
Lung Microenvironment Among Patients with Nontuberculous Mycobacterial Pulmonary Disease by Metagenomic Sequencing Technique

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Subjects
2.2. Metagenomic Sequencing
2.3. Immunological Phenotyping and Cytokine Analysis
2.4. Data Analysis
3. Results
3.1. Clinical Characteristics of Participants
3.2. Microbiome Diversity Reduction Associated with Immunosuppression
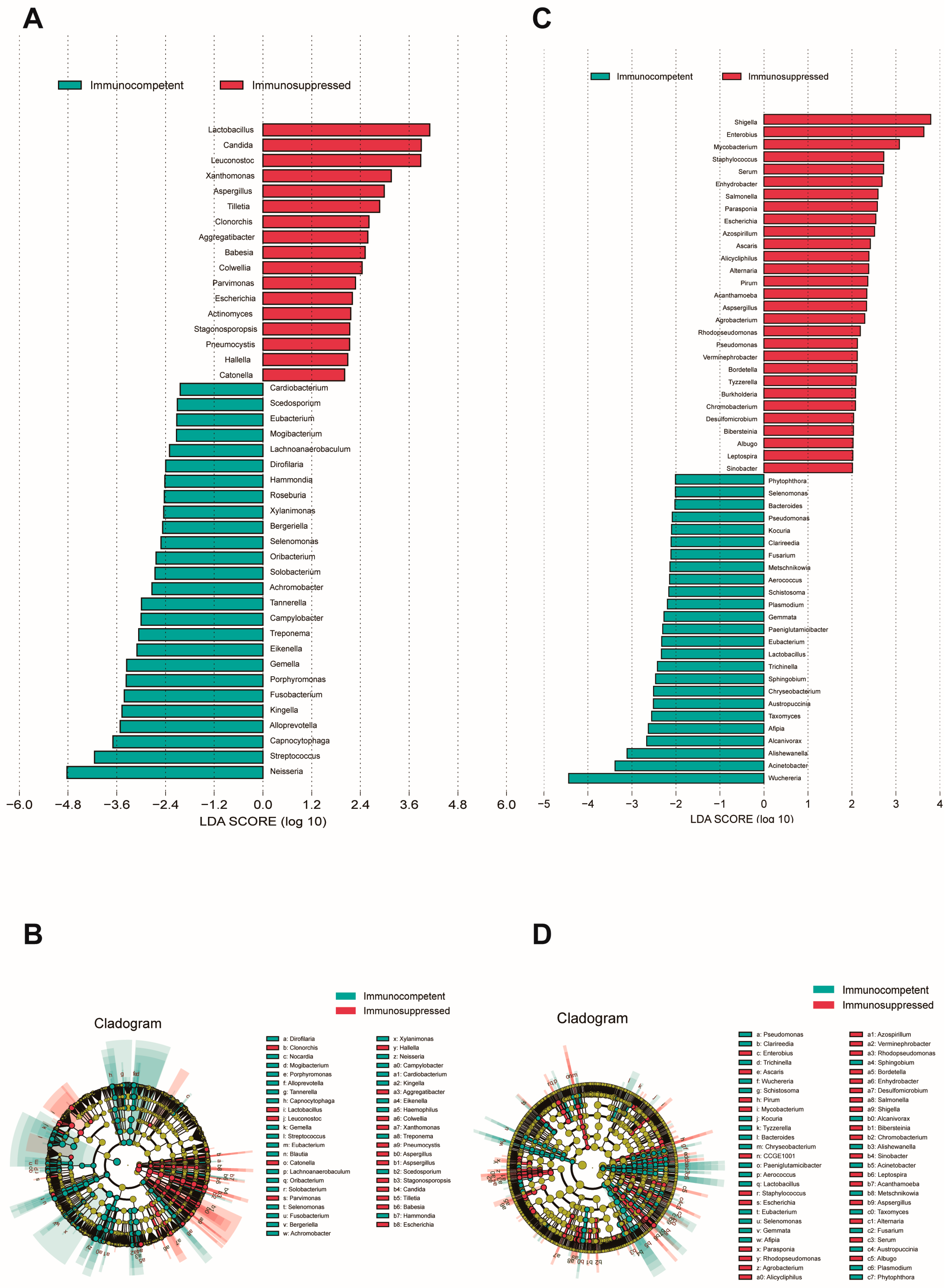
3.3. Differential Microbial Analysis Associated with Immunosuppression
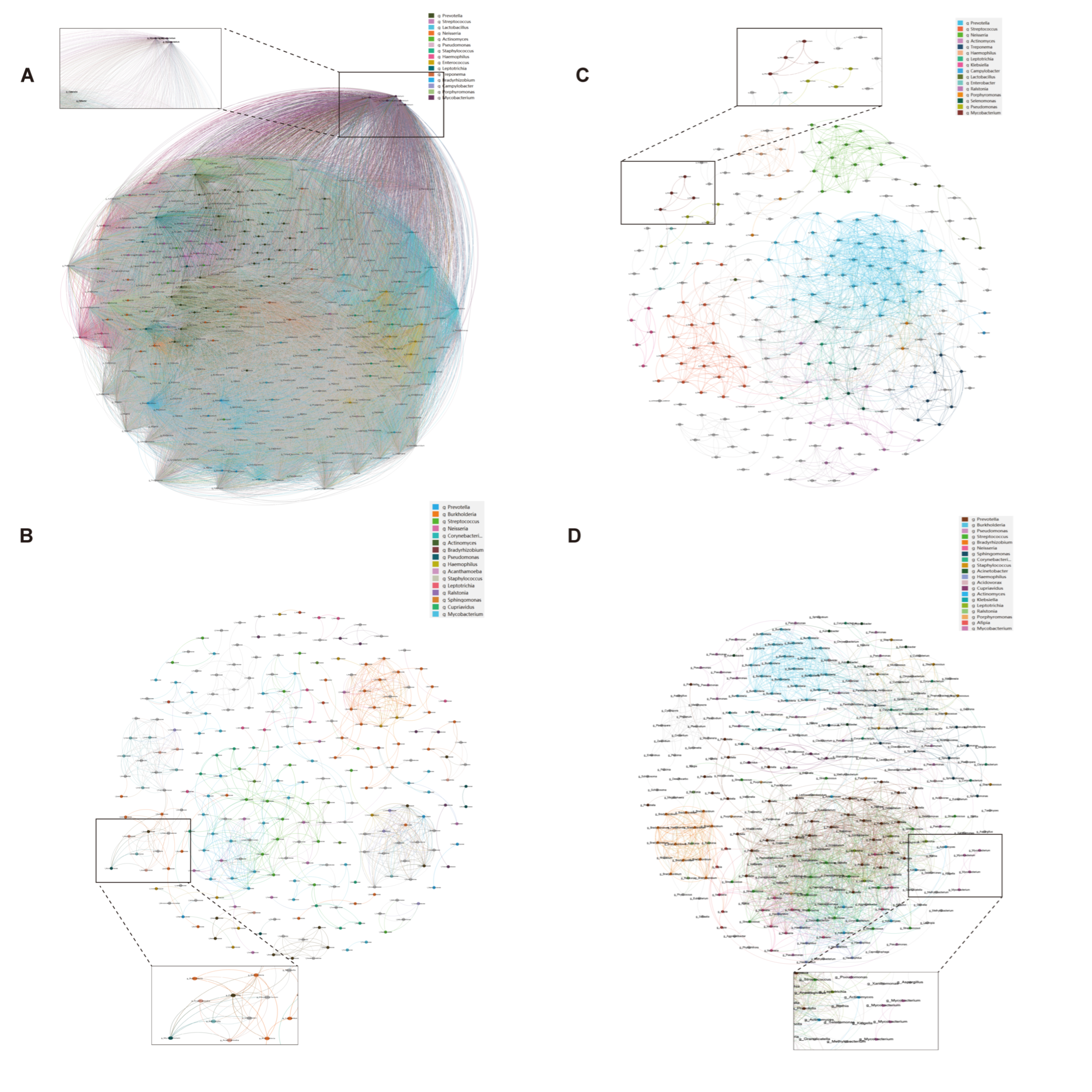
3.4. Tighter Co-Occurrence Network Structure Associated with Immunosuppression
3.5. Association Between Microbial Community and Host Immune Status
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NTM-PD | Nontuberculous mycobacterial pulmonary disease |
| BALF | Bronchoalveolar lavage fluid |
| PCoA | Principal Coordinates Analysis |
| LEfSe | Linear discriminant analysis Effect Size |
| NTM | Nontuberculosis Mycobacterium |
| NGS | Next-Generation Sequencing |
| ANCOVA | Analysis of Covariance |
| BH | Benjamini–Hochberg |
| COPD | Chronic Obstructive Pulmonary Disease |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Immunosuppressed | Immunocompetent | p-Value |
|---|---|---|---|
| BALF specimens | |||
| Nodes | 250 | 311 | 0.028 * |
| Edges | 825 | 2122 | <0.001 *** |
| Positive correlations | 824 | 2122 | <0.001 *** |
| Negative correlations | 1 | 0 | 0.317 |
| Average degree | 6.60 | 13.65 | <0.001 *** |
| Average path length | 2.89 | 2.83 | 0.245 |
| Network diameter | 8.06 | 9.51 | 0.032 * |
| Network density | 0.027 | 0.044 | 0.015 * |
| Clustering coefficient | 0.791 | 0.476 | <0.001 *** |
| Sputum specimens | |||
| Nodes | 354 | 243 | 0.008 ** |
| Edges | 62449 | 1375 | <0.001 *** |
| Positive correlations | 41403 | 1375 | <0.001 *** |
| Negative correlations | 21046 | 0 | <0.001 *** |
| Average degree | 352.82 | 11.32 | <0.001 *** |
| Average path length | 0.007 | 1.77 | <0.001 *** |
