

Molecular Genetic Architecture of Morbid Obesity in Russian Children
 , , , ,
, , , ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
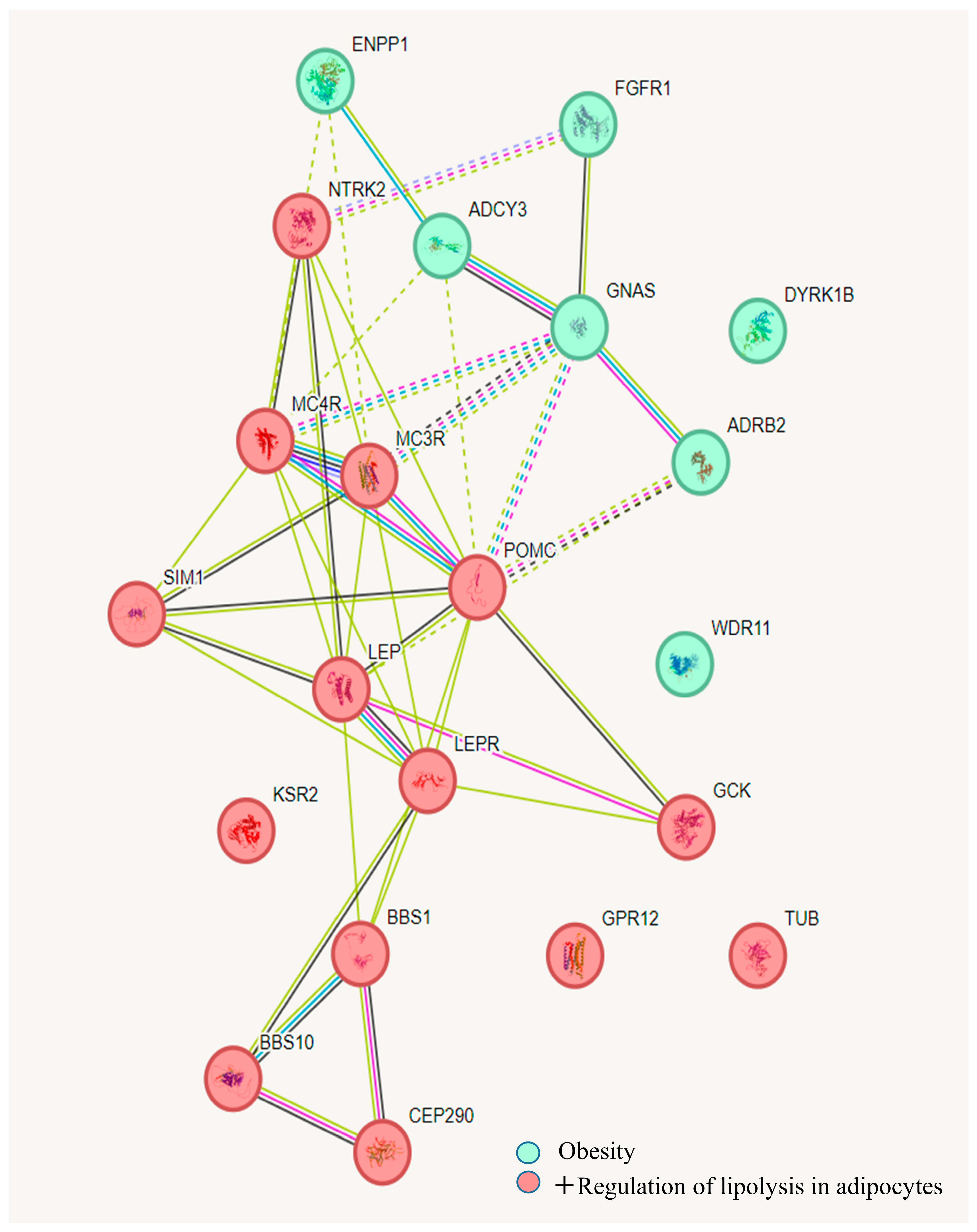
3. Results
- Associated with obesity;
- Associated with diabetes and insulin resistance;
- Associated with other comorbid conditions, typically intellectual developmental disorders.
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vasyukova, O.V. Obesity in Children and Adolescents: Diagnosis Criteria. Obes. Metab. 2019, 16, 70–73. [Google Scholar] [CrossRef]
- WHO Latest Data Shows Southern European Countries Have Highest Rate of Childhood Obesity. Available online: https://dev-cms.who.int/europe/news/item/24-05-2018-latest-data-shows-southern-european-countries-have-highest-rate-of-childhood-obesity (accessed on 20 January 2025).
