
Investigating the Prognostic Role of Telomerase-Related Cellular Senescence Gene Signatures in Breast Cancer Using Machine Learning


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection and Preprocessing
2.2. Identification and Functional Annotation of Telomere-Related Cellular Senescence Genes in Breast Cancer
2.3. Integration of Machine Learning Algorithms to Identify Prognostic Gene Signatures
2.4. Clinical Correlation Assessment, Nomogram Modeling, and Comparative Performance Analysis
2.5. Assessment of the Risk Score Signature for Immunotherapy Response and Prognostic Evaluation
2.6. Correlation Analysis Between Signature Genes and Stromal Scores
2.7. Biological Mechanisms and Mutational Analysis of Risk Score Features
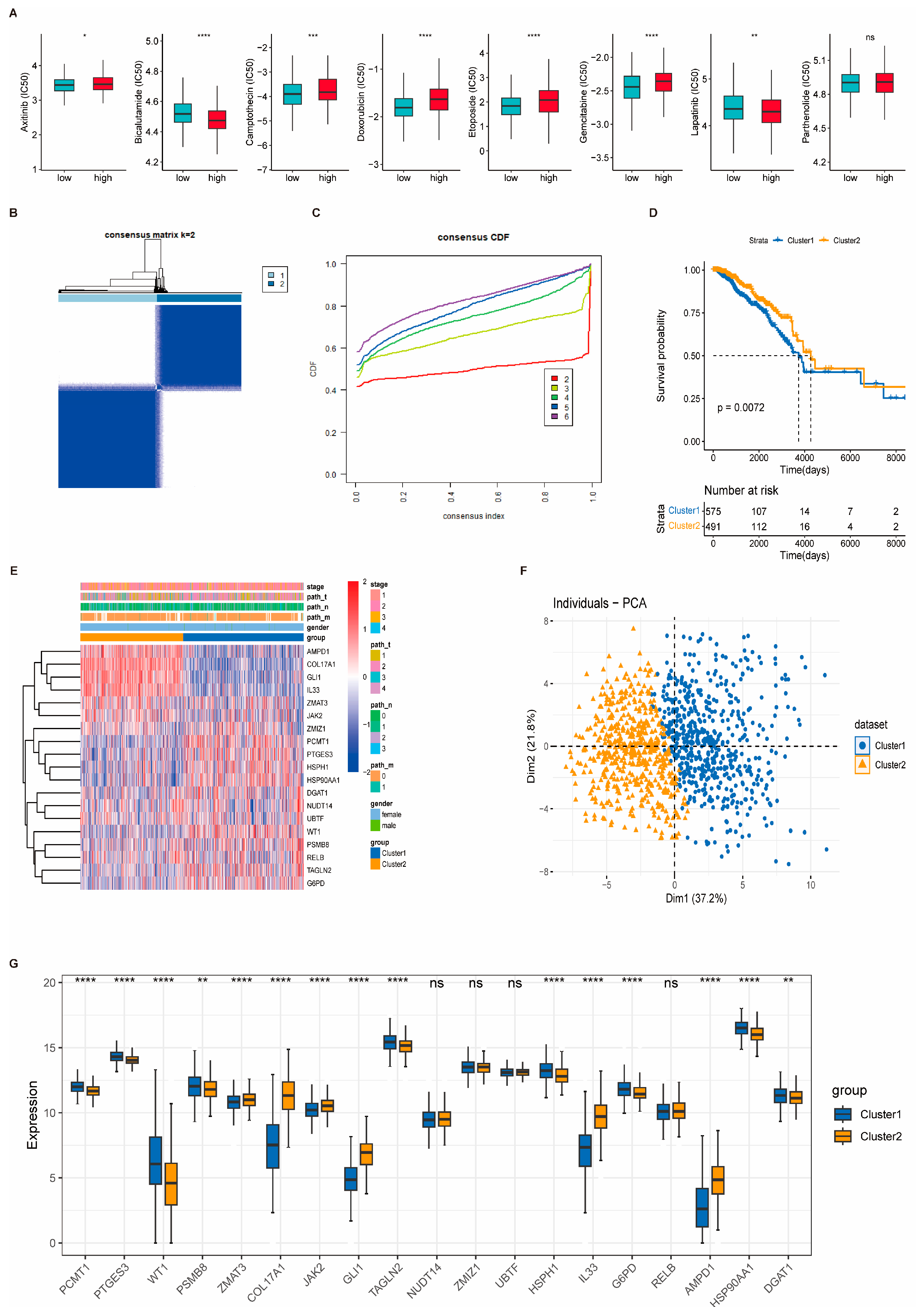
2.8. Chemotherapeutic Drug Sensitivity
2.9. Consensus Clustering Identifies Subgroups of Breast Cancer
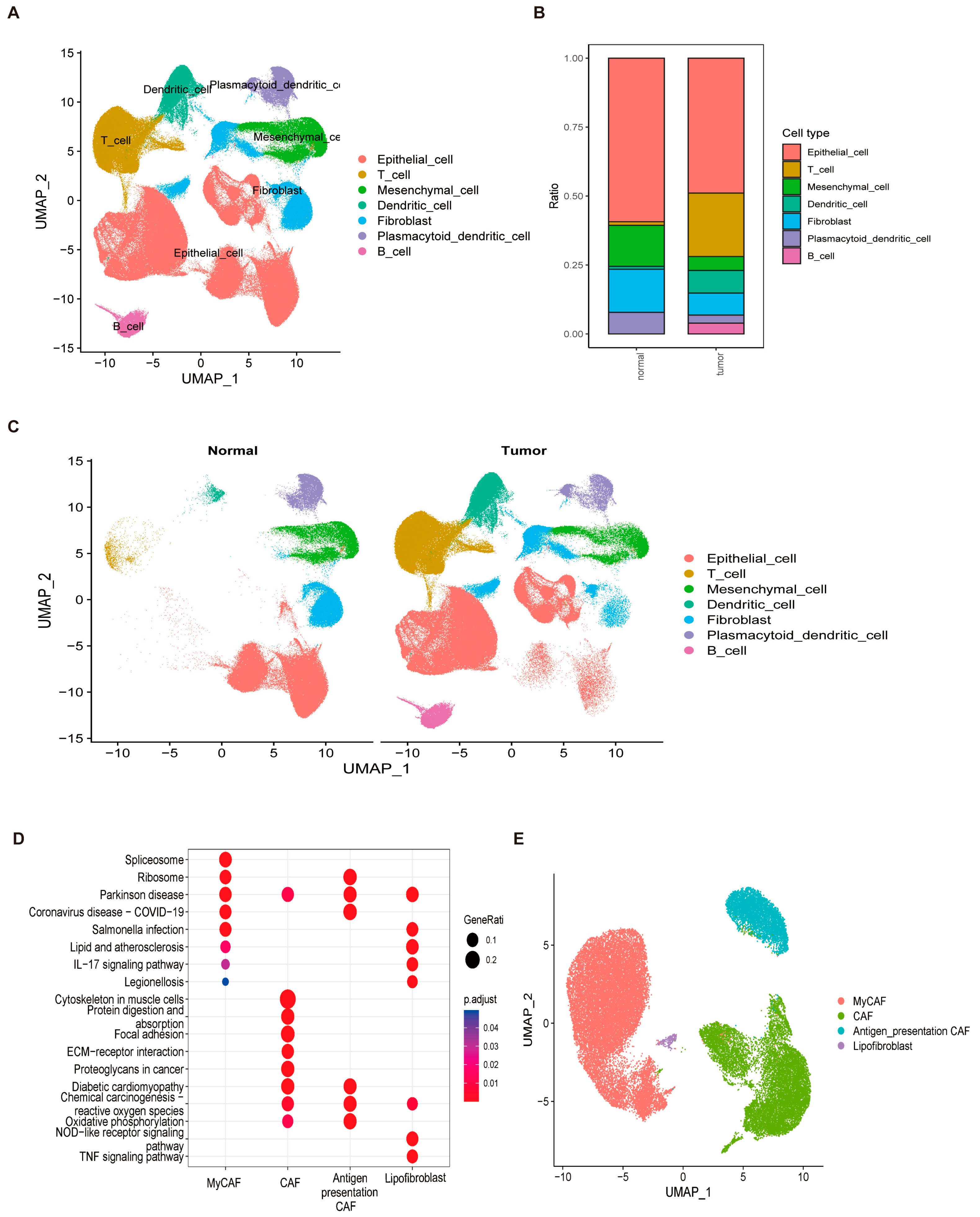
2.10. ScRNA-Seq Data Processing and Cell Subpopulation Analysis
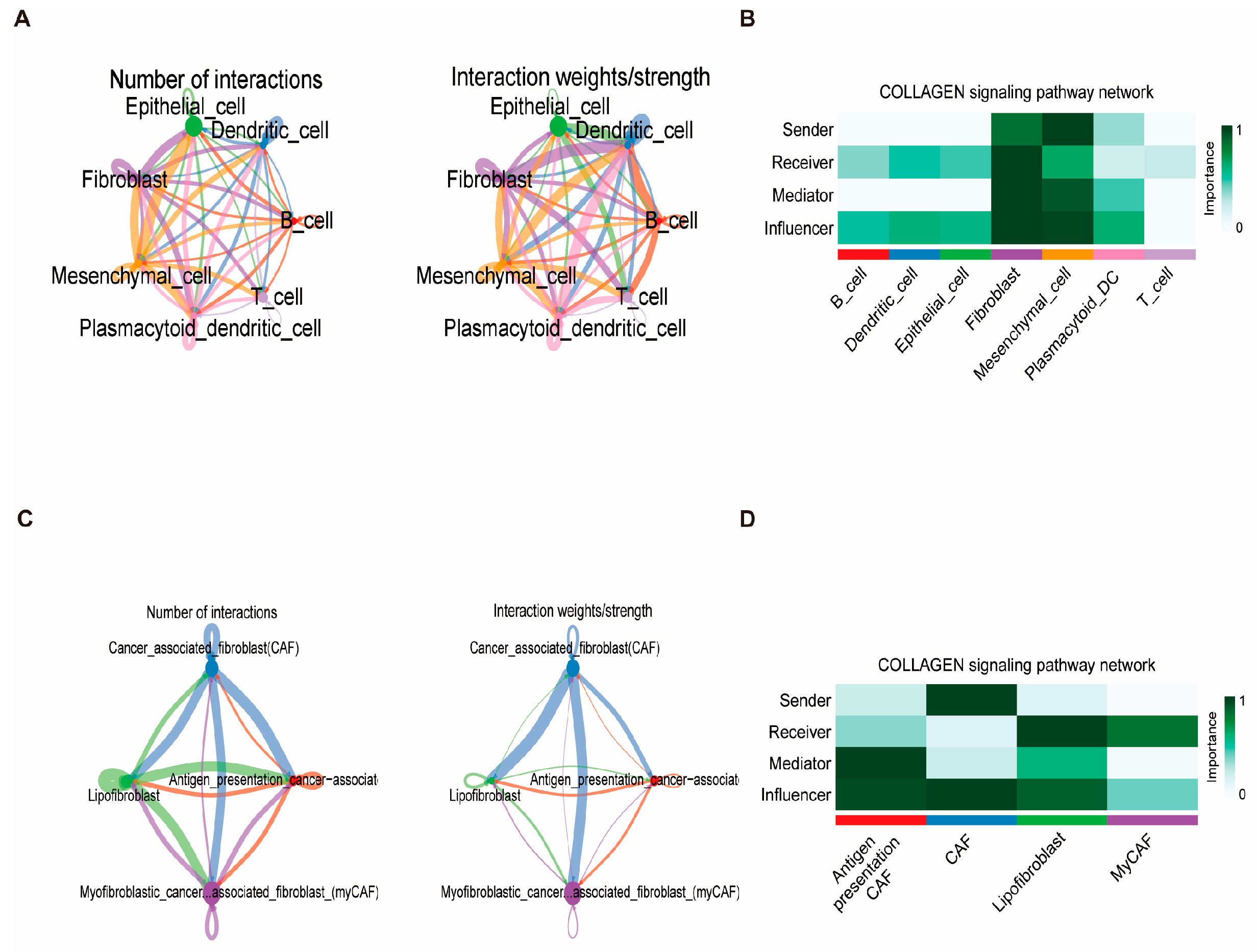
2.11. Cell Communication and Pseudo-Time Analysis Immunohistochemistry Validation via the HPA Database
2.12. Immunohistochemical Validation of Key Genes Using the Human Protein Atlas
2.13. Statistical Methods
3. Results
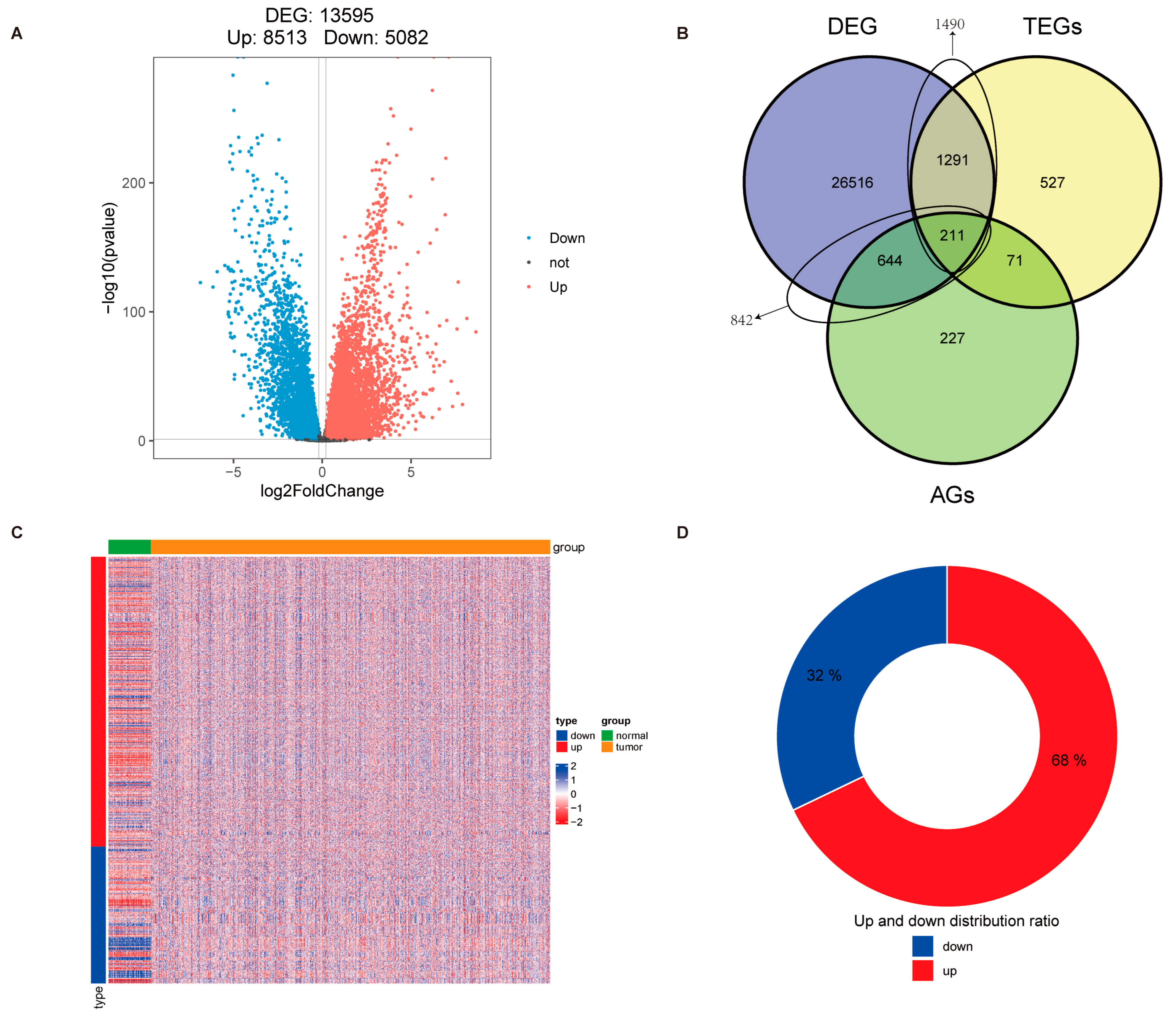
3.1. Identification of Telomere-Related Cellular Senescence Genes with Significant Differences in Breast Cancer and Enrichment Analysis of Intersection Genes
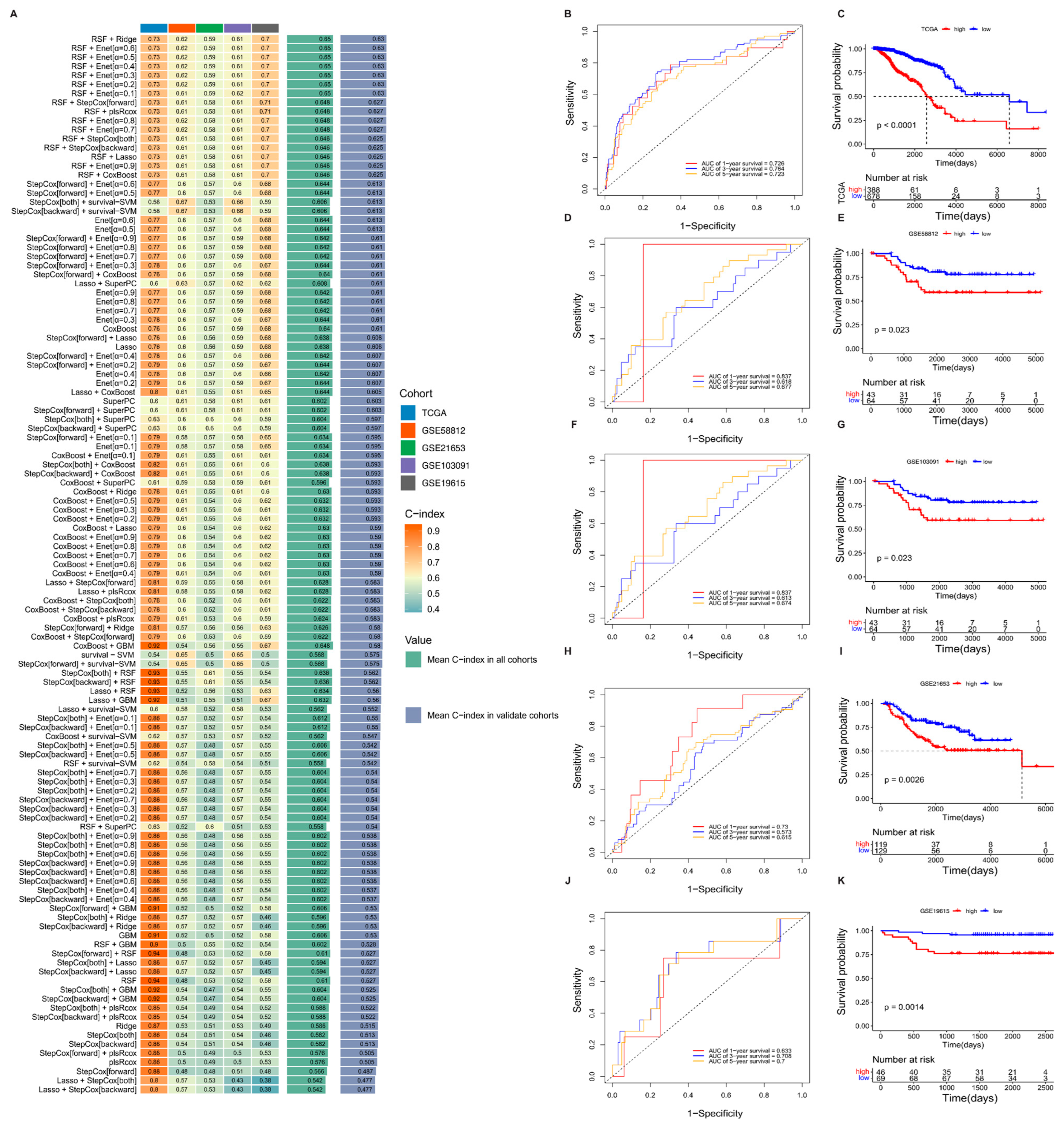
3.2. Machine Learning Development and Validation of Breast Cancer Consensus Gene Signature
3.3. Association Between Risk Score and Clinical Characteristics
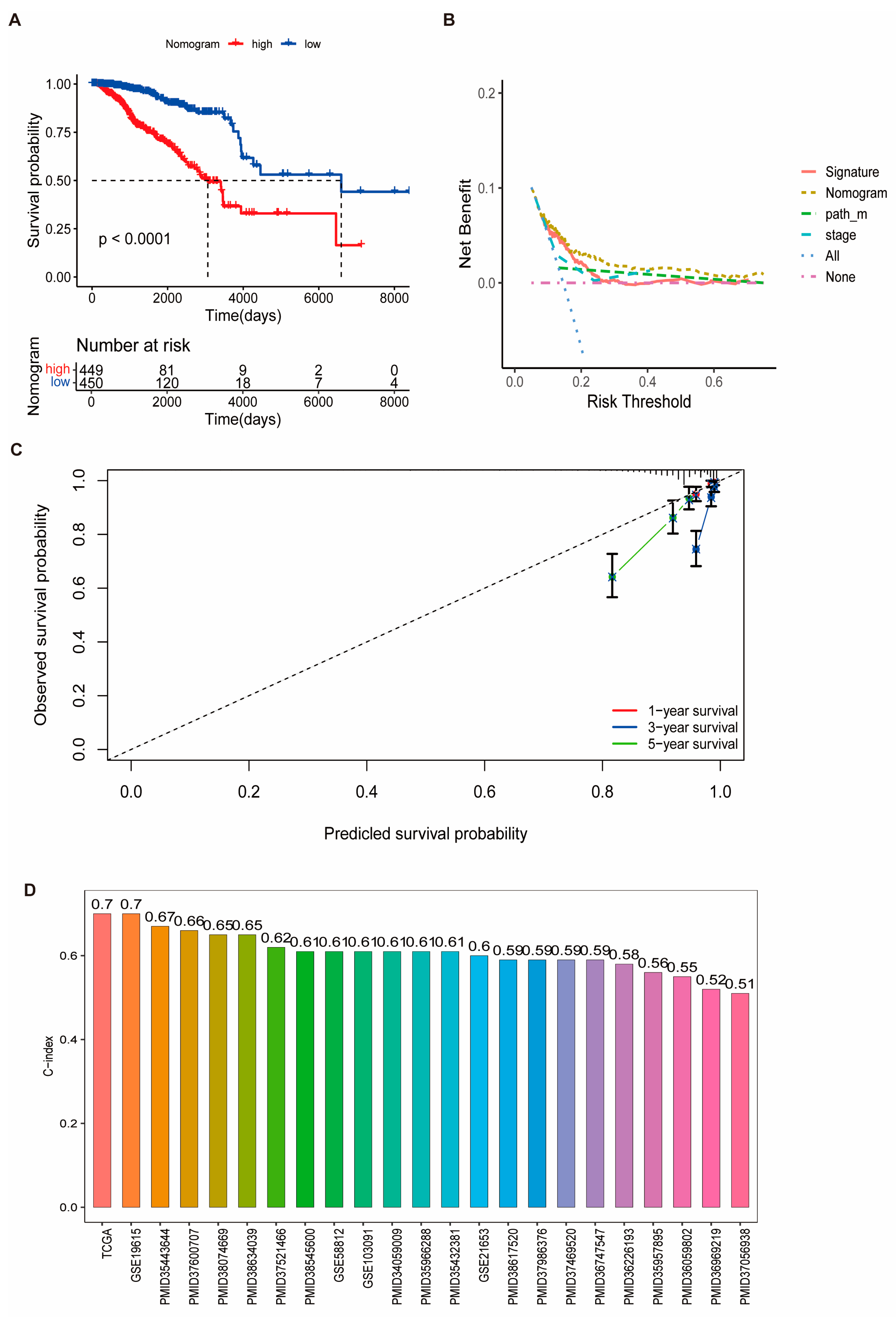
3.4. Prognostic Performance of the Risk Signature and Nomogram Construction
3.5. Immune Correlation of Risk Score Signatures as Predictive Biomarkers for Immunotherapy Response and Associated Biological Mechanisms
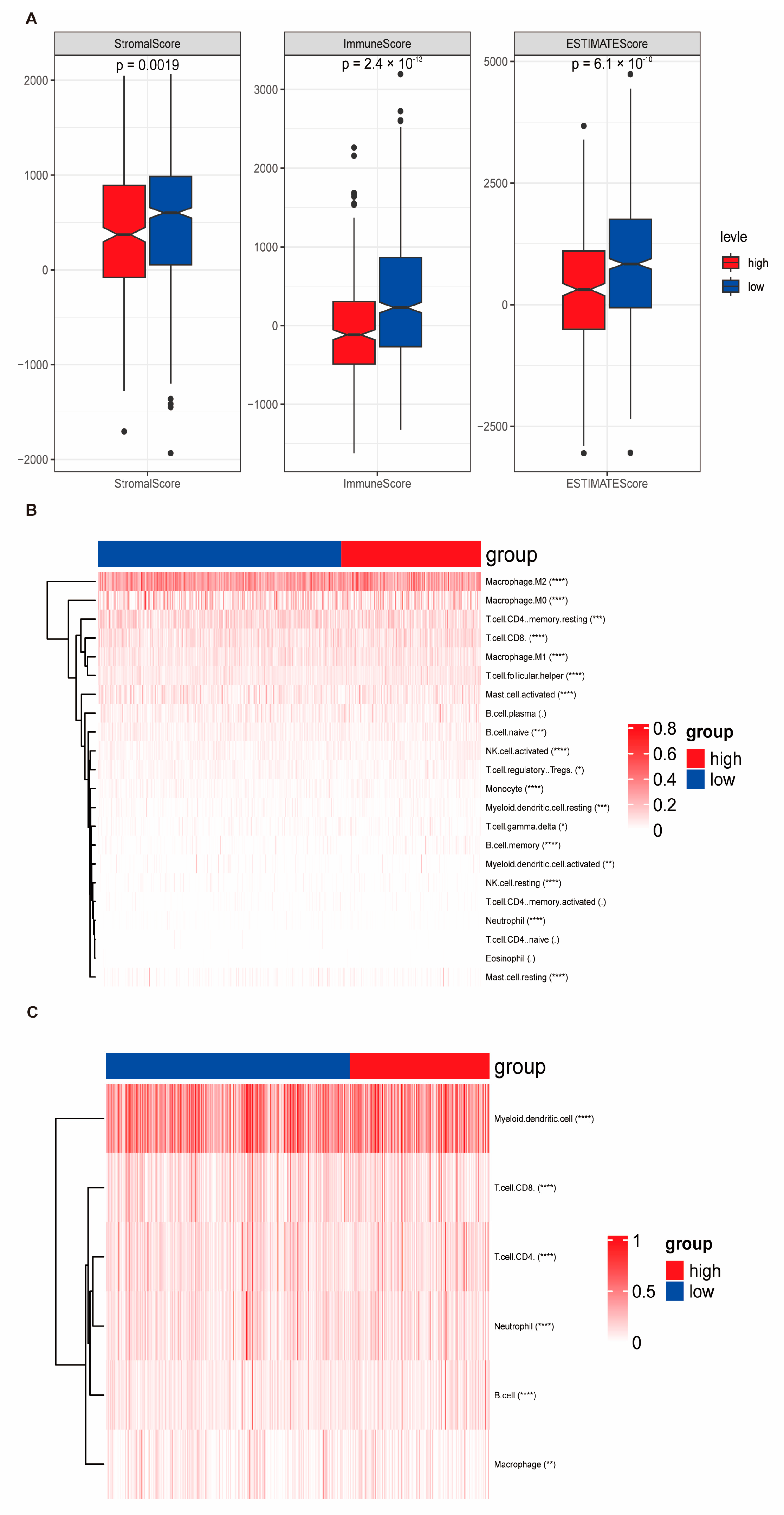
3.6. Stratification Functional Characterization of Risk Score and Immune Landscape
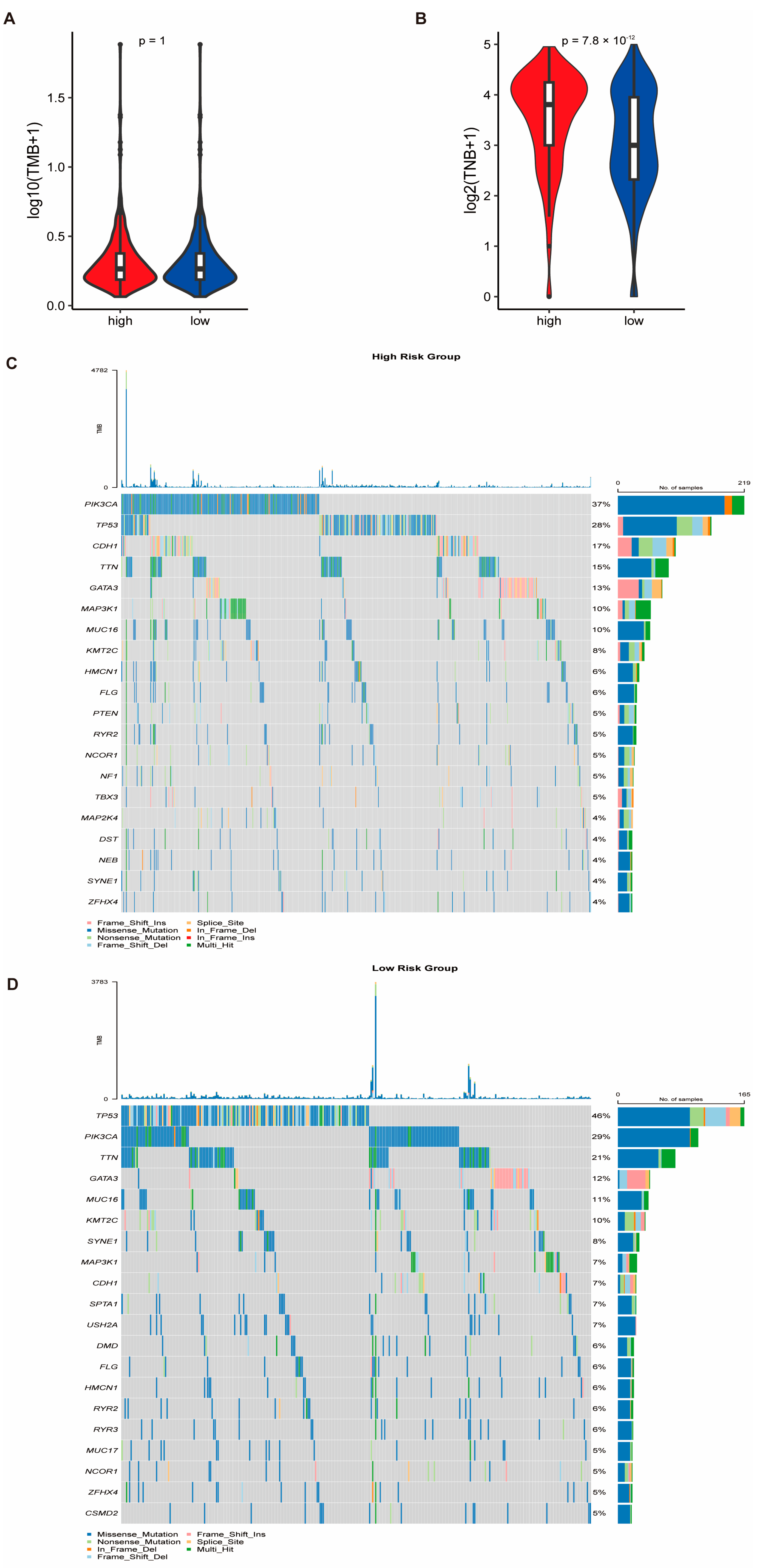
3.7. Genomic Alterations and Drug Sensitivity Prediction Based on Risk Stratification
3.8. Consensus Clustering Identifies Molecular Subtypes of Breast Cancer
3.9. Cellular Heterogeneity and Identification of Fibroblasts as a Key Cell Type in Breast Cancer
3.10. Fibroblast Differentiation Trajectory and Intercellular Communication
3.11. IHC-Based Validation of Candidate Prognostic Genes in Normal and Tumor Breast Tissues