| Network diameter | 0.018 | 4.49 | <0.001 *** |
References
- Nguyen, M.-V.H.; Haas, M.K.; Kasperbauer, S.H.; Calado Nogueira de Moura, V.; Eddy, J.J.; Mitchell, J.D.; Khare, R.; Griffith, D.E.; Chan, E.D.; Daley, C.L. Nontuberculous Mycobacterial Pulmonary Disease: Patients, Principles, and Prospects. Clin. Infect. Dis. 2024, 79, e27–e47. [Google Scholar]
- Piersimoni, C.; Scarparo, C. Pulmonary infections associated with non-tuberculous mycobacteria in immunocompetent patients. Lancet Infect. Dis. 2008, 8, 323–334. [Google Scholar] [PubMed]
- Turenne, C.Y. Nontuberculous mycobacteria: Insights on taxonomy and evolution. Infect. Genet. Evol. 2019, 72, 159–168. [Google Scholar] [PubMed]
- Iseman, M.D.; Marras, T.K. The importance of nontuberculous mycobacterial lung disease. Am. J. Respir. Crit. Care Med. 2008, 178, 999–1000. [Google Scholar] [PubMed]
- Lin, C.; Russell, C.; Soll, B.; Chow, D.; Bamrah, S.; Brostrom, R.; Kim, W.; Scott, J.; Bankowski, M.J. Increasing Prevalence of Nontuberculous Mycobacteria in Respiratory Specimens from US-Affiliated Pacific Island Jurisdictions. Emerg. Infect. Dis. 2018, 24, 485–491. [Google Scholar] [PubMed]
- Li, R.; Li, J.; Zhou, X. Lung microbiome: New insights into the pathogenesis of respiratory diseases. Signal Transduct. Target. Ther. 2024, 9, 19. [Google Scholar]
- Al-Houqani, M.; Jamieson, F.; Mehta, M.; Chedore, P.; May, K.; Marras, T.K. Aging, COPD, and other risk factors do not explain the increased prevalence of pulmonary Mycobacterium avium complex in Ontario. Chest 2012, 141, 190–197. [Google Scholar] [CrossRef]
- Whiteside, S.A.; McGinniss, J.E.; Collman, R.G. The lung microbiome: Progress and promise. J. Clin. Investig. 2021, 131, e150473. [Google Scholar]
- Rastogi, S.; Briken, V. Interaction of Mycobacteria with Host Cell Inflammasomes. Front. Immunol. 2022, 13, 791136. [Google Scholar]
- Prasla, Z.; Sutliff, R.L.; Sadikot, R.T. Macrophage Signaling Pathways in Pulmonary Nontuberculous Mycobacteria Infections. Am. J. Respir. Cell Mol. Biol. 2020, 63, 144–151. [Google Scholar] [CrossRef]
