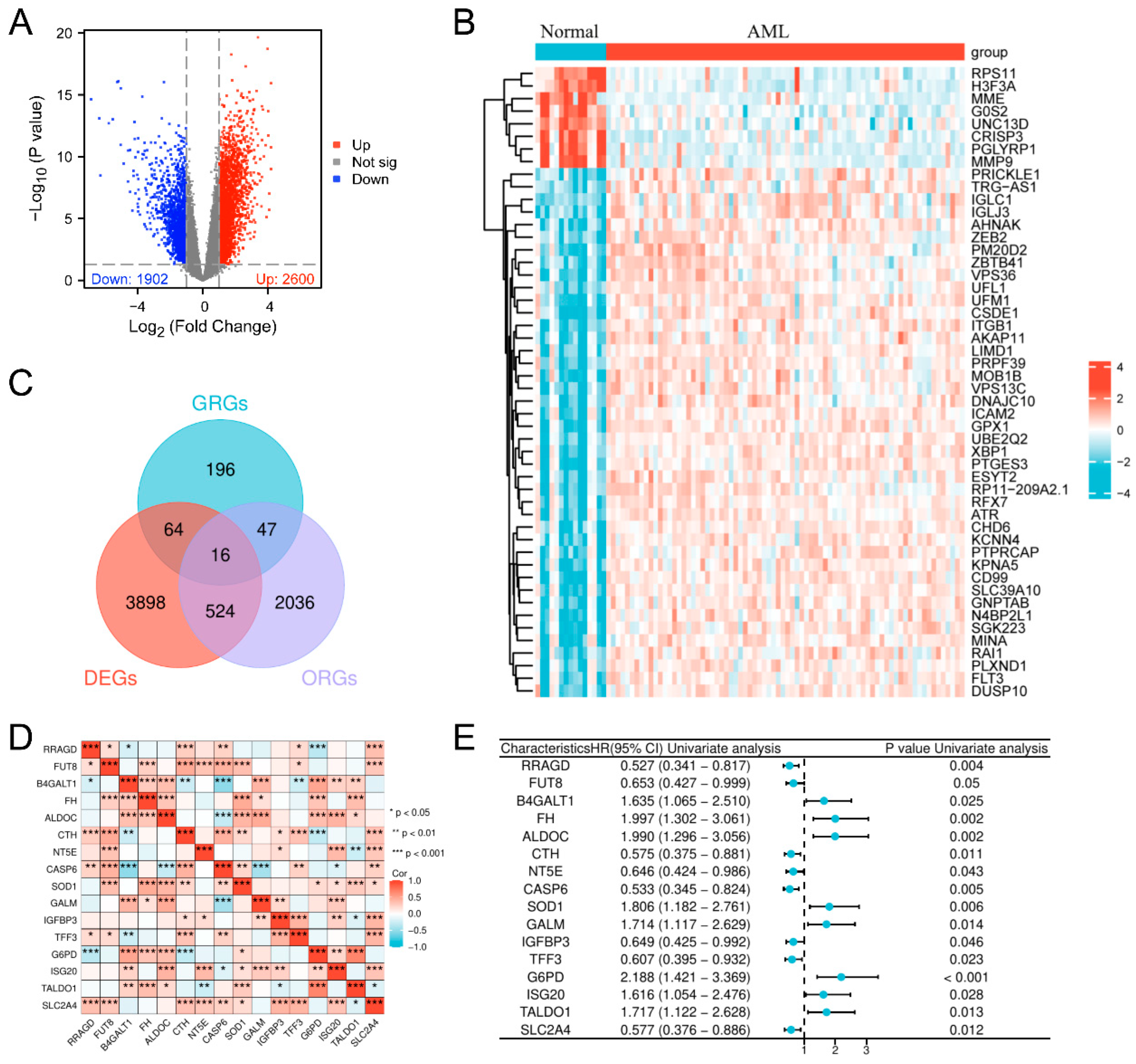
3.1. Screening of Glycolysis-Related Prognostic Genes in AML
3.2. Development of a Prognostic Risk Evaluation Model Integrating Glycolysis-Dependent Genes in AML
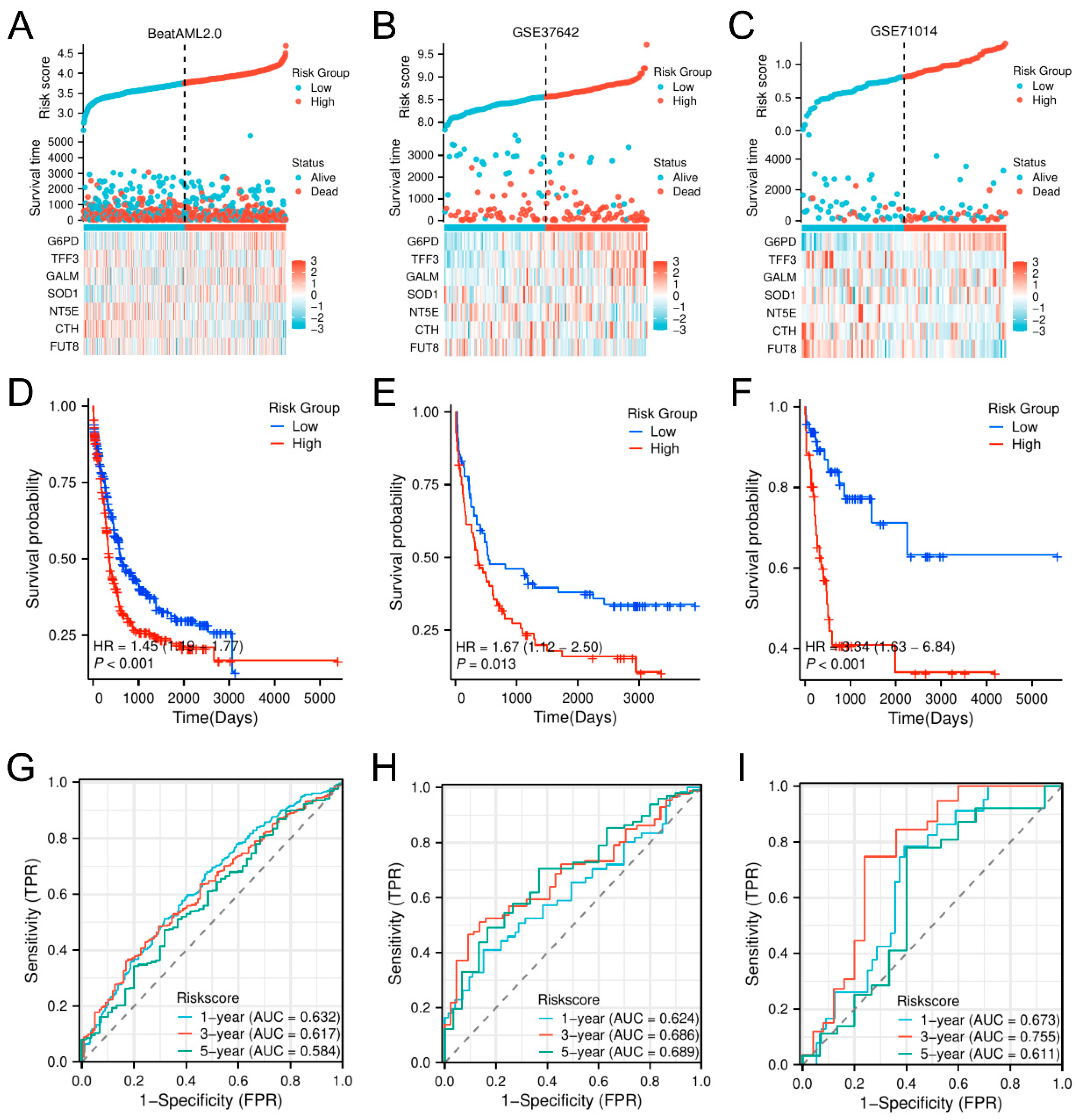
3.3. Validation and Prognostic Assessment of the Glycolysis-Related Prognostic Model (GPM) for Overall Survival in AML Across Multiple Independent Datasets
3.4. Enhancement of AML Prognosis Prediction by Combining the Glycolysis-Related Model with the ELN Classification
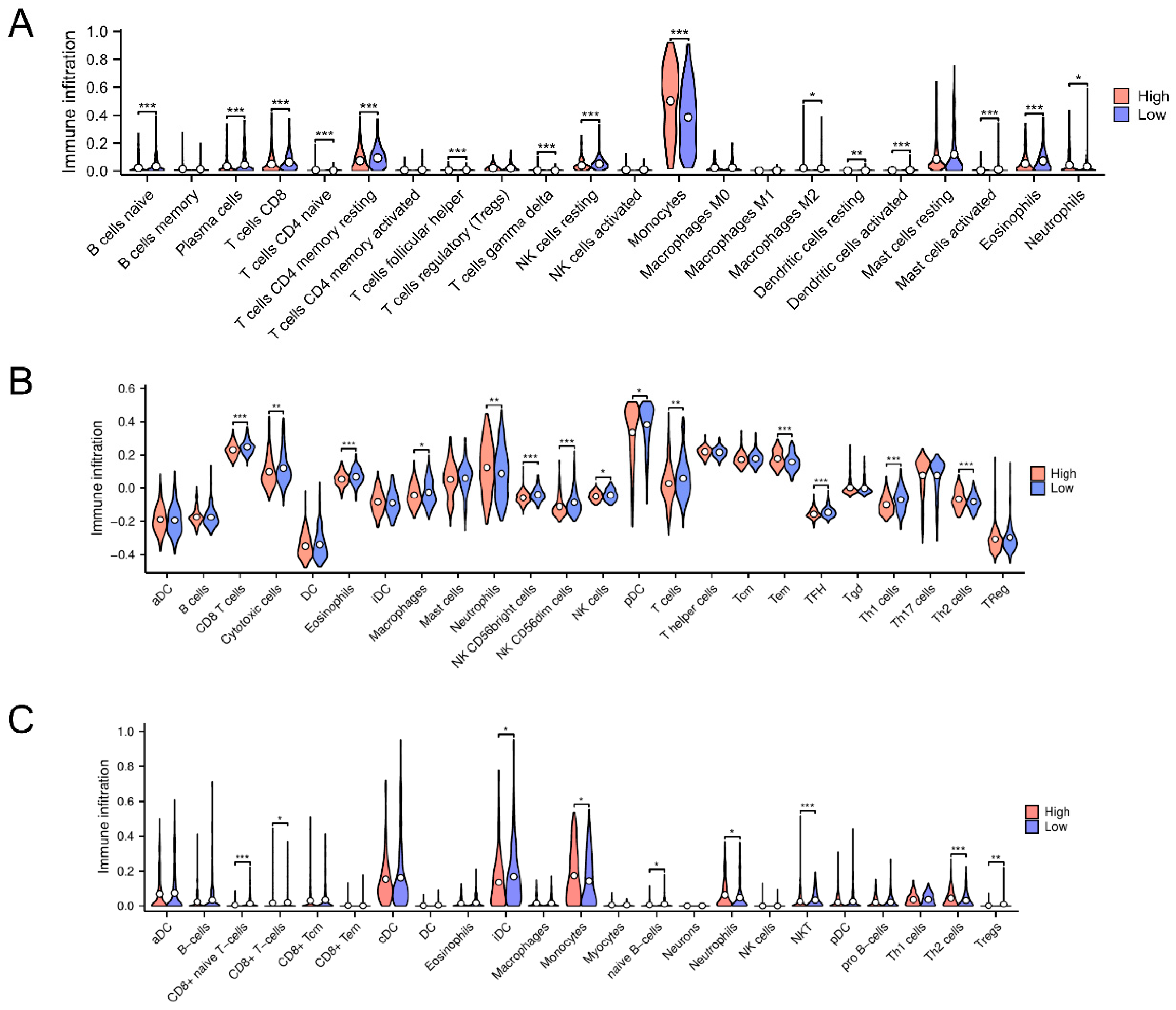
3.5. Immune Microenvironmental Variances Between the High-Risk and Low-Risk Classifications Uncover Immune Downregulation and Anti-Oncogenic Activity
3.6. Identification of Potential Therapeutic Agents for AML Using Prognostic Model-Based Drug Sensitivity Analysis
3.7. Differential Gene Expression and Functional Enrichment Analyses Reveal IL-6 as a Key Mediator in High-Risk AML
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute Myeloid Leukemia: Current Progress and Future Directions. Blood Cancer J. 2021, 11, 1–25. [Google Scholar] [CrossRef]
- Tettamanti, S.; Pievani, A.; Biondi, A.; Dotti, G.; Serafini, M. Catch Me If You Can: How AML and Its Niche Escape Immunotherapy. Leukemia 2022, 36, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Wei, A.H.; Löwenberg, B. Towards Precision Medicine for AML. Nat. Rev. Clin. Oncol. 2021, 18, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Sauerer, T.; Velázquez, G.F.; Schmid, C. Relapse of Acute Myeloid Leukemia after Allogeneic Stem Cell Transplantation: Immune Escape Mechanisms and Current Implications for Therapy. Mol. Cancer 2023, 22, 180. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and Management of AML in Adults: 2017 ELN Recommendations from an International Expert Panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and Management of AML in Adults: 2022 Recommendations from an International Expert Panel on Behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Hoff, F.W.; Blum, W.G.; Huang, Y.; Welkie, R.L.; Swords, R.T.; Traer, E.; Stein, E.M.; Lin, T.L.; Archer, K.J.; Patel, P.A.; et al. Beat-AML 2024 ELN–Refined Risk Stratification for Older Adults with Newly Diagnosed AML given Lower-Intensity Therapy. Blood Adv. 2024, 8, 5297–5305. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why Do Cancers Have High Aerobic Glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Warburg, O. On Respiratory Impairment in Cancer Cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. Available online: https://www.science.org/doi/abs/10.1126/science.123.3191.309 (accessed on 23 December 2024).
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- Yang, M.; Soga, T.; Pollard, P. Oncometabolites: Linking Altered Metabolism with Cancer. J. Clin. Invest. 2013, 123, 3652–3658. Available online: https://www.jci.org/articles/view/67228 (accessed on 23 December 2024). [CrossRef] [PubMed]
- Vander Heiden, M.G. Targeting Cancer Metabolism: A Therapeutic Window Opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. Available online: https://www.nature.com/articles/nrd3504 (accessed on 23 December 2024). [CrossRef]
- Groves, A.M.; Win, T.; Haim, S.B.; Ell, P.J. Non-[18F]FDG PET in Clinical Oncology. Lancet Oncol. 2007, 8, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, I.; Kohno, B. 18 FDG-PET/CT: 21st Century Approach to Leukemic Tumors in 124 Cases. Am. J. Hematol. 2016, 91, 379–384. [Google Scholar] [CrossRef]
- Herst, P.M.; Howman, R.A.; Neeson, P.J.; Berridge, M.V.; Ritchie, D.S. The Level of Glycolytic Metabolism in Acute Myeloid Leukemia Blasts at Diagnosis Is Prognostic for Clinical Outcome. J. Leukoc. Biol. 2011, 89, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-L.; Wang, J.-H.; Zhao, A.-H.; Xu, X.; Wang, Y.-H.; Chen, T.-L.; Li, J.-M.; Mi, J.-Q.; Zhu, Y.-M.; Liu, Y.-F.; et al. A Distinct Glucose Metabolism Signature of Acute Myeloid Leukemia with Prognostic Value. Blood 2014, 124, 1645–1654. [Google Scholar] [CrossRef]