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Howell, A.; Anderson, A.S.; Clarke, R.B.; Duffy, S.W.; Evans, D.G.; Garcia-Closas, M.; Gescher, A.J.; Key, T.J.; Saxton, J.M.; Harvie, M.N. Risk determination and prevention of breast cancer. Breast Cancer Res. 2014, 16, 446. [Google Scholar]
- Coughlin, S.S. Social determinants of breast cancer risk, stage, and survival. Breast Cancer Res Treat 2019, 177, 537–548. [Google Scholar] [PubMed]
- Benz, C.C. Impact of aging on the biology of breast cancer. Crit. Rev. Oncol. Hematol. 2008, 66, 65–74. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef]
- Blackburn, E.H. Telomere states and cell fates. Nature 2000, 408, 53–56. [Google Scholar] [CrossRef]
- Shay, J.W. Telomeres and aging. Curr. Opin. Cell Biol. 2018, 52, 1–7. [Google Scholar] [CrossRef]
- Maciejowski, J.; de Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef]
- Martinez-Delgado, B.; Gallardo, M.; Tanic, M.; Yanowsky, K.; Inglada-Perez, L.; Barroso, A.; Rodriguez-Pinilla, M.; Cañamero, M.; Blasco, M.A.; Benitez, J. Short telomeres are frequent in hereditary breast tumors and are associated with high tumor grade. Breast Cancer Res. Treat. 2013, 141, 231–242. [Google Scholar] [CrossRef]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef]
- de Lange, T. Telomeres and senescence: Ending the debate. Science 1998, 279, 334–335. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Si, J.; Li, Q.; Zhang, W.; He, J. Unlocking the potential of senescence-related gene signature as a diagnostic and prognostic biomarker in sepsis: Insights from meta-analyses, single-cell RNA sequencing, and in vitro experiments. Aging 2024, 16, 3989–4013. [Google Scholar] [CrossRef]
- Chambers, C.R.; Ritchie, S.; Pereira, B.A.; Timpson, P. Overcoming the senescence-associated secretory phenotype (SASP): A complex mechanism of resistance in the treatment of cancer. Mol. Oncol. 2021, 15, 3242–3255. [Google Scholar] [CrossRef]
- Asri, H.; Mousannif, H.; Moatassime, H.A.; Noel, T. Using Machine Learning Algorithms for Breast Cancer Risk Prediction and Diagnosis. Procedia Comput. Sci. 2016, 83, 1064–1069. [Google Scholar] [CrossRef]
- Binder, A.; Bockmayr, M.; Hägele, M.; Wienert, S.; Heim, D.; Hellweg, K.; Ishii, M.; Stenzinger, A.; Hocke, A.; Denkert, C.; et al. Morphological and molecular breast cancer profiling through explainable machine learning. Nat. Mach. Intell. 2021, 3, 355–366. [Google Scholar] [CrossRef]