- Kartalija, M.; Ovrutsky, A.R.; Bryan, C.L.; Pott, G.B.; Fantuzzi, G.; Thomas, J.; Strand, M.J.; Bai, X.; Ramamoorthy, P.; Rothman, M.S.; et al. Patients with nontuberculous mycobacterial lung disease exhibit unique body and immune phenotypes. Am. J. Respir. Crit. Care Med. 2013, 187, 197–205. [Google Scholar] [PubMed]
- Sulaiman, I.; Wu, B.G.; Li, Y.; Scott, A.S.; Malecha, P.; Scaglione, B.; Wang, J.; Basavaraj, A.; Chung, S.; Bantis, K.; et al. Evaluation of the airway microbiome in nontuberculous mycobacteria disease. Eur. Respir. J. 2018, 52, e1800810. [Google Scholar]
- Chai, J.; Han, X.; Mei, Q.; Liu, T.; Walline, J.H.; Xu, J.; Liu, Y.; Zhu, H. Clinical Characteristics and Mortality of Non-tuberculous Mycobacterial Infection in Immunocompromised vs. Immunocompetent Hosts. Front. Med. 2022, 9, e884446. [Google Scholar]
- Griffith, D.E.; Aksamit, T.; Brown-Elliott, B.A.; Catanzaro, A.; Daley, C.; Gordin, F.; Holland, S.M.; Horsburgh, R.; Huitt, G.; Iademarco, M.F.; et al. An official ATS/IDSA statement: Diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am. J. Respir. Crit. Care Med. 2007, 175, 367–416. [Google Scholar] [PubMed]
- Daley, C.L.; Iaccarino, J.M.; Lange, C. Treatment of Nontuberculous Mycobacterial Pulmonary Disease: An Official ATS/ERS/ESCMID/IDSA Clinical Practice Guideline. Clin. Infect. Dis. 2020, 71, e1–e36. [Google Scholar]
- Azoulay, E.; Russell, L.; Van de Louw, A.; Metaxa, V.; Bauer, P.; Povoa, P.; Montero, J.G.; Loeches, I.M.; Mehta, S.; Puxty, K.; et al. Diagnosis of severe respiratory infections in immunocompromised patients. Intensive Care Med. 2020, 46, 298–314. [Google Scholar]
- Marušić, A.; Katalinić-Janković, V.; Popović-Grle, S.; Janković, M.; Mažuranić, I.; Puljić, I.; Milić, H.S. Mycobacterium xenopi pulmonary disease—Epidemiology and clinical features in non-immunocompromised patients. J. Infect. 2009, 58, 108–112. [Google Scholar]