- Simsek, T.; Kocabas, F.; Zheng, J.; Deberardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The Distinct Metabolic Profile of Hematopoietic Stem Cells Reflects Their Location in a Hypoxic Niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting Mitochondrial Oxidative Phosphorylation Eradicates Therapy-Resistant Chronic Myeloid Leukemia Stem Cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 Inhibition Targets Oxidative Phosphorylation and Selectively Eradicates Quiescent Human Leukemia Stem Cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef]
- van Gils, N.; Denkers, F.; Smit, L. Escape from Treatment; the Different Faces of Leukemic Stem Cells and Therapy Resistance in Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 659253. [Google Scholar] [CrossRef]
- Huang, Z.-W.; Zhang, X.-N.; Zhang, L.; Liu, L.-L.; Zhang, J.-W.; Sun, Y.-X.; Xu, J.-Q.; Liu, Q. STAT5 Promotes PD-L1 Expression by Facilitating Histone Lactylation to Drive Immunosuppression in Acute Myeloid Leukemia. Signal Transduct. Target. Ther. 2023, 8, 391. Available online: https://www.nature.com/articles/s41392-023-01605-2 (accessed on 17 December 2024). [PubMed]
- Hammon, K.; Renner, K.; Althammer, M.; Voll, F.; Babl, N.; Decking, S.-M.; Siska, P.J.; Matos, C.; Conejo, Z.E.C.; Mendes, K.; et al. D-2-Hydroxyglutarate Supports a Tolerogenic Phenotype with Lowered Major Histocompatibility Class II Expression in Non-Malignant Dendritic Cells and Acute Myeloid Leukemia Cells. Haematologica 2024, 109, 2500–2514. [Google Scholar] [CrossRef]
- Cui, L.; Cheng, Z.; Liu, Y.; Dai, Y.; Pang, Y.; Jiao, Y.; Ke, X.; Cui, W.; Zhang, Q.; Shi, J.; et al. Overexpression of PDK2 and PDK3 Reflects Poor Prognosis in Acute Myeloid Leukemia. Cancer Gene Ther. 2020, 27, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Li, J.; Hong, M.; Zhu, Y.; Zhao, H.; Xie, Y.; Huang, J.; Lian, Y.; Li, Y.; Wang, S.; et al. TIGAR Cooperated with Glycolysis to Inhibit the Apoptosis of Leukemia Cells and Associated with Poor Prognosis in Patients with Cytogenetically Normal Acute Myeloid Leukemia. J. Hematol. Oncol.J Hematol Oncol 2016, 9, 128. [Google Scholar] [CrossRef]
