

Multi-Omics Integration Reveals Mitochondrial Gene Regulation as a Determinant of Tuberculosis Susceptibility: A Mendelian Randomization Approach

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
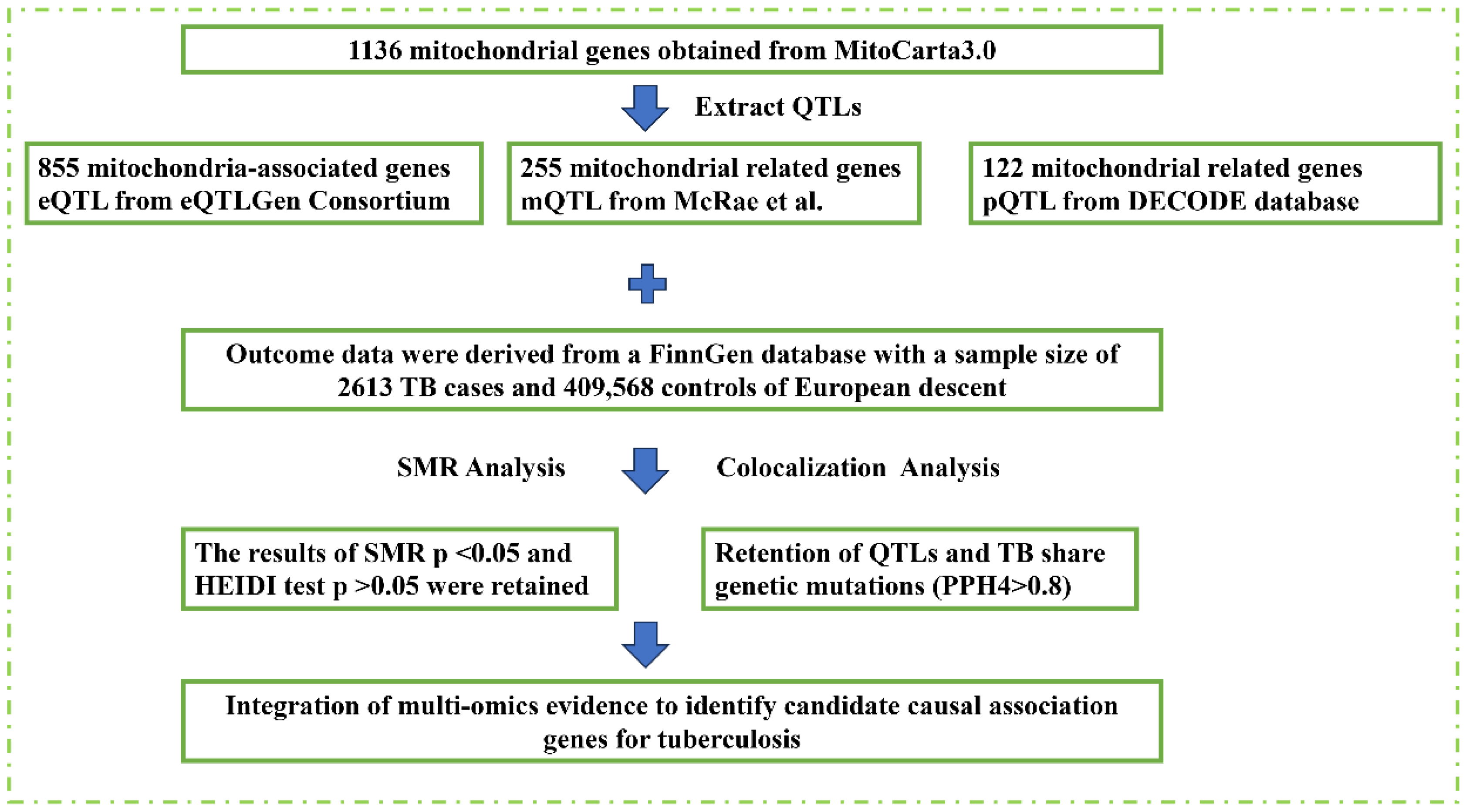
2.1. Data Sources and Quality Control
2.2. Mendelian Randomization and Colocalization Analysis
2.3. Differential Expression and Functional Enrichment Analysis
2.4. Feature Gene Analysis and Validation
2.5. Immune Cell Infiltration Analysis
3. Results
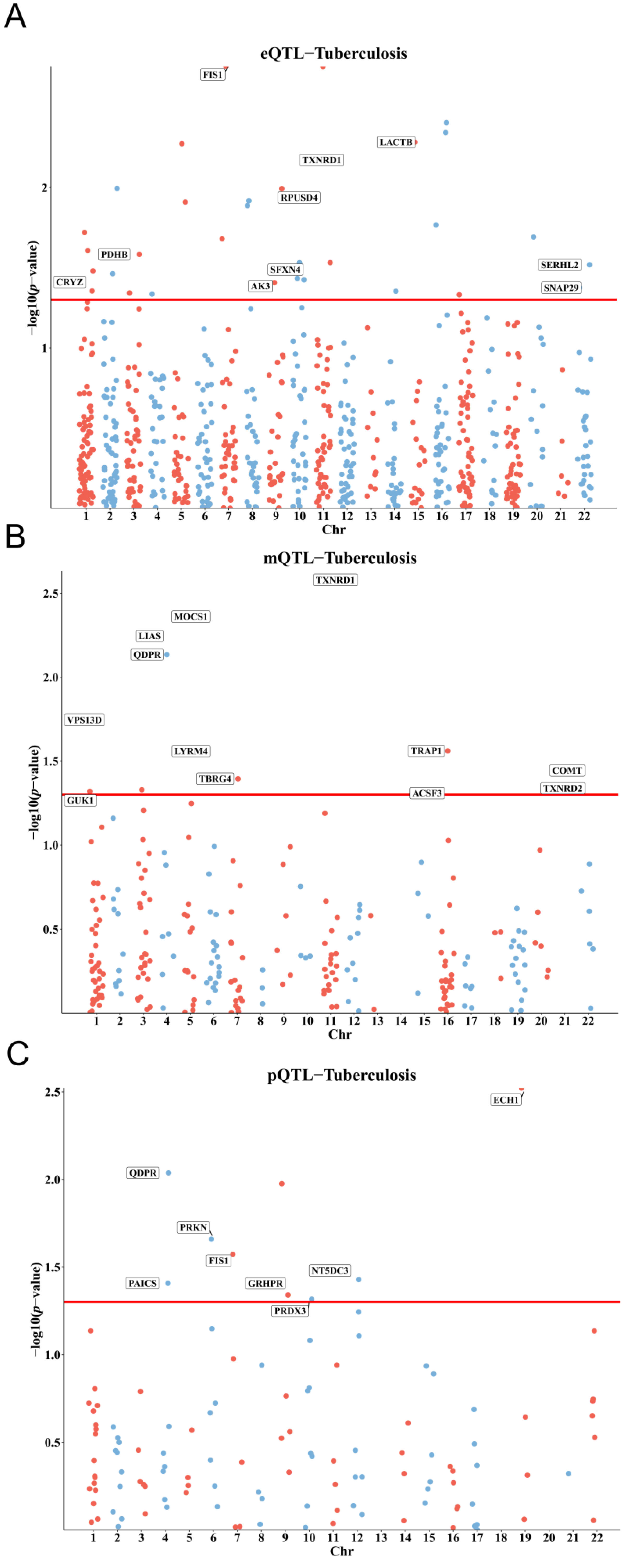
3.1. Genetic, Epigenetic, and Protein-Level Associations of Mitochondrial Genes with TB Susceptibility
3.2. Colocalization and Integration of Multi-Omics Evidence for Causal Genes
3.3. Differential Expression Analysis and Identification of Key Mitochondrial Genes
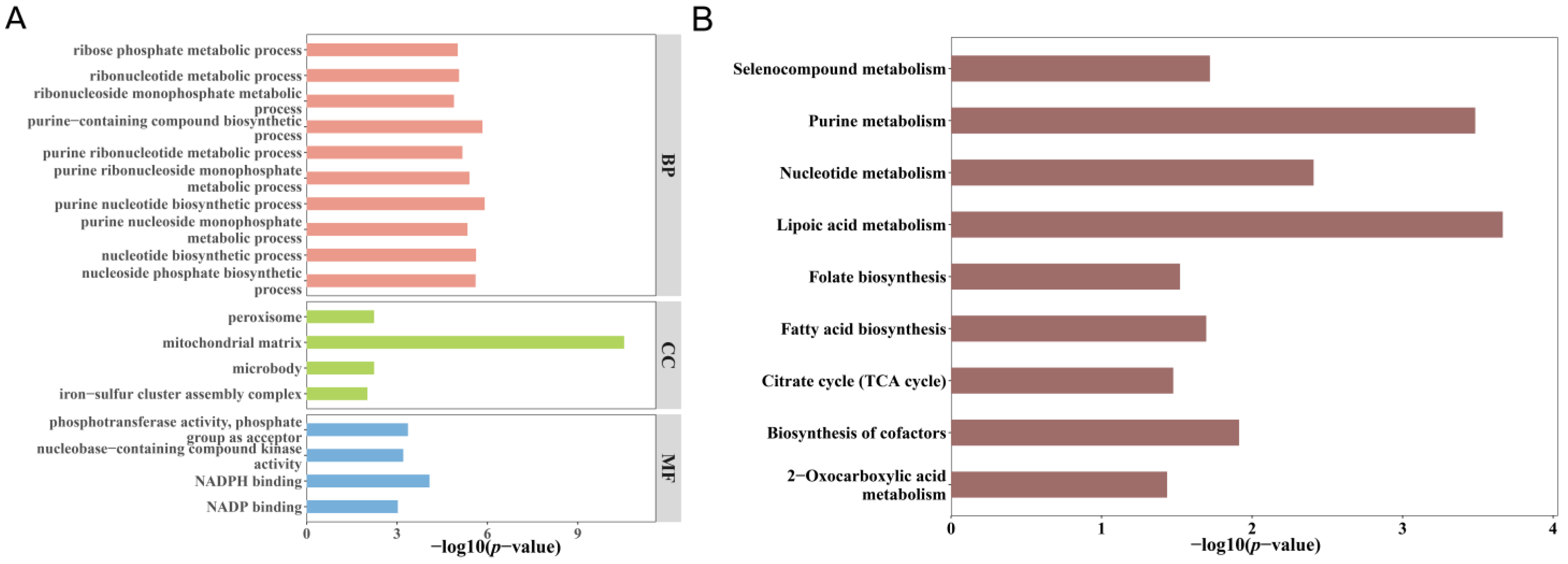
3.4. Functional Enrichment of Overlapping Genes
3.5. Identification and Validation of Key Predictive Genes
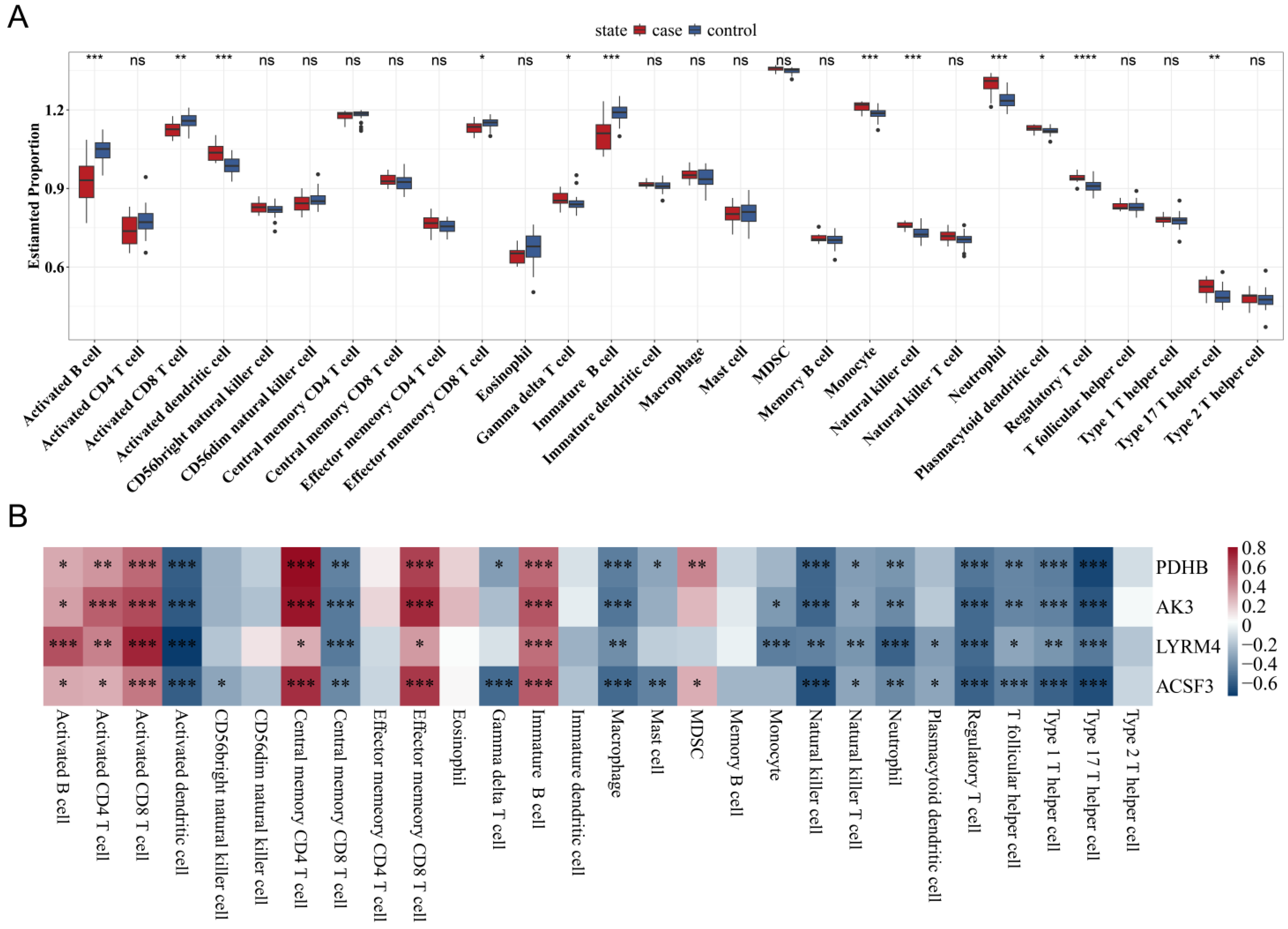
3.6. Correlation Between Mitochondrial Gene Expression and Immune Cell Infiltration
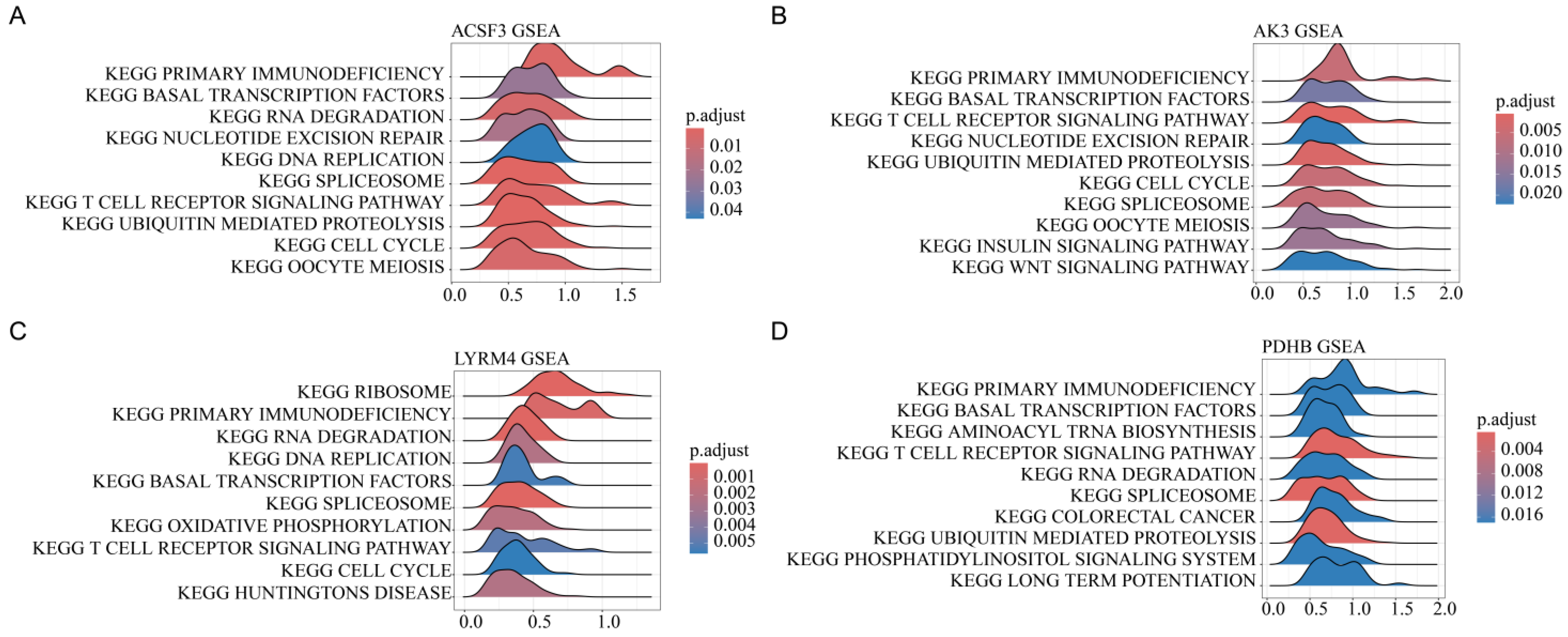
3.7. Pathway Enrichment Analysis of Mitochondrial Gene Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lange, C.; Aaby, P.; Behr, M.A.; Donald, P.R.; Kaufmann, S.H.E.; Netea, M.G.; Mandalakas, A.M. 100 years of Mycobacterium bovis bacille Calmette-Guerin. Lancet Infect. Dis. 2022, 22, e2–e12. [Google Scholar] [CrossRef] [PubMed]
- Bagcchi, S. WHO’s Global Tuberculosis Report 2022. Lancet Microbe 2023, 4, e20. [Google Scholar] [CrossRef] [PubMed]
- Abhimanyu Longlax, S.C.; Nishiguchi, T.; Ladki, M.; Sheikh, D.; Martinez, A.L.; Mace, E.M.; Grimm, S.L.; Caldwell, T.; Varela, A.P. TCA metabolism regulates DNA hypermethylation in LPS and Mycobacterium tuberculosis-induced immune tolerance. Proc. Natl. Acad. Sci. USA 2024, 121, e1890126175. [Google Scholar]