- Liu, Q.; Du, J.; An, H.; Li, X.; Guo, D.; Li, J.; Gong, W.; Liang, J. Clinical characteristics of patients with non-tuberculous mycobacterial pulmonary disease: A seven-year follow-up study conducted in a certain tertiary hospital in Beijing. Front. Cell Infect. Microbiol. 2023, 13, 1205225. [Google Scholar]
- Rofael, S.A.; Brown, J.; Lipman, M.C.; Lowe, D.M.; Spratt, D.; Quaderi, S.; Hurst, J.R.; McHugh, T.D. Impact of prophylactic and ’rescue pack’ antibiotics on the airway microbiome in chronic lung disease. BMJ Open Respir. Res. 2023, 10, e001335. [Google Scholar]
- Kuster, S.P.; Rudnick, W.; Shigayeva, A.; Green, K.; Baqi, M.; Gold, W.L.; Lovinsky, R.; Muller, M.P.; Powis, J.E.; Rau, N.; et al. Previous Antibiotic Exposure and Antimicrobial Resistance in Invasive Pneumococcal Disease: Results from Prospective Surveillance. Clin. Infect. Dis. 2014, 59, 944–952. [Google Scholar] [CrossRef]
- Diel, R.; Jacob, J.; Lampenius, N.; Loebinger, M.; Nienhaus, A.; Rabe, K.F.; Ringshausen, F.C. Burden of non-tuberculous mycobacterial pulmonary disease in Germany. Eur. Respir. J. 2017, 49, e1602109. [Google Scholar] [CrossRef]
- Li, N.; Cai, Q.; Miao, Q.; Song, Z.; Fang, Y.; Hu, B. High-Throughput Metagenomics for Identification of Pathogens in the Clinical Settings. Small Methods 2021, 5, 2000792. [Google Scholar] [CrossRef] [PubMed]
- Cossarizza, A.; Chang, H.D.; Radbruch, A.; Abrignani, S.; Addo, R.; Akdis, M.; Andrä, I.; Andreata, F.; Annunziato, F.; Arranz, E.; et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies (third edition). Eur. J. Immunol. 2021, 51, 2708–3145. [Google Scholar] [CrossRef] [PubMed]
- Tighe, P.J.; Ryder, R.R.; Todd, I.; Fairclough, L.C. ELISA in the multiplex era: Potentials and pitfalls. Proteom. Clin. Appl. 2015, 9, 406–422. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, G.; Jiang, S.; Liu, Y.X. Wekemo Bioincloud: A user-friendly platform for meta-omics data analyses. Imeta 2024, 3, e175. [Google Scholar] [CrossRef]
- Huang, H. linkET: Everything Is Linkable, R Package Version 0.0.3, R Foundation: Vienna, Austria, 2021.
