
Ramp Sequence May Explain Synonymous Variant Association with Alzheimer’s Disease in the Paired Immunoglobulin-like Type 2 Receptor Alpha (PILRA)

 ,
,  , , , , , and
, , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Identifying AD-Associated Genetic Variants
2.2. Identifying Ramp Sequences
2.3. Biological Assessment of Ramp Sequence Effects in PILRA
2.4. Transfection of Wildtype and Mutant Transcripts
2.5. qPCR Protocol
2.6. ELISA Protocol
3. Results
3.1. Ramp Sequence Variation Caused by Exonic GWAS Hits
3.2. PILRA Ramp Sequence
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| CHO | Chinese hamster ovary |
| RQC | Ribosome-associated protein quality control |
References
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of Genes and Environments for Explaining Alzheimer Disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Bellenguez, C.; Kucukali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef] [PubMed]
- Balin, B.J.; Hudson, A.P. Etiology and Pathogenesis of Late-Onset Alzheimer’s Disease. Curr. Allergy Asthma Rep. 2014, 14, 417. [Google Scholar] [CrossRef]
- Marioni, R.E.; Harris, S.E.; Zhang, Q.; McRae, A.F.; Hagenaars, S.P.; Hill, W.D.; Davies, G.; Ritchie, C.W.; Gale, C.R.; Starr, J.M.; et al. GWAS on family history of Alzheimer’s disease. Transl. Psychiatry 2018, 8, 99. [Google Scholar] [CrossRef]
- Jun, G.; Naj, A.C.; Beecham, G.W.; Wang, L.S.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Ertekin-Taner, N.; Fallin, M.D.; Friedland, R.; et al. Meta-analysis Confirms CR1, CLU, and PICALM as Alzheimer Disease Risk Loci and Reveals Interactions with APOE Genotypes. Arch. Neurol. 2010, 67, 1473–1484. [Google Scholar] [CrossRef]
- Hu, X.; Pickering, E.; Liu, Y.C.; Hall, S.; Fournier, H.; Katz, E.; Dechairo, B.; John, S.; van Eerdewegh, P.; Soares, H.; et al. Meta-Analysis for Genome-Wide Association Study Identifies Multiple Variants at the BIN1 Locus Associated with Late-Onset Alzheimer’s Disease. PLoS ONE 2011, 6, e16616. [Google Scholar] [CrossRef]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef]
- Ridge, P.G.; Hoyt, K.B.; Boehme, K.; Mukherjee, S.; Crane, P.K.; Haines, J.L.; Mayeux, R.; Farrer, L.A.; Pericak-Vance, M.A.; Schellenberg, G.D.; et al. Assessment of the genetic variance of late-onset Alzheimer’s disease. Neurobiol. Aging 2016, 41, 200.e13–200.e20. [Google Scholar] [CrossRef]
- Andrews, S.J.; Renton, A.E.; Fulton-Howard, B.; Podlesny-Drabiniok, A.; Marcora, E.; Goate, A.M. The complex genetic architecture of Alzheimer’s disease: Novel insights and future directions. EBioMedicine 2023, 90, 104511. [Google Scholar] [CrossRef]
- Escott-Price, V.; Sims, R.; Bannister, C.; Harold, D.; Vronskaya, M.; Majounie, E.; Badarinarayan, N.; Morgan, K.; Passmore, P.; Holmes, C.; et al. Common polygenic variation enhances risk prediction for Alzheimer’s disease. Brain 2015, 138 Pt 12, 3673–3684. [Google Scholar] [CrossRef]
- Bakulski, K.M.; Vadari, H.S.; Faul, J.D.; Heeringa, S.G.; Kardia, S.L.R.; Langa, K.M.; Smith, J.A.; Manly, J.J.; Mitchell, C.M.; Benke, K.S.; et al. Cumulative Genetic Risk and APOE ε4 Are Independently Associated With Dementia Status in a Multiethnic, Population-Based Cohort. Neurol. Genet. 2021, 7, e576. [Google Scholar] [CrossRef] [PubMed]
- Bredesen, D.E. Metabolic profiling distinguishes three subtypes of Alzheimer’s disease. Aging 2015, 7, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.; Verhagen, C.; Hernandez-Cabrera, J.A.; Cavallin, L.; Guo, C.J.; Ekman, U.; Muehlboeck, J.S.; Simmons, A.; Barroso, J.; Wahlund, L.O.; et al. Distinct subtypes of Alzheimer’s disease based on patterns of brain atrophy: Longitudinal trajectories and clinical applications. Sci. Rep. 2017, 7, 46263. [Google Scholar] [CrossRef] [PubMed]