- Krug, S.; Gupta, M.; Kumar, P.; Feller, L.; Ihms, E.A.; Kang, B.G.; Srikrishna, G.; Dawson, T.M.; Dawson, V.L.; Bishai, W.R. Inhibition of host PARP1 contributes to the anti-inflammatory and antitubercular activity of pyrazinamide. Nat. Commun. 2023, 14, 8161. [Google Scholar] [CrossRef]
- Murphy, M.P.; O’Neill, L. A break in mitochondrial endosymbiosis as a basis for inflammatory diseases. Nature 2024, 626, 271–279. [Google Scholar] [CrossRef]
- Monzel, A.S.; Enriquez, J.A.; Picard, M. Multifaceted mitochondria: Moving mitochondrial science beyond function and dysfunction. Nat. Metab. 2023, 5, 546–562. [Google Scholar] [CrossRef]
- Hu, M.M.; Shu, H.B. Mitochondrial DNA-triggered innate immune response: Mechanisms and diseases. Cell. Mol. Immunol. 2023, 20, 1403–1412. [Google Scholar] [CrossRef]
- Andrews, J.T.; Zhang, Z.; Prasad, G.; Huey, F.; Nazarova, E.V.; Wang, J.; Ranaraja, A.; Weinkopff, T.; Li, L.-X.; Mu, S.; et al. Metabolically active neutrophils represent a permissive niche for Mycobacterium tuberculosis. Mucosal Immunol. 2024, 17, 825–842. [Google Scholar] [CrossRef]
- Cumming, B.M.; Addicott, K.W.; Maruri, F.; Pillay, V.; Asmal, R.; Moodley, S.; Barreto-Durate, B.; Araújo-Pereira, M.; Mazibuko, M.; Mhlane, Z.; et al. Longitudinal mitochondrial bioenergetic signatures of blood monocytes and lymphocytes improve during treatment of drug-susceptible pulmonary tuberculosis patients Monocyte/lymphocyte bioenergetic signatures post-TB treatment. Front. Immunol. 2024, 15, 1465448. [Google Scholar] [CrossRef]
- Gao, Y.; Mi, N.; Wu, W.; Zhao, Y.; Fan, F.; Liao, W.; Ming, Y.; Guan, W.; Bai, C. Transfer of inflammatory mitochondria via extracellular vesicles from M1 macrophages induces ferroptosis of pancreatic beta cells in acute pancreatitis. J. Extracell. Vesicles 2024, 13, e12410. [Google Scholar] [CrossRef]