- Ryu, M.J.; Han, J.; Kim, S.J.; Lee, M.J.; Ju, X.; Lee, Y.L.; Son, J.H.; Cui, J.; Jang, Y.; Chung, W.; et al. PTEN/AKT Signaling Mediates Chemoresistance in Refractory Acute Myeloid Leukemia through Enhanced Glycolysis. Oncol. Rep. 2019, 42, 2149–2158. [Google Scholar] [CrossRef]
- Wang, B.; Wang, X.; Hou, D.; Huang, Q.; Zhan, W.; Chen, C.; Liu, J.; You, R.; Xie, J.; Chen, P.; et al. Exosomes Derived from Acute Myeloid Leukemia Cells Promote Chemoresistance by Enhancing Glycolysis-Mediated Vascular Remodeling. J. Cell. Physiol. 2019, 234, 10602–10614. [Google Scholar] [CrossRef]
- Chen, S.; Tao, Y.; Wang, Q.; Ren, J.; Jing, Y.; Huang, J.; Zhang, L.; Li, R. Glucose Induced-AKT/mTOR Activation Accelerates Glycolysis and Promotes Cell Survival in Acute Myeloid Leukemia. Leuk. Res. 2023, 128, 107059. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Bottomly, D.; Long, N.; Schultz, A.R.; Kurtz, S.E.; Tognon, C.E.; Johnson, K.; Abel, M.; Agarwal, A.; Avaylon, S.; Benton, E.; et al. Integrative Analysis of Drug Response and Clinical Outcome in Acute Myeloid Leukemia. Cancer Cell 2022, 40, 850–864.e9. [Google Scholar] [CrossRef]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef]
- Friedman, J.H.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Zwaan, C.M.; Kolb, E.A.; Reinhardt, D.; Abrahamsson, J.; Adachi, S.; Aplenc, R.; De Bont, E.S.J.M.; De Moerloose, B.; Dworzak, M.; Gibson, B.E.S.; et al. Collaborative Efforts Driving Progress in Pediatric Acute Myeloid Leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 2949–2962. [Google Scholar] [CrossRef]
- Totiger, T.M.; Ghoshal, A.; Zabroski, J.; Sondhi, A.; Bucha, S.; Jahn, J.; Feng, Y.; Taylor, J. Targeted Therapy Development in Acute Myeloid Leukemia. Biomedicines 2023, 11, 641. [Google Scholar] [CrossRef]
- Ravandi, F. Relapsed Acute Myeloid Leukemia: Why Is There No Standard of Care? Best Pract. Res. Clin. Haematol. 2013, 26, 253–259. [Google Scholar] [CrossRef]