- Dykens, E.M.; Maxwell, M.A.; Pantino, E.; Kossler, R.; Roof, E. Assessment of hyperphagia in Prader-Willi syndrome. Obesity 2007, 15, 1816–1826. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Kopytina, D.A.; Vasyukova, O.V.; Salakhov, R.R.; Okorokov, P.L.; Kopytina, E.V.; Nagaeva, E.V.; Khusainova, R.I.; Minniakhmetov, I.R.; Popov, S.V.; Bezlepkina, O.B.; et al. Identification of Novel Pathogenic Variants in the Gnas Gene in Children with Morbid Obesity and Pseudohypoparathyroidism. Obes. Metab. 2024, 21, 412–424. [Google Scholar] [CrossRef]
- Ludwig, D.S.; Ebbeling, C.B. The Carbohydrate-Insulin Model of Obesity: Beyond “Calories in, Calories Out”. JAMA Intern. Med. 2018, 178, 1098–1103. [Google Scholar] [CrossRef]
- Singh, R.K.; Kumar, P.; Mahalingam, K. Molecular Genetics of Human Obesity: A Comprehensive Review. Comptes Rendus Biol. 2017, 340, 87–108. [Google Scholar] [CrossRef]
- Keller, M.; Svensson, S.I.A.; Rohde-Zimmermann, K.; Kovacs, P.; Böttcher, Y. Genetics and Epigenetics in Obesity: What Do We Know so Far? Curr. Obes. Rep. 2023, 12, 482–501. [Google Scholar] [CrossRef]
- Martins, L.B.; Monteze, N.M.; Calarge, C.; Ferreira, A.V.M.; Teixeira, A.L. Pathways Linking Obesity to Neuropsychiatric Disorders. Nutrition 2019, 66, 16–21. [Google Scholar] [CrossRef]
- Kelly, A.S.; Barlow, S.E.; Rao, G.; Inge, T.H.; Hayman, L.L.; Steinberger, J.; Urbina, E.M.; Ewing, L.J.; Daniels, S.R. Severe Obesity in Children and Adolescents: Identification, Associated Health Risks, and Treatment Approaches: A Scientific Statement from the American Heart Association. Circulation 2013, 128, 1689–1712. [Google Scholar] [CrossRef]
- Serra-Juhé, C.; Martos-Moreno, G.; Bou de Pieri, F.; Flores, R.; Chowen, J.A.; Pérez-Jurado, L.A.; Argente, J. Heterozygous Rare Genetic Variants in Non-Syndromic Early-Onset Obesity. Int. J. Obes. 2020, 44, 830–841. [Google Scholar] [CrossRef]
- George, A.; Navi, S.; Nanda, P.M.; Daniel, R.; Anand, K.; Banerjee, S.; Panigrahi, I.; Dayal, D. Clinical and Molecular Characterisation of Children with Monogenic Obesity: A Case Series. Pediatr. Endocrinol. Diabetes Metab. 2024, 30, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Kernohan, K.D.; Boycott, K.M. The Expanding Diagnostic Toolbox for Rare Genetic Diseases. Nat. Rev. Genet. 2024, 25, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.J.; Chaves, E.; Ariza, A.J.; Thaker, V.V.; Cho, C.C.; Binns, H.J. Exploring Genetic Testing for Rare Disorders of Obesity: Experience and Perspectives of Pediatric Weight Management Providers. Child. Obes. 2024, 20, 451–458. [Google Scholar] [CrossRef]
- Nordang, G.B.N.; Busk, Ø.L.; Tveten, K.; Hanevik, H.I.; Fell, A.K.M.; Hjelmesæth, J.; Holla, Ø.L.; Hertel, J.K. Next-Generation Sequencing of the Monogenic Obesity Genes LEP, LEPR, MC4R, PCSK1 and POMC in a Norwegian Cohort of Patients with Morbid Obesity and Normal Weight Controls. Mol. Genet. Metab. 2017, 121, 51–56. [Google Scholar] [CrossRef]
- Akıncı, A.; Turkkahraman, D.; Tekedereli, I.; Ozer, L.; Evren, B.; Şahin, I.; Kalkan, T.; Curek, Y.; Camtosun, E.; Döğer, E.; et al. Novel Mutations in Obesity-Related Genes in Turkish Children with Non-Syndromic Early Onset Severe Obesity: A Multicentre Study. J. Clin. Res. Pediatr. Endocrinol. 2019, 11, 341–349. [Google Scholar] [CrossRef]