- Jiang, T.; Zhu, K.; Kang, G.; Wu, G.; Wang, L.; Tan, Y. Infectious viruses and neurodegenerative diseases: The mitochondrial defect hypothesis. Rev. Med. Virol. 2024, 34, e2565. [Google Scholar] [CrossRef] [PubMed]
- Headley, C.A.; Gautam, S.; Olmo-Fontanez, A.; Garcia-Vilanova, A.; Dwivedi, V.; Schami, A.; Weintraub, S.; Tsao, P.S.; Torrelles, J.B.; Turner, J. Mitochondrial Transplantation Promotes Protective Effector and Memory CD4(+) T Cell Response During Mycobacterium Tuberculosis Infection and Diminishes Exhaustion and Senescence in Elderly CD4(+) T cells. Adv. Sci. 2024, 11, e2401077. [Google Scholar] [CrossRef] [PubMed]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef] [PubMed]
- Vosa, U.; Claringbould, A.; Westra, H.J.; Bonder, M.J.; Deelen, P.; Zeng, B.; Kirsten, H.; Saha, A.; Kreuzhuber, R.; Yazar, S.; et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 2021, 53, 1300–1310. [Google Scholar] [CrossRef]
- Mcrae, A.F.; Marioni, R.E.; Shah, S.; Yang, J.; Powell, J.E.; Harris, S.E.; Gibson, J.; Henders, A.K.; Bowdler, L.; Painter, J.N.; et al. Identification of 55,000 Replicated DNA Methylation QTL. Sci. Rep. 2018, 8, 17605. [Google Scholar] [CrossRef]
- Ferkingstad, E.; Sulem, P.; Atlason, B.A.; Sveinbjornsson, G.; Styrmisdottir, E.L.; Gunnarsdottir, K.; Helgason, A.; Oddsson, A.; Halldorsson, B.V.; Jensson, B.O.; et al. Large-scale integration of the plasma proteome with genetics and disease. Nat. Genet. 2021, 53, 1712–1721. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef]
- Timmers, P.; Wilson, J.F.; Joshi, P.K.; Deelen, J. Multivariate genomic scan implicates novel loci and haem metabolism in human ageing. Nat. Commun. 2020, 11, 3570. [Google Scholar] [CrossRef]