- Chelakkot, C.; Chelakkot, V.S.; Shin, Y.; Song, K. Modulating Glycolysis to Improve Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 2606. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, M.; Ózsvári, B.; Sotgia, F.; Lisanti, M.P. High ATP Production Fuels Cancer Drug Resistance and Metastasis: Implications for Mitochondrial ATP Depletion Therapy. Front. Oncol. 2021, 11, 740720. [Google Scholar] [CrossRef]
- Wang, T.; Ma, F.; Qian, H.-L. Defueling the Cancer: ATP Synthase as an Emerging Target in Cancer Therapy. Mol. Ther. Oncolytics 2021, 23, 82–95. [Google Scholar] [CrossRef]
- Gregory, M.A.; D’Alessandro, A.; Alvarez-Calderon, F.; Kim, J.; Nemkov, T.; Adane, B.; Rozhok, A.I.; Kumar, A.; Kumar, V.; Pollyea, D.A.; et al. ATM/G6PD-Driven Redox Metabolism Promotes FLT3 Inhibitor Resistance in Acute Myeloid Leukemia. Proc. Natl. Acad. Sci. USA 2016, 113, E6669–E6678. [Google Scholar] [CrossRef]
- Wang, J.; Qiao, Y.; Sun, M.; Sun, H.; Xie, F.; Chang, H.; Wang, Y.; Song, J.; Lai, S.; Yang, C.; et al. FTO Promotes Colorectal Cancer Progression and Chemotherapy Resistance via Demethylating G6PD/PARP1. Clin. Transl. Med. 2022, 12, e772. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-C.; Wu, Y.-H.; Yen, W.-C.; Liu, H.-Y.; Hwang, T.-L.; Stern, A.; Chiu, D.T.-Y. The Redox Role of G6PD in Cell Growth, Cell Death, and Cancer. Cells 2019, 8, 1055. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Y.; Lu, W.; Li, J.; Yu, S.; Brown, E.J.; Stanger, B.Z.; Rabinowitz, J.D.; Yang, X. G6PD-Mediated Increase in de Novo NADP+ Biosynthesis Promotes Antioxidant Defense and Tumor Metastasis. Sci. Adv. 2022, 8, eabo0404. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Chen, Z.; Wu, K.; Huang, S.M.; Wu, W.L.; LeBoeuf, S.E.; Pillai, R.G.; Rabinowitz, J.D.; Papagiannakopoulos, T. Activation of the NRF2 Antioxidant Program Sensitizes Tumors to G6PD Inhibition. Sci. Adv. 2021, 7, eabk1023. Available online: https://www.science.org/doi/full/10.1126/sciadv.abk1023 (accessed on 9 January 2025). [CrossRef]