- Foucan, L.; Larifla, L.; Durand, E.; Rambhojan, C.; Armand, C.; Michel, C.T.; Billy, R.; Dhennin, V.; De Graeve, F.; Rabearivelo, I.; et al. High Prevalence of Rare Monogenic Forms of Obesity in Obese Guadeloupean Afro-Caribbean Children. J. Clin. Endocrinol. Metab. 2018, 103, 539–545. [Google Scholar] [CrossRef]
- Trevellin, E.; Granzotto, M.; Host, C.; Grisan, F.; De Stefani, D.; Grinzato, A.; Lefkimmiatis, K.; Pagano, C.; Rizzuto, R.; Vettor, R. A Novel Loss of Function Melanocortin-4-Receptor Mutation (MC4R-F313Sfs∗29) in Morbid Obesity. J. Clin. Endocrinol. Metab. 2021, 106, 736–749. [Google Scholar] [CrossRef]
- Saeed, S.; Bonnefond, A.; Manzoor, J.; Shabir, F.; Ayesha, H.; Philippe, J.; Durand, E.; Crouch, H.; Sand, O.; Ali, M.; et al. Genetic Variants in LEP, LEPR, and MC4R Explain 30% of Severe Obesity in Children from a Consanguineous Population. Obesity 2015, 23, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, I.; Haris, B.; Al-Barazenji, T.; Vasudeva, D.; Tomei, S.; Al Azwani, I.; Dauleh, H.; Shehzad, S.; Chirayath, S.; Mohamadsalih, G.; et al. Understanding the Genetics of Early-Onset Obesity in a Cohort of Children from Qatar. J. Clin. Endocrinol. Metab. 2023, 108, 3201–3213. [Google Scholar] [CrossRef]
- Saeed, S.; Bonnefond, A.; Tamanini, F.; Mirza, M.U.; Manzoor, J.; Janjua, Q.M.; Din, S.M.; Gaitan, J.; Milochau, A.; Durand, E.; et al. Loss-of-Function Mutations in ADCY3 Cause Monogenic Severe Obesity. Nat. Genet. 2018, 50, 175–179. [Google Scholar] [CrossRef]
- Andres-Hernando, A.; Jensen, T.J.; Kuwabara, M.; Orlicky, D.J.; Cicerchi, C.; Li, N.; Roncal-Jimenez, C.A.; Garcia, G.E.; Ishimoto, T.; Maclean, P.S.; et al. Vasopressin Mediates Fructose-Induced Metabolic Syndrome by Activating the V1b Receptor. JCI Insight 2021, 6, e140848. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, K.A.; Wheeler, E.; Morris, A.P.; Ricketts, S.L.; Hallmans, G.; Rolandsson, O.; Daly, A.; Wasson, J.; Permutt, A.; Hattersley, A.T.; et al. Detailed Investigation of the Role of Common and Low-Frequency WFS1 Variants in Type 2 Diabetes Risk. Diabetes 2010, 59, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Ivask, M.; Volke, V.; Raasmaja, A.; Kõks, S. High-Fat Diet Associated Sensitization to Metabolic Stress in Wfs1 Heterozygous Mice. Mol. Genet. Metab. 2021, 134, 203–211. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Patient Diagnosis and Comorbidities | Number (N) | Mean Age and Standard Deviation (μ ± SD) | SDS ± SD |
|---|---|---|---|
| Total cohort | 163 | 11.00 ± 4.00 | 3.95 ± 0.89 |
| Morbid obesity | 59 | 8.00 ± 4.56 | 4.03 ± 0.80 |
| Obesity with insulin resistance or diabetes | 39 | 13.00 ± 3.08 | 3.91 ± 0.73 |
| Obesity with other comorbid conditions | 65 | 8.00 ± 4.70 | 3.91 ± 0.11 |
| Patient | Nosology Associated with the Gene | Inheritance Type | Gene, Variant | ID | Zygosity | |
|---|---|---|---|---|---|---|
| 1 | Morbid obesity due to leptin deficiency (OMIM: 614962) | AR | LEP (c.309C>A, p.Asn103Lys) | rs28954113 | Homozygote | |
| 2 | LEP (c.175G>T, p.Gly59Cys) | Not reported | Heterozygote | |||
| 3 | Morbid obesity (OMIM: 614963) | AR | LEPR (c.131_132insTGAC:p.Y44fs/c.2588_2589del:p.S863fs) | rs757358893/Not reported | Compound heterozygote | |
| 4 | LEPR (c.1630A>G, p.Lys544Glu/c.133_136dup, p.Tyr46Ter) | Not reported | Compound heterozygote | |||
| 5 | Early-onset obesity (OMIM: 601665) | AR | POMC (c.616G>T, p.Glu206Ter/c.599_604dup, p.Ala201_Gln202insArgAla) | rs202127120/rs762710034 | Compound heterozygote | |
| 6 | Bardet-Biedl syndrome, type 1 (OMIM: 209900) | AR | BBS1 (c.235G>A, p.Glu79Lys/c.47+114T>C) | rs138744839/Not reported | Compound heterozygote | |