- Eppig, J.S.; Edmonds, E.C.; Campbell, L.; Sanderson-Cimino, M.; Delano-Wood, L.; Bondi, M.W.; for the Alzheimer’s Disease Neuroimaging Initiative. Statistically derived subtypes and associations with cerebrospinal fluid and genetic biomarkers in mild cognitive impairment: A latent profile analysis. J. Int. Neuropsychol. Soc. 2017, 23, 564–576. [Google Scholar] [CrossRef]
- Squitti, R.; Ventriglia, M.; Gennarelli, M.; Colabufo, N.A.; El Idrissi, I.G.; Bucossi, S.; Mariani, S.; Rongioletti, M.; Zanetti, O.; Congiu, C. Non-ceruloplasmin copper distincts subtypes in Alzheimer’s disease: A genetic study of ATP7B frequency. Mol. Neurobiol. 2017, 54, 671–681. [Google Scholar] [CrossRef]
- Mao, Y.-F.; Guo, Z.-Y.; Pu, J.-L.; Chen, Y.-X.; Zhang, B.-R. Association of CD33 and MS4A cluster variants with Alzheimer’s disease in East Asian populations. Neurosci. Lett. 2015, 609, 235–239. [Google Scholar] [CrossRef]
- Mann, U.M.; Mohr, E.; Gearing, M.; Chase, T.N. Heterogeneity in Alzheimer’s disease: Progression rate segregated by distinct neuropsychological and cerebral metabolic profiles. J. Neurol. Neurosurg. Psychiatry 1992, 55, 956–959. [Google Scholar] [CrossRef]
- Na, H.K.; Kang, D.R.; Kim, S.; Seo, S.W.; Heilman, K.M.; Noh, Y.; Na, D.L. Malignant progression in parietal-dominant atrophy subtype of Alzheimer’s disease occurs independent of onset age. Neurobiol. Aging 2016, 47, 149–156. [Google Scholar] [CrossRef]
- Park, J.Y.; Na, H.K.; Kim, S.; Kim, H.; Kim, H.J.; Seo, S.W.; Na, D.L.; Han, C.E.; Seong, J.K.; Alzheimer’s Disease Neuroimaging Initiative. Robust Identification of Alzheimer’s Disease subtypes based on cortical atrophy patterns. Sci. Rep. 2017, 7, 43270. [Google Scholar] [CrossRef]
- Persson, K.; Eldholm, R.S.; Barca, M.L.; Cavallin, L.; Ferreira, D.; Knapskog, A.B.; Selbaek, G.; Braekhus, A.; Saltvedt, I.; Westman, E.; et al. MRI-assessed atrophy subtypes in Alzheimer’s disease and the cognitive reserve hypothesis. PLoS ONE 2017, 12, e0186595. [Google Scholar] [CrossRef]
- Varol, E.; Sotiras, A.; Davatzikos, C. HYDRA: Revealing heterogeneity of imaging and genetic patterns through a multiple max-margin discriminative analysis framework. NeuroImage 2017, 145, 346–364. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Mez, J.; Trittschuh, E.H.; Saykin, A.J.; Gibbons, L.E.; Fardo, D.W.; Wessels, M.; Bauman, J.; Moore, M.; Choi, S.-E.; et al. Genetic data and cognitively defined late-onset Alzheimer’s disease subgroups. Mol. Psychiatry 2018, 25, 2942–2951. [Google Scholar] [CrossRef]
- Warren, J.D.; Fletcher, P.D.; Golden, H.L. The paradox of syndromic diversity in Alzheimer disease. Nat. Rev. Neurol. 2012, 8, 451–464. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S.; et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539–547. [Google Scholar] [CrossRef]
- Bondareff, W.; Mountjoy, C.Q.; Roth, M.; Rossor, M.N.; Iversen, L.L.; Reynolds, G.P. Age and histopathologic heterogeneity in Alzheimer’s disease: Evidence for subtypes. Arch. Gen. Psychiatry 1987, 44, 412–417. [Google Scholar] [CrossRef]
- Crane, P.K.; Trittschuh, E.; Mukherjee, S.; Saykin, A.J.; Sanders, R.E.; Larson, E.B.; McCurry, S.M.; McCormick, W.; Bowen, J.D.; Grabowski, T.; et al. Incidence of cognitively defined late-onset Alzheimer’s dementia subgroups from a prospective cohort study. Alzheimer’s Dement. 2017, 13, 1307–1316. [Google Scholar] [CrossRef]
- Cummings, J.L. Cognitive and behavioral heterogeneity in Alzheimer’s disease: Seeking the neurobiological basis. Neurobiol. Aging 2000, 21, 845–861. [Google Scholar] [CrossRef]