- Lou, S.-J.; Hou, M.-F.; Chang, H.-T.; Chiu, C.-C.; Lee, H.-H.; Yeh, S.-C.J.; Shi, H.-Y. Machine Learning Algorithms to Predict Recurrence within 10 Years after Breast Cancer Surgery: A Prospective Cohort Study. Cancers 2020, 12, 3817. [Google Scholar] [CrossRef]
- Sammut, S.-J.; Crispin-Ortuzar, M.; Chin, S.-F.; Provenzano, E.; Bardwell, H.A.; Ma, W.; Cope, W.; Dariush, A.; Dawson, S.-J.; Abraham, J.E.; et al. Multi-omic machine learning predictor of breast cancer therapy response. Nature 2022, 601, 623–629. [Google Scholar] [CrossRef]
- Sugimoto, M.; Hikichi, S.; Takada, M.; Toi, M. Machine learning techniques for breast cancer diagnosis and treatment: A narrative review. Ann. Breast Surg. 2021, 7. [Google Scholar] [CrossRef]
- Goodin, D.A.; Chau, E.; Zheng, J.; O’Connell, C.; Tiwari, A.; Xu, Y.; Niravath, P.; Chen, S.-H.; Godin, B.; Frieboes, H.B. Characterization of the Breast Cancer Liver Metastasis Microenvironment via Machine Learning Analysis of the Primary Tumor Microenvironment. Cancer Res. Commun. 2024, 4, 2846–2857. [Google Scholar] [CrossRef]
- Guo, X.; Cao, Y.; Shi, X.; Xing, J.; Feng, C.; Wang, T. Evaluating the prognostic potential of telomerase signature in breast cancer through advanced machine learning model. Front. Immunol. 2024, 15, 1462953. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Jézéquel, P.; Loussouarn, D.; Guérin-Charbonnel, C.; Campion, L.; Vanier, A.; Gouraud, W.; Lasla, H.; Guette, C.; Valo, I.; Verrièle, V.; et al. Gene-expression molecular subtyping of triple-negative breast cancer tumours: Importance of immune response. Breast Cancer Res. 2015, 17, 43. [Google Scholar] [CrossRef] [PubMed]
- Sabatier, R.; Finetti, P.; Adelaide, J.; Guille, A.; Borg, J.P.; Chaffanet, M.; Lane, L.; Birnbaum, D.; Bertucci, F. Down-regulation of ECRG4, a candidate tumor suppressor gene, in human breast cancer. PLoS ONE 2011, 6, e27656. [Google Scholar] [CrossRef]
- Jézéquel, P.; Kerdraon, O.; Hondermarck, H.; Guérin-Charbonnel, C.; Lasla, H.; Gouraud, W.; Canon, J.L.; Gombos, A.; Dalenc, F.; Delaloge, S.; et al. Identification of three subtypes of triple-negative breast cancer with potential therapeutic implications. Breast Cancer Res. 2019, 21, 65. [Google Scholar] [CrossRef]
- Li, Y.; Zou, L.; Li, Q.; Haibe-Kains, B.; Tian, R.; Li, Y.; Desmedt, C.; Sotiriou, C.; Szallasi, Z.; Iglehart, J.D.; et al. Amplification of LAPTM4B and YWHAZ contributes to chemotherapy resistance and recurrence of breast cancer. Nat. Med. 2010, 16, 214–218. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Liu, D.; Qin, Z.; Yi, B.; Xie, H.; Liang, Y.; Zhu, L.; Yang, K.; Xu, Y.; Zhang, H. Telomerase related molecular subtype and risk model reveal immune activity and evaluate prognosis and immunotherapy response in prostate cancer. Cancer Cell Int. 2024, 24, 294. [Google Scholar] [CrossRef]