- Ju, H.-Q.; Lu, Y.-X.; Wu, Q.-N.; Liu, J.; Zeng, Z.-L.; Mo, H.-Y.; Chen, Y.; Tian, T.; Wang, Y.; Kang, T.-B.; et al. Disrupting G6PD-Mediated Redox Homeostasis Enhances Chemosensitivity in Colorectal Cancer. Oncogene 2017, 36, 6282–6292. Available online: https://www.nature.com/articles/onc2017227 (accessed on 9 January 2025). [CrossRef]
- Naville, A.-S.; Lazaro, E.; Boutin, J.; Prot-Leurent, C.; Mansier, O.; Richard, E.; Augis, V.; Weinmann, L.; Fuster, V.; Vial, J.-P.; et al. Acquired Glucose 6-phosphate Dehydrogenase (G6PD) Deficiency in a Patient with Chronic Myelomonocytic Leukemia. Br. J. Haematol. 2022, 197, e45. Available online: https://openurl.ebsco.com/EPDB%3Agcd%3A3%3A17754243/detailv2?sid=ebsco%3Aplink%3Ascholar&id=ebsco%3Agcd%3A156737254&crl=c&link_origin=scholar.google.com.hk (accessed on 14 March 2025). [CrossRef] [PubMed]
- Im, S.; Yoo, C.; Jung, J.-H.; Choi, H.J.; Yoo, J.; Kang, C.S. Reduced Expression of TFF1 and Increased Expression of TFF3 in Gastric Cancer: Correlation with Clinicopathological Parameters and Prognosis. Int. J. Med. Sci. 2012, 10, 133–140. [Google Scholar] [CrossRef]
- May, F.E.B.; Westley, B.R. TFF3 Is a Valuable Predictive Biomarker of Endocrine Response in Metastatic Breast Cancer. Endocr. Relat. Cancer 2015, 22, 465–479. [Google Scholar] [CrossRef]
- Cui, H.-Y.; Wang, S.-J.; Song, F.; Cheng, X.; Nan, G.; Zhao, Y.; Qian, M.-R.; Chen, X.; Li, J.-Y.; Liu, F.-L.; et al. CD147 Receptor Is Essential for TFF3-Mediated Signaling Regulating Colorectal Cancer Progression. Signal Transduct. Target. Ther. 2021, 6, 268. Available online: https://www.nature.com/articles/s41392-021-00677-2 (accessed on 9 January 2025). [CrossRef]
- Leung, W.K.; Yu, J.; Chan, F.K.L.; To, K.F.; Chan, M.W.Y.; Ebert, M.P.A.; Ng, E.K.W.; Chung, S.C.S.; Malfertheiner, P.; Sung, J.J.Y. Expression of Trefoil Peptides (TFF1, TFF2, and TFF3) in Gastric Carcinomas, Intestinal Metaplasia, and Non-neoplastic Gastric Tissues. J. Pathol. 2002, 197, 582–588. Available online: https://pathsocjournals.onlinelibrary.wiley.com/doi/abs/10.1002/path.1147 (accessed on 9 January 2025). [CrossRef]