- Giambartolomei, C.; Vukcevic, D.; Schadt, E.E.; Franke, L.; Hingorani, A.D.; Wallace, C.; Plagnol, V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014, 10, e1004383. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, T.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. ClusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Alderden, J.; Pepper, G.A.; Wilson, A.; Whitney, J.D.; Richardson, S.; Butcher, R.; Jo, Y.; Cummins, M.R. Predicting Pressure Injury in Critical Care Patients: A Machine-Learning Model. Am. J. Crit. Care 2018, 27, 461–468. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef]
- Cai, S.; Zhao, M.; Zhou, B.; Yoshii, A.; Bugg, D.; Villet, O.; Sahu, A.; Olson, G.S.; Davis, J.; Tian, R. Mitochondrial dysfunction in macrophages promotes inflammation and suppresses repair after myocardial infarction. J. Clin. Investig. 2023, 133, e159498. [Google Scholar] [CrossRef]
- Rossmann, M.P.; Dubois, S.M.; Agarwal, S.; Zon, L.I. Mitochondrial function in development and disease. Dis. Models Mech. 2021, 14, dmm048912. [Google Scholar] [CrossRef]
- Guo, H.; Wang, Q.; Ghneim, K.; Wang, L.; Rampanelli, E.; Holley-Guthrie, E.; Cheng, L.; Garrido, C.; Margolis, D.M.; Eller, L.A.; et al. Multi-omics analyses reveal that HIV-1 alters CD4(+) T cell immunometabolism to fuel virus replication. Nat. Immunol. 2021, 22, 423–433. [Google Scholar] [CrossRef]
- Ahamad, N.; Gupta, S.; Parashar, D. Using Omics to Study Leprosy, Tuberculosis, and Other Mycobacterial Diseases. Front. Cell. Infect. Microbiol. 2022, 12, 792617. [Google Scholar] [CrossRef]
- Rajwani, R.; Galata, C.; Lee, A.; So, P.-K.; Leung, K.S.S.; Tam, K.K.G.; Shehzad, S.; Ng, T.T.L.; Zhu, L.; Lao, H.Y.; et al. A multi-omics investigation into the mechanisms of hyper-virulence in Mycobacterium tuberculosis. Virulence 2022, 13, 1088–1100. [Google Scholar] [CrossRef]