- Lv, Y.; Wu, L.; Jian, H.; Zhang, C.; Lou, Y.; Kang, Y.; Hou, M.; Li, Z.; Li, X.; Sun, B.; et al. Identification and characterization of aging/senescence-induced genes in osteosarcoma and predicting clinical prognosis. Front. Immunol. 2022, 13, 997765. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Liang, J.Y.; Wang, D.S.; Lin, H.C.; Chen, X.X.; Yang, H.; Zheng, Y.; Li, Y.H. A Novel Ferroptosis-related Gene Signature for Overall Survival Prediction in Patients with Hepatocellular Carcinoma. Int. J. Biol. Sci. 2020, 16, 2430–2441. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhang, H.; Wu, W.; Zhou, R.; Li, S.; Wang, Z.; Dai, Z.; Zhang, L.; Liu, F.; Liu, Z.; et al. Machine learning-based identification of tumor-infiltrating immune cell-associated lncRNAs for improving outcomes and immunotherapy responses in patients with low-grade glioma. Theranostics 2022, 12, 5931–5948. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, L.; Weng, S.; Guo, C.; Dang, Q.; Xu, H.; Wang, L.; Lu, T.; Zhang, Y.; Sun, Z.; et al. Machine learning-based integration develops an immune-derived lncRNA signature for improving outcomes in colorectal cancer. Nat. Commun. 2022, 13, 816. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Yu, S.-L.; Chen, C.-H.; Chang, G.-C.; Chen, C.-Y.; Yuan, A.; Cheng, C.-L.; Wang, C.-H.; Terng, H.-J.; Kao, S.-F.; et al. A Five-Gene Signature and Clinical Outcome in Non–Small-Cell Lung Cancer. New Engl. J. Med. 2007, 356, 11–20. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e814. [Google Scholar] [CrossRef]
- Huang, R.-H.; Hong, Y.-K.; Du, H.; Ke, W.-Q.; Lin, B.-B.; Li, Y.-L. A machine learning framework develops a DNA replication stress model for predicting clinical outcomes and therapeutic vulnerability in primary prostate cancer. J. Transl. Med. 2023, 21, 20. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Yang, K.; Dinasarapu, A.R.; Reis, E.S.; Deangelis, R.A.; Ricklin, D.; Subramaniam, S.; Lambris, J.D. CMAP: Complement Map Database. Bioinformatics 2013, 29, 1832–1833. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, Z. Bioinformatic Analysis for Potential Biomarkers and Therapeutic Targets of T2DM-related MI. Int. J. Gen. Med. 2021, 14, 4337–4347. [Google Scholar] [CrossRef]