- Charlson, E.S.; Diamond, J.M.; Bittinger, K.; Fitzgerald, A.S.; Yadav, A.; Haas, A.R.; Bushman, F.D.; Collman, R.G. Lung-enriched organisms and aberrant bacterial and fungal respiratory microbiota after lung transplant. Am. J. Respir. Crit. Care Med. 2012, 186, 536–545. [Google Scholar] [CrossRef]
- Singh, S.; Pragman, A.A.; Segal, L.N. Balancing Benefits and Risks: Do Inhaled Corticosteroids Modify the Lung Microbiome? Am. J. Respir. Crit. Care Med. 2021, 204, 1117–1119. [Google Scholar] [CrossRef]
- Mourad, A.; Baker, A.W.; Stout, J.E. Reduction in Expected Survival Associated with Nontuberculous Mycobacterial Pulmonary Disease. Clin. Infect. Dis. 2021, 72, e552–e557. [Google Scholar] [CrossRef]
- Labaki, W.W.; Rosenberg, S.R. Chronic Obstructive Pulmonary Disease. Ann. Intern. Med. 2020, 173, itc17–itc32. [Google Scholar] [CrossRef]
- Jiang, L.; Xin, J.; Liang, L.; Xia, M.; Li, J.; Tong, J.; Huang, C.; Li, T. Enhanced diagnosis of pulmonary tuberculosis through nucleotide MALDI-TOF MS analysis of BALF: A retrospective clinical study. Sci. Rep. 2024, 14, 18416. [Google Scholar]
- Chalmers, J.D.; Smith, M.P.; McHugh, B.J.; Doherty, C.; Govan, J.R.; Hill, A.T. Short- and long-term antibiotic treatment reduces airway and systemic inflammation in non-cystic fibrosis bronchiectasis. Am. J. Respir. Crit. Care Med. 2012, 186, 657–665. [Google Scholar] [PubMed]
- Russell, D.G.; Cardona, P.J.; Kim, M.J.; Allain, S.; Altare, F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat. Immunol. 2009, 10, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Silva Miranda, M.; Breiman, A.; Allain, S.; Deknuydt, F.; Altare, F. The tuberculous granuloma: An unsuccessful host defence mechanism providing a safety shelter for the bacteria? Clin. Dev. Immunol. 2012, 2012, 139127. [Google Scholar] [CrossRef] [PubMed]
- Awuh, J.A.; Flo, T.H. Molecular basis of mycobacterial survival in macrophages. Cell. Mol. Life Sci. 2017, 74, 1625–1648. [Google Scholar]
- Dickson, R.P.; Huffnagle, G.B. The Lung Microbiome: New Principles for Respiratory Bacteriology in Health and Disease. PLoS Pathog. 2015, 11, e1004923. [Google Scholar]