| 7 | Bardet-Biedl syndrome, type 10 (OMIM: 615987) | AR | BBS10 (c.263T>C, p.Ile88Thr/c.145C>T, p.Arg49Trp) | Not reported/rs768933093 | Compound heterozygote | |
| 8 | Obesity (OMIM: 618406) | AD | MC4R (c.380C>T, p.Ser127Leu) | rs13447331 | Heterozygote | |
| 9 | MC4R (c.731C>A, p.Ala244Glu) | rs13447335 | Heterozygote | |||
| 10 | MC4R (c.202G>T, p.Ala68Ser) | rs1360504571 | Heterozygote | |||
| 11 | Obesity (OMIM: 618406) | AD | MC4R (c.731C>A, p.Ala244Glu) | rs13447335 | Heterozygote | |
| Non-insulin-dependent diabetes mellitus with late onset (OMIM: 125853), MODY2 (OMIM: 125851) | AD | GCK (c.821A>G, p.Asp274Gly) | Not reported | Heterozygote | ||
| 12 | Obesity (OMIM: 618406) | AD | MC4R (c.750_751del, p.Ile251TrpfsTer34) | Not reported | Heterozygote | |
| Hartsfield syndrome (OMIM: 615465); hypogonadotropic hypogonadism 2 with or without anosmia (OMIM: 147950); Jackson-Weiss syndrome (OMIM: 123150); osteoglophonic dysplasia (OMIM: 166250); Pfeiffer syndrome (OMIM: 101600); trigonocephaly 1 (OMIM: 190440) | AD | FGFR1 (c.1497_1501del, p.Leu500GlnfsTer41) | Not reported | Heterozygote | ||
| 13 | Obesity (OMIM: 602025) | AD | MC3R (c.508G>T, p.Val170Phe) | rs971796328 | Heterozygote | |
| Hypogonadotropic hypogonadism, 14 (OMIM: 614858) | AD | WDR11 (c.607A>G, p.Ile203Val) | Not reported | Heterozygote | ||
| 14 | Obesity (PMID: 29273807, 24209692) | AD | KSR2 (c.1858G>A, p.Glu620Lys) | Not reported | Heterozygote | |
| Obesity (OMIM: 601665); Cole disease (OMIM: 615522) | AD, AR | ENPP1 (c.155C>T, p.Pro52Leu) | rs1408308764 | Heterozygote | ||
| 15 | Obesity (OMIM: 601665), Cole disease (OMIM: 615522) | AD, AR | ENPP1 (c.2028G>T, p.Gln676His) | Not reported | Heterozygote | |
| 16 | Abdominal obesity and metabolic syndrome 3 (OMIM: 615812) | AD | DYRK1B (c.619C>T, p.Arg207Cys) | rs141849649 | Heterozygote | |
| 17 | DYRK1B (c.533G>A, p.Arg178Gln) | rs1402482186 | Heterozygote | |||
| 18 | DYRK1B (c.1627C>A, p.Pro543Thr) | rs752860992 | Heterozygote | |||
| 19 | Dyslipidemia and obesity (PMID: 34174278, 16887097) | AD, AR | GPR12 (c.166A>G, p.Thr56Ala) | rs1209054859 | Heterozygote | |
| 20 | Severe early-onset obesity (PMID: 30991789, 28472148, 23778139, 23778136) | AD | SIM1 (c.125del, p.Ile42LysfsTer11) | Not reported | Heterozygote | |
| 21 | Obesity and type 2 diabetes (OMIM: 109690) | Multifactorial | ADRB2 (c.524G>A, p.Arg175Gln) | Not reported | Heterozygote | |
| 22 | Retinal dystrophy with obesity (OMIM: 616188) | AR | TUB (c.8G>T, p.Gly3Val) | Not reported | Heterozygote | |
| 23 | Obesity (OMIM: 617885) | AR | ADCY3 (c.2906G>A, p.Arg969Gln) | rs371465590 | Heterozygote | |
| 24 | Pseudohypoparathyroidism Ia, Ib, Ic (OMIM: 103580, 603233, 612462) | AD | GNAS (c.493C>T, p.Arg165Cys) | rs137854532 | Heterozygote | |
| 25 | AD | GNAS (c.586-18_591del) | Not reported | Heterozygote | ||
| 26 | Bardet–Biedl syndrome 14; Joubert syndrome 5; Meckel syndrome 4; Senior-Loken syndrome 6 (OMIM: 610142) | AR | CEP290 (c.4267C>T, p.Arg1423Cys) | rs1353027764 | Heterozygote | |
| 27 | Epileptic encephalopathy with obesity, hyperphagia, and developmental delay (OMIM: 600456) | AD | NTRK2 (c.2143C>T, p.Arg715Trp) | Not reported | Heterozygote | |
| 28 | Developmental delay, obesity, short stature | de novo | Duplication of 17q segment, 679,017 bp | Not reported | Heterozygote | |
| 29 | Prader–Willi syndrome (OMIM: 176270) | de novo | Deletion of 15q segment, 5,933,764 bp | Not reported | Heterozygote | |