- Geeleher, P.; Cox, N.; Huang, R.S. pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 2014, 9, e107468. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e3529. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Li, T.; Xu, Y.; Zhang, X.; Li, F.; Bai, J.; Chen, J.; Jiang, W.; Yang, K.; Ou, Q.; et al. CellMarker 2.0: An updated database of manually curated cell markers in human/mouse and web tools based on scRNA-seq data. Nucleic Acids Res. 2023, 51, D870–D876. [Google Scholar] [CrossRef]
- Franzén, O.; Gan, L.M.; Björkegren, J.L.M. PanglaoDB: A web server for exploration of mouse and human single-cell RNA sequencing data. Database 2019, 2019, baz046. [Google Scholar] [CrossRef]
- Luo, J.; Deng, M.; Zhang, X.; Sun, X. ESICCC as a systematic computational framework for evaluation, selection, and integration of cell-cell communication inference methods. Genome Res. 2023, 33, 1788–1805. [Google Scholar] [CrossRef]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef]
- Alabdallah, A.; Ohlsson, M.; Pashami, S.; Rögnvaldsson, T. The Concordance Index decomposition: A measure for a deeper understanding of survival prediction models. Artif. Intell. Med. 2024, 148, 102781. [Google Scholar]
- Henderson, I.C.; Patek, A.J. The relationship between prognostic and predictive factors in the management of breast cancer. Breast Cancer Res. Treat. 1998, 52, 261–288. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Paik, S.; Shak, S.; Tang, G.; Kim, C.; Baker, J.; Cronin, M.; Baehner, F.L.; Walker, M.G.; Watson, D.; Park, T.; et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med. 2004, 351, 2817–2826. [Google Scholar] [CrossRef]
- Sparano, J.A.; Paik, S. Development of the 21-gene assay and its application in clinical practice and clinical trials. J. Clin. Oncol. 2008, 26, 721–728. [Google Scholar] [CrossRef]
- Lashen, A.; Toss, M.S.; Fadhil, W.; Oni, G.; Madhusudan, S.; Rakha, E. Evaluation oncotype DX(®) 21-gene recurrence score and clinicopathological parameters: A single institutional experience. Histopathology 2023, 82, 755–766. [Google Scholar] [CrossRef]
- Saha, A.; Harowicz, M.R.; Grimm, L.J.; Kim, C.E.; Ghate, S.V.; Walsh, R.; Mazurowski, M.A. A machine learning approach to radiogenomics of breast cancer: A study of 922 subjects and 529 DCE-MRI features. Br. J. Cancer 2018, 119, 508–516. [Google Scholar] [CrossRef]
- Hatzis, C.; Pusztai, L.; Valero, V.; Booser, D.J.; Esserman, L.; Lluch, A.; Vidaurre, T.; Holmes, F.; Souchon, E.; Wang, H.; et al. A Genomic Predictor of Response and Survival Following Taxane-Anthracycline Chemotherapy for Invasive Breast Cancer. JAMA 2011, 305, 1873–1881. [Google Scholar] [CrossRef]