- Ferrer, M.; Méndez-García, C.; Rojo, D.; Barbas, C.; Moya, A. Antibiotic use and microbiome function. Biochem. Pharmacol. 2017, 134, 114–126. [Google Scholar] [CrossRef]
- Dickson, R.P.; Singer, B.H.; Newstead, M.W.; Falkowski, N.R.; Erb-Downward, J.R.; Standiford, T.J.; Huffnagle, G.B. Enrichment of the lung microbiome with gut bacteria in sepsis and the acute respiratory distress syndrome. Nat. Microbiol. 2016, 1, 16113. [Google Scholar]
- Dickson, R.P.; Erb-Downward, J.R.; Freeman, C.M.; McCloskey, L.; Falkowski, N.R.; Huffnagle, G.B.; Curtis, J.L. Bacterial Topography of the Healthy Human Lower Respiratory Tract. mBio 2017, 8, e02287-16. [Google Scholar]
- Wei, N.; Lu, J.; Dong, Y.; Li, S. Profiles of Microbial Community and Antibiotic Resistome in Wild Tick Species. mSystems 2022, 7, e0003722. [Google Scholar]
- Kahl, L.J.; Stremmel, N.; Esparza-Mora, M.A.; Wheatley, R.M.; MacLean, R.C.; Ralser, M. Interkingdom interactions between Pseudomonas aeruginosa and Candida albicans affect clinical outcomes and antimicrobial responses. Curr. Opin. Microbiol. 2023, 75, 102368. [Google Scholar]
- Kunst, H.; Wickremasinghe, M.; Wells, A.; Wilson, R. Nontuberculous mycobacterial disease and Aspergillus-related lung disease in bronchiectasis. Eur. Respir. J. 2006, 28, 352–357. [Google Scholar] [PubMed]
- Fayos, M.; Silva, J.T.; López-Medrano, F.; Aguado, J.M. Non-Tuberculous Mycobacteria and Aspergillus Lung Co-Infection: Systematic Review. J. Clin. Med. 2022, 11, e5619. [Google Scholar] [CrossRef]
- Gaastra, W.; Kusters, J.G.; Van Duijkeren, E.; Lipman, L.J.A. Escherichia fergusonii . Vet. Microbiol. 2014, 172, 7–12. [Google Scholar] [PubMed]
- Henkle, E.; Winthrop, K.L. Nontuberculous mycobacteria infections in immunosuppressed hosts. Clin. Chest Med. 2015, 36, 91–99. [Google Scholar]
- Andréjak, C.; Nielsen, R.; Thomsen, V.Ø.; Duhaut, P.; Sørensen, H.T.; Thomsen, R.W. Chronic respiratory disease, inhaled corticosteroids and risk of non-tuberculous mycobacteriosis. Thorax 2013, 68, 256–262. [Google Scholar]


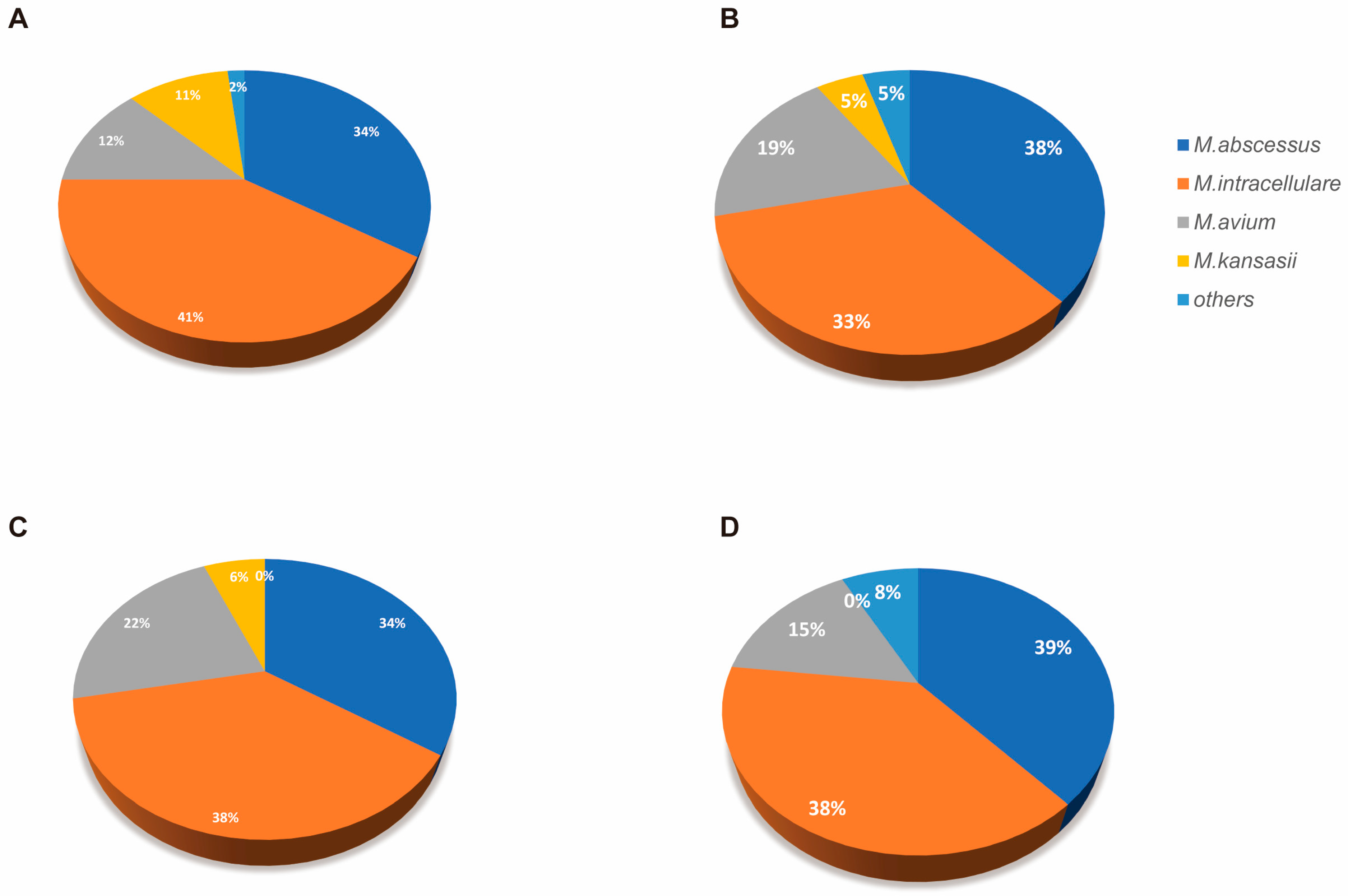