- Sumathipala, D.; Stromme, P.; Fattahi, Z.; Lüders, T.; Sheng, Y.; Kahrizi, K.; Einarsen, I.H.; Sloan, J.L.; Najmabadi, H.; Heuvel, L.v.D.; et al. ZBTB11 dysfunction: Spectrum of brain abnormalities, biochemical signature and cellular consequences. Brain 2022, 145, 2602–2616. [Google Scholar] [CrossRef]
- Sun, R.; Kang, X.; Zhao, Y.; Wang, Z.; Wang, R.; Fu, R.; Li, Y.; Hu, Y.; Wang, Z.; Shan, W.; et al. Sirtuin 3-mediated deacetylation of acyl-CoA synthetase family member 3 by protocatechuic acid attenuates non-alcoholic fatty liver disease. Br. J. Pharmacol. 2020, 177, 4166–4180. [Google Scholar] [CrossRef]
- Rogne, P.; Dulko-Smith, B.; Goodman, J.; Rosselin, M.; Grundström, C.; Hedberg, C.; Nam, K.; Sauer-Eriksson, A.E.; Wolf-Watz, M. Structural Basis for GTP versus ATP Selectivity in the NMP Kinase AK3. Biochemistry 2020, 59, 3570–3581. [Google Scholar] [CrossRef]
- Jiang, C.; Lu, Y.; Zhu, R.; Zong, Y.; Huang, Y.; Wang, D.; Da, Z.; Yu, B.; Shen, L.; Cao, Q. Pyruvate dehydrogenase beta subunit (Pdhb) promotes peripheral axon regeneration by regulating energy supply and gene expression. Exp. Neurol. 2023, 363, 114368. [Google Scholar] [CrossRef]
- Jiang, X.; Ji, S.; Yuan, F.; Li, T.; Cui, S.; Wang, W.; Ye, X.; Wang, R.; Chen, Y.; Zhu, S. Pyruvate dehydrogenase B regulates myogenic differentiation via the FoxP1-Arih2 axis. J. Cachexia Sarcopenia Muscle 2023, 14, 606–621. [Google Scholar] [CrossRef]
- Pang, Y.; Tan, G.; Yang, X.; Lin, Y.; Chen, Y.; Zhang, J.; Xie, T.; Zhou, H.; Fang, J.; Zhao, Q.; et al. Iron-sulphur cluster biogenesis factor LYRM4 is a novel prognostic biomarker associated with immune infiltrates in hepatocellular carcinoma. Cancer Cell Int. 2021, 21, 463. [Google Scholar] [CrossRef]
- Pisu, D.; Huang, L.; Narang, V.; Theriault, M.; Lê-Bury, G.; Lee, B.; Lakudzala, A.E.; Mzinza, D.T.; Mhango, D.V.; Mitini-Nkhoma, S.C.; et al. Single cell analysis of M. tuberculosis phenotype and macrophage lineages in the infected lung. J. Exp. Med. 2021, 218, e20210615. [Google Scholar] [CrossRef]