- Kourou, K.; Exarchos, T.P.; Exarchos, K.P.; Karamouzis, M.V.; Fotiadis, D.I. Machine learning applications in cancer prognosis and prediction. Comput. Struct. Biotechnol. J. 2015, 13, 8–17. [Google Scholar]
- Ma, S.; Kosorok, M.R. Detection of gene pathways with predictive power for breast cancer prognosis. BMC Bioinform. 2010, 11, 1. [Google Scholar] [CrossRef]
- Hernández Borrero, L.J.; El-Deiry, W.S. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2021, 1876, 188556. [Google Scholar]
- Gasco, M.; Shami, S.; Crook, T. The p53 pathway in breast cancer. Breast Cancer Res. 2002, 4, 70. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Wu, X.; Chen, S.; Lu, C. Amyloid precursor protein promotes the migration and invasion of breast cancer cells by regulating the MAPK signaling pathway. Int. J. Mol. Med. 2020, 45, 162–174. [Google Scholar] [CrossRef]
- Wu, G.S. The functional Interactions Between the MAPK and p53 Signaling Pathways. Cancer Biol. Ther. 2004, 3, 156–161. [Google Scholar] [CrossRef]
- Cui, S.; Feng, J.; Tang, X.; Lou, S.; Guo, W.; Xiao, X.; Li, S.; Chen, X.; Huan, Y.; Zhou, Y.; et al. The prognostic value of tumor mutation burden (TMB) and its relationship with immune infiltration in breast cancer patients. Eur. J. Med. Res. 2023, 28, 90. [Google Scholar] [CrossRef]
- Silwal-Pandit, L.; Langerød, A.; Børresen-Dale, A.L. TP53 Mutations in Breast and Ovarian Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026252. [Google Scholar] [CrossRef]
- Silwal-Pandit, L.; Vollan, H.K.; Chin, S.F.; Rueda, O.M.; McKinney, S.; Osako, T.; Quigley, D.A.; Kristensen, V.N.; Aparicio, S.; Børresen-Dale, A.L.; et al. TP53 mutation spectrum in breast cancer is subtype specific and has distinct prognostic relevance. Clin. Cancer Res. 2014, 20, 3569–3580. [Google Scholar] [CrossRef]
- Børresen-Dale, A.L. TP53 and breast cancer. Hum. Mutat. 2003, 21, 292–300. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Chen, L.; Yang, L.; Yao, L.; Kuang, X.Y.; Zuo, W.J.; Li, S.; Qiao, F.; Liu, Y.R.; Cao, Z.G.; Zhou, S.L.; et al. Characterization of PIK3CA and PIK3R1 somatic mutations in Chinese breast cancer patients. Nat. Commun. 2018, 9, 1357. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Dotto, G.P.; Weinberg, R.A.; Ariza, A. Malignant transformation of mouse primary keratinocytes by Harvey sarcoma virus and its modulation by surrounding normal cells. Proc. Natl. Acad. Sci. USA 1988, 85, 6389–6393. [Google Scholar] [CrossRef]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes. Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef]