| Patient | Nosology Associated with the Gene | Inheritance Type | Gene, Variant | ID | Zygosity |
|---|---|---|---|---|---|
| 1 | Diabetes mellitus, non-insulin-dependent (OMIM: 125853); permanent neonatal diabetes mellitus, type 3, with or without neurological features (OMIM: 618857); MODY12 (OMIM: 600509) | AD | ABCC8 (c.3867G>A, p.Met1289Ile) | rs1467178918 | Heterozygote |
| 2 | Diabetes mellitus, non-insulin-dependent (OMIM: 125853); permanent neonatal diabetes mellitus, type 3, with or without neurological features (OMIM: 618857); MODY12 (OMIM: 600509) | AD | ABCC8 (c.560T>A, p.Val187Asp) | rs137852672 | Heterozygote |
| Obesity due to proopiomelanocortin deficiency (OMIM: 609734) | AR | POMC (c.394C>G, p.Pro132Ala) | rs8192606 | Heterozygote | |
| 3 | Predisposition to obesity (OMIM: 617885) | AR | ADCY3 (c.3380C>G, p.Thr1127Ser) | rs772212679 | Heterozygote |
| Diabetes mellitus and familial hyperinsulinemic hypoglycemia, 2 (OMIM: 600937) | AD | KCNJ11 (c.628A>G, p.Ile210Val) | Not reported | Heterozygote | |
| 4 | Nephrogenic diabetes insipidus 1 (OMIM: 304800); nephrogenic syndrome of inappropriate antidiuresis (OMIM: 300539) | X-linked | AVPR2 (c.262G>T, p.Val88Leu) | Not reported | Heterozygote |
| 5 | Non-insulin-dependent diabetes mellitus (OMIM: 604641) | AD | MAPK8IP1 (c.409T>G, p.Ser137Ala) | rs1398556817 | Heterozygote |
| Non-insulin-dependent diabetes mellitus (OMIM: 138160) | AD | SLC2A2 (c.1039G>A, p.Ala347Thr) | rs776435170 | Heterozygote | |
| 6 | MODY2 (OMIM: 125851); non-insulin-dependent diabetes mellitus with late onset (OMIM: 125853); familial hyperinsulinemic hypoglycemia, 3 (OMIM: 602485) | AD | GCK (c.12_13del, p.Asp4GlufsTer47) | rs1483208659 | Heterozygote |
| 7 | Wolfram syndrome 1 (OMIM: 222300) | AR | WFS1 (c.1943G>A, p.Trp648Ter/c.1244T>A, p.Val415Asp) | Not reported/rs150465110 | Compound heterozygote |
| Patient | Syndrome Name | Gene, Variant | Inheritance Type | Description in the Literature |
|---|---|---|---|---|
| 1 | Helsmoortel–van der Aa syndrome (OMIM: 615873) | ADNP (c.2368G>C, p.Ala790Pro) | AD | Not reported |
| 2 | Curie–Isidor syndrome; CURI (OMIM: 619762) | BAP1 (c.1971G>C, p.Arg657Ser) | AD | Not reported |
| 3 | Menkes–Hennekam syndrome 1; Rubinstein–Taybi syndrome 1 (OMIM: 600140) | CREBBP (c.3233C>T, p.Ser1078Leu) | AD | rs942848385 |
| 4 | Schuurs–Hoeijmakers syndrome (OMIM: 615009) | PACS1 (c.755C>T, p.Ser252Phe) | AD | Not reported |
| 5 | Coffin–Siris syndrome 1 (OMIM: 135900) | ARID1B deletion of 2734 bp | AD | Not reported |
| 6 | Chung–Jansen syndrome (OMIM: 617991) | PHIP (c.685T>G, p.Ser229Ala) | AD | Not reported |
| 7 | Coffin–Siris syndrome (OMIM: 135900) | Deletion of chromosome 6 segment, 2734 bp | AD | Not reported |
| 8 | Radio–Tartaglia syndrome (OMIM: 619312, RATARS) | SPEN (c.9354C>G, p.His3118Gln) | AD | Not reported |
| 9 | Hypertriglyceridemia, type 2 (OMIM: 619324) | CREB3L3 (c.732dup, p.Lys245GlufsTer130) | AD | rs780374391 |
| 10 | Familial partial lipodystrophy, type 6 (OMIM: 615980) | LIPE (c.883+1G>A/c.1419+32C>T) | AR | rs199830207, rs373163203 |
| 11 | Familial partial lipodystrophy, type 4 (OMIM: 613877) | PLIN1 (c.445G>T, p.Gly149Trp) | AD | rs200235164 |
| 12 | Spastic paraplegia with intellectual disability, nystagmus, and obesity (SINO) (OMIM: 615759) | KIDINS220 (c.5128C>T, p.Gln1710Ter) | AD | Not reported |