- Ahmad, F.; Rani, A.; Alam, A.; Zarin, S.; Pandey, S.; Singh, H.; Hasnain, S.E.; Ehtesham, N.Z. Macrophage: A Cell With Many Faces and Functions in Tuberculosis. Front. Immunol. 2022, 13, 747799. [Google Scholar] [CrossRef]
- Schurz, H.; Naranbhai, V.; Yates, T.A.; Gilchrist, J.J.; Parks, T.; Dodd, P.J.; Möller, M.; Hoal, E.G.; Morris, A.P.; Hill, A.V.; et al. Multi-ancestry meta-analysis of host genetic susceptibility to tuberculosis identifies shared genetic architecture. eLife 2024, 13, e84394. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | pQTL | mQTL | eQTL | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| p_SMR | p_HEIDI | OR (95% CI) | p_SMR | p_HEIDI | probeID | OR (95% CI) | p_SMR | p_HEIDI | OR (95% CI) | |
| QDPR | 0.00917 | 0.718 | 1.2473 (1.0563 to 1.4729) | 0.0073 | 0.2330 | cg08808571 | 0.8901 (0.8174 to 0.9692) | |||
| 0.0071 | 0.2580 | cg26689483 | 0.9173 (0.8614 to 0.9768) | |||||||
| PAICS | 0.0391 | 0.623 | 2.7788 (1.0526 to 7.3362) | |||||||
| PRKN | 0.0219 | 0.082 | 0.1479 (0.0289 to 0.7579) | |||||||
| FIS1 | 0.0267 | 0.811 | 1.5163 (1.0491 to 2.1914) | 0.0018 | 0.4049 | 1.3601 (1.1218 to 1.6489) | ||||
| GRHPR | 0.0456 | 0.132 | 1.2484 (1.0043 to 1.5519) | |||||||
| PRDX3 | 0.0482 | 0.966 | 4.2439 (1.0116 to 17.8040) | |||||||
| NT5DC3 | 0.0372 | 0.082 | 1.4193 (1.0209 to 1.9731) | |||||||
| ECH1 | 0.0030 | 0.636 | 1.3526 (1.1079 to 1.6512) | |||||||
| ACSF3 | 0.0490 | 0.1480 | cg02193283 | 1.2622 (1.0010 to 1.5916) | ||||||
| ACSF3 | 0.0490 | 0.4880 | cg05104581 | 1.2576 (1.0010 to 1.5800) | ||||||
| COMT | 0.0359 | 0.9760 | cg10122187 | 1.1841 (1.0111 to 1.3866) | ||||||
| GUK1 | 0.0479 | 0.2270 | cg12796841 | 1.2462 (1.0021 to 1.5497) | ||||||