- Gentric, G.; Mieulet, V.; Mechta-Grigoriou, F. Heterogeneity in Cancer Metabolism: New Concepts in an Old Field. Antioxid. Redox Signal 2017, 26, 462–485. [Google Scholar] [CrossRef]
- Costa, A.; Scholer-Dahirel, A.; Mechta-Grigoriou, F. The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin. Cancer Biol. 2014, 25, 23–32. [Google Scholar] [CrossRef]
- Benyahia, Z.; Dussault, N.; Cayol, M.; Sigaud, R.; Berenguer-Daizé, C.; Delfino, C.; Tounsi, A.; Garcia, S.; Martin, P.M.; Mabrouk, K.; et al. Stromal fibroblasts present in breast carcinomas promote tumor growth and angiogenesis through adrenomedullin secretion. Oncotarget 2017, 8, 15744–15762. [Google Scholar] [CrossRef]
- Toullec, A.; Gerald, D.; Despouy, G.; Bourachot, B.; Cardon, M.; Lefort, S.; Richardson, M.; Rigaill, G.; Parrini, M.C.; Lucchesi, C.; et al. Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Mol. Med. 2010, 2, 211–230. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e410. [Google Scholar] [CrossRef]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef]
- Paulsson, J.; Rydén, L.; Strell, C.; Frings, O.; Tobin, N.P.; Fornander, T.; Bergh, J.; Landberg, G.; Stål, O.; Östman, A. High expression of stromal PDGFRβ is associated with reduced benefit of tamoxifen in breast cancer. J. Pathol. Clin. Res. 2017, 3, 38–43. [Google Scholar] [CrossRef]
- Ruhland, M.K.; Loza, A.J.; Capietto, A.H.; Luo, X.; Knolhoff, B.L.; Flanagan, K.C.; Belt, B.A.; Alspach, E.; Leahy, K.; Luo, J.; et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Commun. 2016, 7, 11762. [Google Scholar] [CrossRef]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010, 330, 827–830. [Google Scholar] [CrossRef]
- Frydman, J. Folding of newly translated proteins in vivo: The role of molecular chaperones. Annu. Rev. Biochem. 2001, 70, 603–647. [Google Scholar] [CrossRef]
- Crouch, B.T.; Gallagher, J.; Wang, R.; Duer, J.; Hall, A.; Soo, M.S.; Hughes, P.; Haystead, T.; Ramanujam, N. Exploiting heat shock protein expression to develop a non-invasive diagnostic tool for breast cancer. Sci. Rep. 2019, 9, 3461. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Z.; Huang, Y.; Wei, W.; Ning, S.; Li, J.; Liang, X.; Liu, K.; Zhang, L. Plasma HSP90AA1 Predicts the Risk of Breast Cancer Onset and Distant Metastasis. Front. Cell Dev. Biol. 2021, 9, 639596. [Google Scholar] [CrossRef]