| All Patient | Immunocompetent | Immunosuppressed | p-Value | |
|---|---|---|---|---|
| Subjects | 143 | 111 | 32 | |
| Age years, mean ± SD | 59.8 ± 13.5 | 57.9 ± 12.8 | 61.6 ± 14.1 | 0.17 |
| Female | 80 (56) | 68 (61) | 12 (38) | 0.02 |
| BMI, kg/m2 | 21.2 ± 3.8 | 21.4 ± 3.7 | 20.7 ± 4.1 | 0.344 |
| Hypertension, n (%) | 17 (12) | 13 (12) | 4 (13) | 0.75 |
| Diabetes | 9 (6) | 6 (5) | 3 (9) | 0.37 |
| COPD | 11 (8) | 6 (5) | 5 (16) | 0.04 |
| Bronchiectasis | 51 (36) | 43 (39) | 8 (25) | 0.21 |
| Pulmonary co-infection | 19 (13) | 7 (6) | 12 (38) | <0.01 |
| Recent antibiotic use | 44 (31) | 32 (29) | 12 (38) | 0.35 |
| HRCT thorax | ||||
| Cavities | 27 (19) | 20 (18) | 7 (22) | 0.61 |
| Fibrosis | 4 (3) | 2 (2) | 2 (6) | 0.18 |
| Bronchiectasis | 65 (45) | 52 (47) | 13 (41) | 0.57 |
| Thickened airways | 8 (6) | 5 (5) | 3 (9) | 0.36 |
| Nodules | 40 (28) | 31 (28) | 9 (28) | 1 |
| Mucoid impaction | 2 (1) | 1 (1) | 1 (3) | 0.35 |
| Specimen Type | ||||
| Sputum | 111 | 79 | 32 | |
| BALF | 64 | 48 | 16 |
| Specimen Type | Cohort | Number of Specimens | Detection Rate (%) | p-Value | Relative Abundance (%, mean ± SD) | Fold Change | p-Value |
|---|---|---|---|---|---|---|---|
| Sputum | Immunosuppressed | 32 | 65.63 | 0.59 | 0.75 ± 0.87 | 2.13 | 0.01 |
| Immunocompetent | 79 | 70.89 | 1.60 ± 1.98 | ||||
| BALF | Immunosuppressed | 16 | 75.00 | 0.76 | 5.18 ± 1.99 | 0.74 | 0.04 |
| Immunocompetent | 48 | 66.67 | 3.82 ± 2.96 |
| Immunocompetent n = 111 | Immunosuppressed n = 32 | p-Value | |
|---|---|---|---|
| Lymphocyte percentage (n = 132) (%) | |||
| B lymphocyte | 13.8 ± 7.4 | 8.2 ± 5.9 | 0.002 ** |
| T lymphocyte | 69.2 ± 8.5 | 72.4 ± 14.8 | 0.234 |
| CD4+ lymphocyte | 41.2 ± 9.4 | 36.8 ± 14.3 | 0.035 * |
| CD8+ lymphocyte | 24.1 ± 8.6 | 29.3 ± 13.2 | 0.041 * |
| CD4+/CD8+ T lymphocyte ratio | 1.9 ± 0.8 | 1.4 ± 0.9 | 0.015 * |
| NK lymphocyte | 16.4 ± 8.7 | 19.8 ± 13.2 | 0.168 |
| Absolute count of lymphocyte subsets (IQR) (/μL) | 1144.0 (859.0–1642.0) | 1156.9 (704.0–1527.1) | 0.862 |
| B lymphocyte | 151.0 (98.0–307.0) | 94.0 (55.0–166.0) | 0.024 * |