| LIAS | 0.0057 | 0.8520 | cg18147735 | 1.1486 (1.0412 to 1.2670) | ||||||
| LIAS | 0.0059 | 0.6690 | cg25669547 | 1.1797 (1.0487 to 1.3271) | ||||||
| LYRM4 | 0.0275 | 0.6110 | cg00563771 | 0.9275 (0.8675 to 0.9917) | ||||||
| MOCS1 | 0.0043 | 0.9110 | cg10871120 | 1.0976 (1.0296 to 1.1702) | ||||||
| TBRG4 | 0.0403 | 0.9950 | cg07992143 | 0.6720 (0.4596 to 0.9826) | ||||||
| TRAP1 | 0.0275 | 0.6070 | cg02264195 | 0.6152 (0.3994 to 0.9475) | ||||||
| TXNRD1 | 0.0023 | 0.1930 | cg25684105 | 1.4748 (1.1485 to 1.8938) | ||||||
| TXNRD2 | 0.0457 | 0.9660 | cg11182965 | 0.8736 (0.7652 to 0.9974) | ||||||
| VPS13D | 0.0159 | 0.4980 | cg09686497 | 1.2873 (1.0484 to 1.5806) | ||||||
| VPS13D | 0.0201 | 0.3170 | cg24602020 | 1.1231 (1.0183 to 1.2386) | ||||||
| CRYZ | 0.0441 | 0.7625 | 0.8411 (0.7107 to 0.9954) | |||||||
| PDHB | 0.0261 | 0.1126 | 1.2471 (1.0266 to 1.5149) | |||||||
| AK3 | 0.0392 | 0.2904 | 1.3196 (1.0139 to 1.7176) | |||||||
| SFXN4 | 0.0369 | 0.8596 | 0.8345 (0.7041 to 0.9890) | |||||||
| RPUSD4 | 0.0115 | 0.7258 | 0.8342 (0.7248 to 0.9602) | |||||||
| TXNRD1 | 0.0067 | 0.1326 | 1.4405 (1.1066 to 1.8751) | |||||||
| LACTB | 0.0052 | 0.1275 | 1.4885 (1.1261 to 1.9676) | |||||||
| SNAP29 | 0.0421 | 0.7399 | 0.8594 (0.7427 to 0.9946) | |||||||
| SERHL2 | 0.0303 | 0.5571 | 1.5837 (1.0448 to 2.4005) | |||||||
| QTL, quantitative trait loci | ||||||||||
| Gene | p-Value | OR | OR (95% CI) |
|---|---|---|---|
| SERHL2 | 0.001 | 0.0682 | 0.0682 (0.0138 to 0.3369) |
| TBRG4 | 0.0013 | 0.0449 | 0.0449 (0.0068 to 0.2979) |
| LIAS | 0.0014 | 0.0149 | 0.0149 (0.0011 to 0.1978) |
| NT5DC3 | 0.0021 | 0.0025 | 0.0025 (0.0001 to 0.1145) |
| TRAP1 | 0.0023 | 0.1349 | 0.1349 (0.0372 to 0.4891) |
| LYRM4 | 0.0027 | 1.00 × 10−4 | 0.0001 (0.0000 to 0.0381) |
| PAICS | 0.0033 | 0.1412 | 0.1412 (0.0383 to 0.5201) |