| 13 | Leukoencephalopathy with macrocephaly and subcortical cysts (Van der Knaap disease, OMIM: 604004) | MLC1 (c.1027C>G, p.Gln343Glu)/deletion of chromosome 22 segment, 5331 bp | AR | Not reported |
| 14 | Syndromic microphthalmia 1 (OMIM: 309800) and Ogden syndrome (OMIM: 300855) | NAA10 (c.20G>C, p.Arg7Thr) | X-linked | Not reported |
| 15 | Acrodysostosis, type 2, with or without hormone resistance (OMIM: 614613) | PDE4D (c.809-58G>C) | AD | rs1343377101 |
| 16 | Primary pigmented nodular adrenal disease, type 1 (OMIM: 610475) | PDE11A (c.1298C>T, p.Ser433Leu) | AD | rs200330914 |
| 17 | Encephalopathy, type 65, associated with developmental delay and epilepsy (OMIM: 618008) | CYFIP2 (c.3292C>T, p.Arg1098Cys) | AD | Not reported |
| 18 | Intellectual disability with hypotonia, speech impairment, and facial dysmorphism (OMIM: 619556) | TNPO2 (c.771+7_771+14delinsG) | AD | Not reported |
| 19 | Intellectual disability, type 13 (OMIM: 613192) | TRAPPC9 (c.3013C>T, p.Gln1005Ter) | AR | rs913893905 |
| 20 | Intellectual disability, type 30 (OMIM: 616083) | ZMYND11 (c.1109G>A, p.Arg370Gln) | AD | rs767869912 |
| 21 | Intellectual disability, type 39 (OMIM: 616521) | MYT1L (c.1466dup, p.Pro490AlafsTer5) | AD | Not reported |
| 22 | Intellectual disability, type 49, with macrocephaly, obesity, and tall stature (OMIM: 617752) | TRIP12 (c.1349T>A, p.Leu450Ter) | AD | Not reported |
| 23 | Autosomal dominant intellectual disability, type 52; MRD52 (OMIM: 617796) | ASH1L (c.4802A>G, p.Asp1601Gly) | AD | rs373441853 |
| 24 | Intellectual disability, type 72 (OMIM: 618665) | METTL5 (c.362A>G, p.Asp121Gly) | AR | rs760916142 |
| 25 | Autism spectrum disorders | FAT1 (c.4972A>G, p.Thr1658Ala) | AD | rs754752151 |
| KCNJ15 (c.1070A>T, p.Gln357Leu) | ? | Not reported | ||
| 26 | Radio–Tartaglia syndrome (OMIM: 619312, RATARS) | SPEN (c.10704+2T>G) | AD | Not reported |
| Endocrinopathy due to proprotein convertase 1/3 deficiency (OMIM: 600955) | PCSK1 (c.1121G>T, p.Cys374Phe) | AR | Not reported | |
| 27 | Spondyloepimetaphyseal dysplasia, Isidor-Tuten type (OMIM: 618728) | RPL13 (c.578G>A, p.Gly193Asp) | AD | Not reported |
| Chopra–Amiel–Gordon syndrome (OMIM: 619504) | ANKRD17 (c.170T>G, p.Val57Gly) | AD | Not reported | |
| Developmental delay with speech impairment and behavioral abnormalities (OMIM: 619475) | SPTBN1 (c.844G>T, p.Ala282Ser) | AD | Not reported | |
| Autosomal dominant intellectual disability, type 47 (OMIM: 617635) | STAG1 (c.1562A>C, p.Gln521Pro) | AD | Not reported | |
| 28 | X-linked intellectual disability, type 98 (OMIM: 300912) | NEXMIF (c.4313G>A, p.Gly1438Asp) | AD | Not reported |
| Bachmann–Bupp syndrome (macrocephaly, alopecia, and developmental delay) (OMIM: 619075) | ODC1 (c.508T>C, p.Phe170Leu) | AD | rs1197754776 | |
| Noonan syndrome 4 (OMIM: 610733) and/or gingival fibromatosis 1 (OMIM: 135300) | SOS1 (c.2566A>G, p.Ile856Val) | AD | Not reported | |
| 29 | Alström syndrome (OMIM: 203800) | ALMS1 (c.1283G>A, p.Gly428A); (c.4733T>C, p.Ile1578Thr) | AR | rs764312032, rs2103783837 |
| Epileptic encephalopathy 5, developmental delay with/without epilepsy, spastic paraplegia 91, distal hereditary neuronopathy 11 (OMIM: 182810) | SPTAN1 (c.2872-2A>G) | AD | Not reported |
| Gene | cDNA, Variant | Protein, Variant | ACMG Classification | Allele Frequency (gnomAD v4.1.0) | CADD Phred | In-Silico Individual Predictions (Varsome) * | ||
|---|---|---|---|---|---|---|---|---|
| LP/P | Uncertain | LB/B | ||||||