- Shapland, C.; Hsuan, J.J.; Totty, N.F.; Lawson, D. Purification and properties of transgelin: A transformation and shape change sensitive actin-gelling protein. J. Cell Biol. 1993, 121, 1065–1073. [Google Scholar] [CrossRef]
- Yang, L.; Hong, Q.; Xu, S.G.; Kuang, X.Y.; Di, G.H.; Liu, G.Y.; Wu, J.; Shao, Z.M.; Yu, S.J. Downregulation of transgelin 2 promotes breast cancer metastasis by activating the reactive oxygen species/nuclear factor-κB signaling pathway. Mol. Med. Rep. 2019, 20, 4045–4258. [Google Scholar] [CrossRef]
- Yang, D.; Wang, B.; Li, Y.; Zhang, J.; Gong, X.; Qin, H.; Wang, Y.; Zhao, Y.; Wang, Y. HER-2 Expression in Colorectal Cancer and Its Correlation with Immune Cell Infiltration. Biomedicines 2023, 11, 2889. [Google Scholar] [CrossRef]
- Singer, C.F.; Gschwantler-Kaulich, D.; Fink-Retter, A.; Haas, C.; Hudelist, G.; Czerwenka, K.; Kubista, E. Differential gene expression profile in breast cancer-derived stromal fibroblasts. Breast Cancer Res. Treat. 2008, 110, 273–281. [Google Scholar] [CrossRef]
- Finak, G.; Sadekova, S.; Pepin, F.; Hallett, M.; Meterissian, S.; Halwani, F.; Khetani, K.; Souleimanova, M.; Zabolotny, B.; Omeroglu, A.; et al. Gene expression signatures of morphologically normal breast tissue identify basal-like tumors. Breast Cancer Res. 2006, 8, R58. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, X.; Ning, J. Evaluating dynamic and predictive discrimination for recurrent event models: Use of a time-dependent C-index. Biostatistics 2024, 25, 1140–1155. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Liu, H. Investigating the Prognostic Role of Telomerase-Related Cellular Senescence Gene Signatures in Breast Cancer Using Machine Learning. Biomedicines 2025, 13, 826. https://doi.org/10.3390/biomedicines13040826
Li Q, Liu H. Investigating the Prognostic Role of Telomerase-Related Cellular Senescence Gene Signatures in Breast Cancer Using Machine Learning. Biomedicines. 2025; 13(4):826. https://doi.org/10.3390/biomedicines13040826
Chicago/Turabian StyleLi, Qiong, and Hongde Liu. 2025. "Investigating the Prognostic Role of Telomerase-Related Cellular Senescence Gene Signatures in Breast Cancer Using Machine Learning" Biomedicines 13, no. 4: 826. https://doi.org/10.3390/biomedicines13040826
APA StyleLi, Q., & Liu, H. (2025). Investigating the Prognostic Role of Telomerase-Related Cellular Senescence Gene Signatures in Breast Cancer Using Machine Learning. Biomedicines, 13(4), 826. https://doi.org/10.3390/biomedicines13040826