- Xu, J.; Guo, Y.; Ning, W.; Wang, X.; Li, S.; Chen, Y.; Ma, L.; Qu, Y.; Song, Y.; Zhang, H. Comprehensive Analyses of Glucose Metabolism in Glioma Reveal the Glioma-Promoting Effect of GALM. Front. Cell Dev. Biol. 2022, 9, 717182. Available online: https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2021.717182/full (accessed on 9 January 2025).
- Sillar, J.R.; Germon, Z.P.; De Iuliis, G.N.; Dun, M.D. The Role of Reactive Oxygen Species in Acute Myeloid Leukaemia. Int. J. Mol. Sci. 2019, 20, 6003. [Google Scholar] [CrossRef] [PubMed]
- Xavier, A.C.; Taub, J.W. Acute Leukemia in Children with Down Syndrome. Haematologica 2010, 95, 1043–1045. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Re, D.B.; Nagata, T.; Chalazonitis, A.; Jessell, T.M.; Wichterle, H.; Przedborski, S. Astrocytes Expressing ALS-Linked Mutated SOD1 Release Factors Selectively Toxic to Motor Neurons. Nat. Neurosci. 2007, 10, 615–622. Available online: https://www.nature.com/articles/nn1876 (accessed on 9 January 2025).
- Papa, L.; Manfredi, G.; Germain, D. SOD1, an Unexpected Novel Target for Cancer Therapy. Genes Cancer 2014, 5, 15–21. Available online: https://pmc.ncbi.nlm.nih.gov/articles/PMC4063254/ (accessed on 9 January 2025).
- Bullinger, L.; Döhner, K.; Bair, E.; Fröhling, S.; Schlenk, R.F.; Tibshirani, R.; Döhner, H.; Pollack, J.R. Use of Gene-Expression Profiling to Identify Prognostic Subclasses in Adult Acute Myeloid Leukemia. N. Engl. J. Med. 2004, 350, 1605–1616. [Google Scholar] [CrossRef]
- Jakobsen, J.S.; Laursen, L.G.; Schuster, M.B.; Pundhir, S.; Schoof, E.; Ge, Y.; d’Altri, T.; Vitting-Seerup, K.; Rapin, N.; Gentil, C.; et al. Mutant CEBPA Directly Drives the Expression of the Targetable Tumor-Promoting Factor CD73 in AML. Sci. Adv. 2019, 5, eaaw4304. Available online: https://www.science.org/doi/full/10.1126/sciadv.aaw4304 (accessed on 9 January 2025).