| T lymphocyte | 785.0 (553.0–1107.0) | 619.0 (553.0–841.0) | 0.186 |
| CD4+ lymphocyte | 478.0 (344.0–593.0) | 417.0 (272.0–424.0) | 0.042 * |
| CD8+ lymphocyte | 299.0 (186.0–431.0) | 309.0 (115.0–438.0) | 0.984 |
| NK lymphocyte | 177.0 (130.0–244.0) | 187.0 (83.0–363.0) | 0.862 |
| Inflammatory factors (IQR) pg/mL | |||
| TNF-α | 8.2 (5.3–16.8) | 9.7 (5.9–19.2) | 0.286 |
| IL-1 | 5.0 (5.0–5.9) | 5.0 (5.0–11.7) | 0.142 |
| IL-2R | 519.5 (381.0–751.0) | 738.0 (608.0–1192.0) | <0.001 *** |
| IL-6 | 4.2 (2.6–7.9) | 16.9 (6.7–51.4) | <0.001 *** |
| IL-8 | 12.0 (9.0–25.0) | 14.0 (12.0–35.0) | 0.124 |
| IL-10 | 5.0 (5.0–5.0) | 5.0 (5.0–5.0) | 0.856 |
| ESR (mm/h) | 25.0 (13.0–53.0) | 49.0 (19.0–98.0) | 0.008 *** |
| CRP (mg/L) | 3.2 (1.7–16.4) | 21.2 (7.0–119.0) | <0.001 *** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, L.; Chen, Y.; Luan, S.; Yin, X.; Pan, J.; Wang, L.; Yao, Y.; Zhou, C.; Bao, R.; Shen, J.; et al. Lung Microenvironment Among Patients with Nontuberculous Mycobacterial Pulmonary Disease by Metagenomic Sequencing Technique. Biomedicines 2025, 13, 818. https://doi.org/10.3390/biomedicines13040818
Qin L, Chen Y, Luan S, Yin X, Pan J, Wang L, Yao Y, Zhou C, Bao R, Shen J, et al. Lung Microenvironment Among Patients with Nontuberculous Mycobacterial Pulmonary Disease by Metagenomic Sequencing Technique. Biomedicines. 2025; 13(4):818. https://doi.org/10.3390/biomedicines13040818
Chicago/Turabian StyleQin, Le, Yu Chen, Sichun Luan, Xiaoyu Yin, Jue Pan, Leilei Wang, Yumeng Yao, Chunmei Zhou, Rong Bao, Jiajin Shen, and et al. 2025. "Lung Microenvironment Among Patients with Nontuberculous Mycobacterial Pulmonary Disease by Metagenomic Sequencing Technique" Biomedicines 13, no. 4: 818. https://doi.org/10.3390/biomedicines13040818
APA StyleQin, L., Chen, Y., Luan, S., Yin, X., Pan, J., Wang, L., Yao, Y., Zhou, C., Bao, R., Shen, J., Miao, Q., & Hu, B. (2025). Lung Microenvironment Among Patients with Nontuberculous Mycobacterial Pulmonary Disease by Metagenomic Sequencing Technique. Biomedicines, 13(4), 818. https://doi.org/10.3390/biomedicines13040818