| ABCC8 | c.3867G>A | p.Met1289Ile | VUS | 6.203 × 107 | 28.1 | 10 | 9 | 9 |
| c.560T>A | p.Val187Asp | LP | 6.728 × 105 | 27.3 | 6 | 11 | 5 | |
| ADCY3 | c.2906G>A | p.Arg969Gln | VUS | 9.914 × 106 | 22.6 | 2 | 8 | 18 |
| c.3380C>G | p.Thr1127Ser | VUS | 1.921 × 105 | 22.6 | 2 | 3 | 30 | |
| ADRB2 | c.524G>A | p.Arg175Gln | VUS | NA | 22.9 | 2 | 8 | 17 |
| ADNP | c.2368G>C | p.Ala790Pro | VUS | NA | 26.4 | 16 | 10 | 0 |
| ALMS1 | c.1283G>A | p.Gly428A | VUS | 3.718 × 106 | 2.06 | 0 | 1 | 42 |
| c.4733T>C | p.Ile1578Thr | VUS | NA | 0.02 | 0 | 1 | 38 | |
| ANKRD17 | c.170T>G | p.Val57Gly | VUS | NA | 24.5 | 5 | 4 | 16 |
| ASH1L | c.4802A>G | p.Asp1601Gly | VUS | 1.239 × 106 | 26.5 | 0 | 10 | 14 |
| AVPR2 | c.262G>T | p.Val88Leu | LP | NA | 24.8 | 17 | 6 | 0 |
| BAP1 | c.1971G>C | p.Arg657Ser | VUS | NA | 23.1 | 11 | 5 | 11 |
| BBS1 | c.235G>A | p.Glu79Lys | VUS | 0.0012 | 23.8 | 4 | 11 | 5 |
| c.47+114T>C | - | VUS | 6.672 × 107 | 10.3 | - | - | - | |
| BBS10 | c.263T>C | p.Ile88Thr | VUS | NA | 26.6 | 4 | 13 | 6 |
| c.145C>T | p.Arg49Trp | P | 4.71 × 105 | 31.00 | 6 | 12 | 2 | |
| CEP290 | c.4267C>T | p.Arg1423Cys | VUS | 6.886 × 106 | 25.20 | 0 | 8 | 17 |
| CREB3L3 | c.732dup | p.Lys245GlufsTer130 | VUS | 2.782 × 104 | 33.00 | - | - | - |
| CREBBP | c.3233C>T | p.Ser1078Leu | VUS | 1.487 × 105 | 21.7 | 1 | 7 | 22 |
| CYFIP2 | c.3292C>T | p.Arg1098Cys | VUS | 2.478 × 106 | 29.8 | 14 | 3 | 7 |
| DYRK1B | c.619C>T | p.Arg207Cys | VUS | 3.037 × 105 | 31.0 | 9 | 6 | 4 |
| c.533G>A | p.Arg178Gln | VUS | 1.865 × 106 | 24.3 | 6 | 5 | 14 | |
| c.1627C>A | p.Pro543Thr | VUS | NA | 19.98 | 0 | 4 | 28 | |
| ENPP1 | c.155C>T | p.Pro52Leu | VUS | 6.664 × 107 | 24.3 | 4 | 5 | 18 |
| c.2028G>T | p.Gln676His | VUS | NA | 3.13 | 2 | 1 | 39 | |
| FAT1 | c.4972A>G | p.Thr1658Ala | VUS | 2.478 × 106 | 23.9 | 1 | 12 | 11 |
| FGFR1 | c.1497_1501del | p.Leu500GlnfsTer41 | P | NA | - | - | - | - |
| GCK | c.821A>G | p.Asp274Gly | LP | NA | 29.7 | 30 | 4 | 0 |
| c.12_13del | p.Asp4GlufsTer47 | P | 1.239 × 106 | 26.4 | - | - | - | |
| GNAS | c.493C>T | p.Arg165Cys | P | NA | 31.0 | 25 | 5 | 0 |
| c.586-18_591del | P | NA | - | - | - | - | ||
| GPR12 | c.166A>G | p.Thr56Ala | VUS | 1.859 × 106 | 26.3 | 2 | 12 | 7 |
| KCNJ11 | c.628A>G | p.Ile210Val | VUS | NA | 23.3 | 4 | 11 | 4 |
| KCNJ15 | c.1070A>T | p.Gln357Leu | VUS | NA | 22.8 | 1 | 5 | 18 |
| KIDINS220 | c.5128C>T | p.Gln1710Ter | VUS | NA | 41.0 | 7 | 2 | 4 |
| KSR2 | c.1858G>A | p.Glu620Lys | VUS | NA | 22.2 | 2 | 5 | 18 |
| LEP | c.309C>A | p.Asn103Lys | LP | 4.151 × 105 | 21.9 | 8 | 7 | 10 |
| c.175G>T | p.Gly59Cys | VUS | NA | 24.32 | 10 | 10 | 3 | |
| c.131_132insTGAC | p.Y44fs | LP | NA | - | - | - | - | |
| LEPR | c.2588_2589del | p.S863fs | LP | NA | - | - | - | - |
| c.1630A>G | p.Lys544Glu | VUS | NA | 20.5 | 0 | 4 | 24 | |
| c.133_136dup | p.Tyr46Ter | P | 6.196 × 107 | 17.1 | - | - | - | |
| LIPE | c.883+1G>A | - | LP | 7.261 × 105 | 33.0 | 11 | 3 | 0 |
| c.1419+32C>T | - | VUS/LB | 6.109 × 105 | 9.06 | - | - | - | |
| MAPK8IP1 | c.409T>G | p.Ser137Ala | VUS | 1.311 × 105 | 13.5 | 0 | 2 | 44 |
| MC3R | c.508G>T | p.Val170Phe | VUS/LB | NA | 9.34 | 0 | 4 | 24 |
| MC4R | c.380C>T | p.Ser127Leu | LP | 0.00012 | 25.0 | 8 | 7 | 7 |
| c.731C>A | p.Ala244Glu | VUS | 6.196 × 106 | 25.8 | 21 | 7 | 0 | |
| c.202G>T | p.Ala68Ser | VUS | 6.195 × 107 | 25.0 | 9 | 9 | 6 | |
| c.750_751del | p.Ile251TrpfsTer34 | P | 3.284 × 105 | 32.0 | - | - | - | |