- Moore, L.E.; Malats, N.; Rothman, N.; Real, F.X.; Kogevinas, M.; Karami, S.; García-Closas, R.; Silverman, D.; Chanock, S.; Welch, R.; et al. Polymorphisms in One-Carbon Metabolism and Trans-Sulfuration Pathway Genes and Susceptibility to Bladder Cancer. Int. J. Cancer 2007, 120, 2452–2458. [Google Scholar] [CrossRef]
- Lin, Z.; Huang, W.; He, Q.; Li, D.; Wang, Z.; Feng, Y.; Liu, D.; Zhang, T.; Wang, Y.; Xie, M.; et al. FOXC1 Promotes HCC Proliferation and Metastasis by Upregulating DNMT3B to Induce DNA Hypermethylation of CTH Promoter. J. Exp. Clin. Cancer Res. 2021, 40, 50. Available online: https://link.springer.com/article/10.1186/s13046-021-01829-6 (accessed on 9 January 2025).
- Chang, E.Y.-C.; Chang, Y.-C.; Shun, C.-T.; Tien, Y.-W.; Tsai, S.-H.; Hee, S.-W.; Chen, I.-J.; Chuang, L.-M. Inhibition of Prostaglandin Reductase 2, a Putative Oncogene Overexpressed in Human Pancreatic Adenocarcinoma, Induces Oxidative Stress-Mediated Cell Death Involving xCT and CTH Gene Expressions through 15-Keto-PGE2. PLoS ONE 2016, 11, e0147390. Available online: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0147390 (accessed on 9 January 2025). [CrossRef]
- Peleli, M.; Antoniadou, I.; Rodrigues-Junior, D.M.; Savvoulidou, O.; Caja, L.; Katsouda, A.; Ketelhuth, D.F.; Stubbe, J.; Madsen, K.; Moustakas, A.; et al. Cystathionine Gamma-Lyase (CTH) Inhibition Attenuates Glioblastoma Formation. Redox Biol. 2023, 64, 102773. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, E.; Liu, Q.; Burton, C.; Mockabee-Macias, A.; Lester, D.K.; Lau, E. L-Fucose, a Sugary Regulator of Antitumor Immunity and Immunotherapies. Mol. Carcinog. 2022, 61, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Bastian, K.; Scott, E.; Elliott, D.J.; Munkley, J. FUT8 Alpha-(1,6)-Fucosyltransferase in Cancer. Int. J. Mol. Sci. 2021, 22, 455. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Jan, Y.-H.; Juan, Y.-H.; Yang, C.-J.; Huang, M.-S.; Yu, C.-J.; Yang, P.-C.; Hsiao, M.; Hsu, T.-L.; Wong, C.-H. Fucosyltransferase 8 as a Functional Regulator of Nonsmall Cell Lung Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Stefaniuk, P.; Szymczyk, A.; Podhorecka, M. The Neutrophil to Lymphocyte and Lymphocyte to Monocyte Ratios as New Prognostic Factors in Hematological Malignancies—A Narrative Review. Cancer Manag. Res. 2020, 12, 2961–2977. [Google Scholar] [CrossRef]
- Zhang, Y.; Nguyen, T.T.T.; Shang, E.; Mela, A.; Humala, N.; Mahajan, A.; Zhao, J.; Shu, C.; Torrini, C.; Sanchez-Quintero, M.J.; et al. MET Inhibition Elicits PGC1α-Dependent Metabolic Reprogramming in Glioblastoma. Cancer Res. 2020, 80, 30–43. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, R.; Jin, W.; Wang, K. Glycolysis-Driven Prognostic Model for Acute Myeloid Leukemia: Insights into the Immune Landscape and Drug Sensitivity. Biomedicines 2025, 13, 834. https://doi.org/10.3390/biomedicines13040834
Zhang R, Jin W, Wang K. Glycolysis-Driven Prognostic Model for Acute Myeloid Leukemia: Insights into the Immune Landscape and Drug Sensitivity. Biomedicines. 2025; 13(4):834. https://doi.org/10.3390/biomedicines13040834
Chicago/Turabian StyleZhang, Rongsheng, Wen Jin, and Kankan Wang. 2025. "Glycolysis-Driven Prognostic Model for Acute Myeloid Leukemia: Insights into the Immune Landscape and Drug Sensitivity" Biomedicines 13, no. 4: 834. https://doi.org/10.3390/biomedicines13040834
APA StyleZhang, R., Jin, W., & Wang, K. (2025). Glycolysis-Driven Prognostic Model for Acute Myeloid Leukemia: Insights into the Immune Landscape and Drug Sensitivity. Biomedicines, 13(4), 834. https://doi.org/10.3390/biomedicines13040834