| AK3 | 0.0045 | 0.2057 | 0.2057 (0.0692 to 0.6117) |
| TXNRD2 | 0.0122 | 0.0488 | 0.0488 (0.0046 to 0.5178) |
| ACSF3 | 0.0134 | 0.0546 | 0.0546 (0.0055 to 0.5471) |
| PDHB | 0.0136 | 0.2084 | 0.2084 (0.0600 to 0.7245) |
| RPUSD4 | 0.0157 | 0.2447 | 0.2447 (0.0781 to 0.7671) |
| GUK1 | 0.0158 | 3.45 | 3.4500 (1.2623 to 9.4289) |
| CRYZ | 0.0191 | 0.3339 | 0.3339 (0.1334 to 0.8359) |
| QDPR | 0.0431 | 0.2739 | 0.2739 (0.0781 to 0.9603) |
| ECH1 | 0.0681 | 0.2731 | 0.2731 (0.0677 to 1.1014) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, T.; Chen, Y.; Yuan, F.; Ma, Y.; Wang, Q.; Yao, Y.; Cai, S.; Jin, W.; Miao, Q.; Hu, B. Multi-Omics Integration Reveals Mitochondrial Gene Regulation as a Determinant of Tuberculosis Susceptibility: A Mendelian Randomization Approach. Biomedicines 2025, 13, 749. https://doi.org/10.3390/biomedicines13030749
Fang T, Chen Y, Yuan F, Ma Y, Wang Q, Yao Y, Cai S, Jin W, Miao Q, Hu B. Multi-Omics Integration Reveals Mitochondrial Gene Regulation as a Determinant of Tuberculosis Susceptibility: A Mendelian Randomization Approach. Biomedicines. 2025; 13(3):749. https://doi.org/10.3390/biomedicines13030749
Chicago/Turabian StyleFang, Tingting, Yu Chen, Feifei Yuan, Yuyan Ma, Qingqing Wang, Yumeng Yao, Sishi Cai, Wenting Jin, Qing Miao, and Bijie Hu. 2025. "Multi-Omics Integration Reveals Mitochondrial Gene Regulation as a Determinant of Tuberculosis Susceptibility: A Mendelian Randomization Approach" Biomedicines 13, no. 3: 749. https://doi.org/10.3390/biomedicines13030749
APA StyleFang, T., Chen, Y., Yuan, F., Ma, Y., Wang, Q., Yao, Y., Cai, S., Jin, W., Miao, Q., & Hu, B. (2025). Multi-Omics Integration Reveals Mitochondrial Gene Regulation as a Determinant of Tuberculosis Susceptibility: A Mendelian Randomization Approach. Biomedicines, 13(3), 749. https://doi.org/10.3390/biomedicines13030749