| METTL5 | c.362A>G | p.Asp121Gly | P | 1.367 × 105 | 32.0 | 7 | 12 | 3 |
| MLC1 | c.1027C>G | p.Gln343Glu | VUS | 6.372 × 107 | 7.92 | 0 | 4 | 34 |
| MYT1L | c.1466dup | p.Pro490AlafsTer5 | P | NA | - | - | - | - |
| NAA10 | c.20G>C | p.Arg7Thr | LP | NA | 23.3 | 2 | 7 | 17 |
| NEXMIF | c.4313G>A | p.Gly1438Asp | VUS | 8.256 × 107 | 24.3 | 1 | 3 | 19 |
| NTRK2 | c.2143C>T | p.Arg715Trp | LP | NA | 28.7 | 26 | 5 | 0 |
| ODC1 | c.508T>C | p.Phe170Leu | VUS/LP | 1.239 × 106 | 31.0 | 21 | 6 | 2 |
| PACS1 | c.755C>T | p.Ser252Phe | LP | NA | 26.7 | 7 | 10 | 6 |
| PCSK1 | c.1121G>T | p.Cys374Phe | LP | NA | 25.8 | 24 | 5 | 0 |
| PDE4D | c.809-58G>C | - | VUS/LB | 5.208 × 106 | 15.84 | - | - | - |
| PDE11A | c.1298C>T | p.Ser433Leu | VUS | 5.778 × 105 | 24.5 | 5 | 8 | 7 |
| PHIP | c.685T>G | p.Ser229Ala | VUS | NA | 26.5 | 5 | 7 | 14 |
| PLIN1 | c.445G>T | p.Gly149Trp | VUS | 7.434 × 106 | 16.52 | 4 | 4 | 20 |
| POMC | c.616G>T | p.Glu206Ter | VUS | 0.00035 | 39.0 | 2 | 4 | 2 |
| c.599_604dup | p.Ala201_Gln202insArgAla | VUS | 0.00035 | 7.15 | - | - | - | |
| c.394C>G | p.Pro132Ala | LB | 0.0019 | 10.1 | 1 | 5 | 24 | |
| RPL13 | c.578G>A | p.Gly193Asp | VUS | NA | 29.4 | 21 | 6 | 0 |
| SIM1 | c.125del | p.Ile42LysfsTer11 | P | NA | - | - | - | - |
| SLC2A2 | c.1039G>A | p.Ala347Thr | VUS | 9.131 × 105 | 13.0 | 0 | 5 | 23 |
| SOS1 | c.2566A>G | p.Ile856Val | VUS | NA | 22.2 | 4 | 9 | 12 |
| SPEN | c.9354C>G | p.His3118Gln | VUS | NA | 19.2 | 0 | 3 | 26 |
| SPEN | c.10704+2T>G | - | LP | NA | 35.0 | 13 | 2 | 0 |
| SPTAN1 | c.2872-2A>G | - | LP | NA | 34.0 | 15 | 2 | 0 |
| SPTBN1 | c.844G>T | p.Ala282Ser | NA | 24.5 | 3 | 11 | 8 | |
| STAG1 | c.1562A>C | p.Gln521Pro | VUS | NA | 27.9 | 9 | 9 | 6 |
| TNPO2 | c.771+7_771+14delinsG | - | VUS | NA | - | - | - | - |
| TRAPPC9 | c.3013C>T | p.Gln1005Ter | P | 6.353 × 107 | 51.0 | 6 | 2 | 2 |
| TRIP12 | c.1349T>A | p.Leu450Ter | P | NA | 39 | 6 | 2 | 2 |
| TUB | c.8G>T | p.Gly3Val | VUS | NA | 27.3 | 3 | 6 | 10 |
| WFS1 | c.1943G>A | p.Trp648Ter | P | 8.05 × 106 | 53.0 | 4 | 3 | 2 |
| c.1244T>A | p.Val415Asp | LP | NA | 24.0 | 12 | 11 | 3 | |
| ZMYND11 | c.1109G>A | p.Arg370Gln | VUS | 5.581 × 106 | 23.0 | - | 5 | 23 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minniakhmetov, I.R.; Khusainova, R.I.; Vasyukova, O.V.; Kopytina, D.A.; Yalaev, B.I.; Salakhov, R.R.; Guseynova, R.M.; Peterkova, V.A.; Mokrysheva, N.G. Molecular Genetic Architecture of Morbid Obesity in Russian Children. Biomedicines 2025, 13, 756. https://doi.org/10.3390/biomedicines13030756
Minniakhmetov IR, Khusainova RI, Vasyukova OV, Kopytina DA, Yalaev BI, Salakhov RR, Guseynova RM, Peterkova VA, Mokrysheva NG. Molecular Genetic Architecture of Morbid Obesity in Russian Children. Biomedicines. 2025; 13(3):756. https://doi.org/10.3390/biomedicines13030756
Chicago/Turabian StyleMinniakhmetov, Ildar R., Rita I. Khusainova, Olga V. Vasyukova, Daria A. Kopytina, Bulat I. Yalaev, Ramil R. Salakhov, Raisat M. Guseynova, Valentina A. Peterkova, and Natalia G. Mokrysheva. 2025. "Molecular Genetic Architecture of Morbid Obesity in Russian Children" Biomedicines 13, no. 3: 756. https://doi.org/10.3390/biomedicines13030756
APA StyleMinniakhmetov, I. R., Khusainova, R. I., Vasyukova, O. V., Kopytina, D. A., Yalaev, B. I., Salakhov, R. R., Guseynova, R. M., Peterkova, V. A., & Mokrysheva, N. G. (2025). Molecular Genetic Architecture of Morbid Obesity in Russian Children. Biomedicines, 13(3), 756. https://doi.org/10.3390/biomedicines13030756