- Larner, A.; Doran, M. Clinical phenotypic heterogeneity of Alzheimer’s disease associated with mutations of the presenilin–1 gene. J. Neurol. 2006, 253, 139–158. [Google Scholar] [CrossRef]
- Pillon, B.; Dubois, B.; Lhermitte, F.; Agid, Y. Heterogeneity of cognitive impairment in progressive supranuclear palsy, Parkinson’s disease, and Alzheimer’s disease. Neurology 1986, 36, 1179. [Google Scholar] [CrossRef]
- Purcell, S.M.; Wray, N.R.; Stone, J.L.; Visscher, P.M.; O’Donovan, M.C.; Sullivan, P.F.; Sklar, P.; Purcell, S.M.; Stone, J.L.; Sullivan, P.F.; et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009, 460, 748–752. [Google Scholar] [CrossRef]
- Lewis, C.M.; Vassos, E. Polygenic risk scores: From research tools to clinical instruments. Genome Med. 2020, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Dittmar, K.A.; Goodenbour, J.M.; Pan, T. Tissue-specific differences in human transfer RNA expression. PLoS Genet. 2006, 2, e221. [Google Scholar] [CrossRef] [PubMed]
- Waldman, Y.Y.; Tuller, T.; Shlomi, T.; Sharan, R.; Ruppin, E. Translation efficiency in humans: Tissue specificity, global optimization and differences between developmental stages. Nucleic Acids Res. 2010, 38, 2964–2974. [Google Scholar] [CrossRef] [PubMed]
- Tuller, T.; Zur, H. Multiple roles of the coding sequence 5’ end in gene expression regulation. Nucleic Acids Res. 2015, 43, 13–28. [Google Scholar] [CrossRef]
- Verma, M.; Choi, J.; Cottrell, K.A.; Lavagnino, Z.; Thomas, E.N.; Pavlovic-Djuranovic, S.; Szczesny, P.; Piston, D.W.; Zaher, H.S.; Puglisi, J.D.; et al. A short translational ramp determines the efficiency of protein synthesis. Nat. Commun. 2019, 10, 5774. [Google Scholar] [CrossRef]
- Miller, J.B.; Brase, L.R.; Ridge, P.G. ExtRamp: A novel algorithm for extracting the ramp sequence based on the tRNA adaptation index or relative codon adaptiveness. Nucleic Acids Res. 2019, 47, 1123–1131. [Google Scholar] [CrossRef]
- Tuller, T.; Veksler-Lublinsky, I.; Gazit, N.; Kupiec, M.; Ruppin, E.; Ziv-Ukelson, M. Composite effects of gene determinants on the translation speed and density of ribosomes. Genome Biol. 2011, 12, R110. [Google Scholar] [CrossRef]
- Tuller, T.; Carmi, A.; Vestsigian, K.; Navon, S.; Dorfan, Y.; Zaborske, J.; Pan, T.; Dahan, O.; Furman, I.; Pilpel, Y. An Evolutionarily Conserved Mechanism for Controlling the Efficiency of Protein Translation. Cell 2010, 141, 344–354. [Google Scholar] [CrossRef]
- Dana, A.; Tuller, T. The effect of tRNA levels on decoding times of mRNA codons. Nucleic Acids Res. 2014, 42, 9171–9181. [Google Scholar] [CrossRef]
- Park, H.; Subramaniam, A.R. Inverted translational control of eukaryotic gene expression by ribosome collisions. PLoS Biol. 2019, 17, e3000396. [Google Scholar] [CrossRef]
- Joazeiro, C.A.P. Mechanisms and functions of ribosome-associated protein quality control. Nat. Rev. Mol. Cell Biol. 2019, 20, 368–383. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, L.M.; Miller, J.B.; Whiting, M.F.; Kauwe, J.S.K.; Ridge, P.G. A comprehensive analysis of the phylogenetic signal in ramp sequences in 211 vertebrates. Sci. Rep. 2021, 11, 622. [Google Scholar] [CrossRef] [PubMed]
- Hodgman, M.W.; Miller, J.B.; Meurs, T.E.; Kauwe, J.S.K. CUBAP: An interactive web portal for analyzing codon usage biases across populations. Nucleic Acids Res. 2020, 48, 11030–11039. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.B.; Meurs, T.E.; Hodgman, M.W.; Song, B.; Miller, K.N.; Ebbert, M.T.W.; Kauwe, J.S.K.; Ridge, P.G. The Ramp Atlas: Facilitating tissue and cell-specific ramp sequence analyses through an intuitive web interface. NAR Genom. Bioinform. 2022, 4, lqac039. [Google Scholar] [CrossRef]
- Wang, J.; Shiratori, I.; Uehori, J.; Ikawa, M.; Arase, H. Neutrophil infiltration during inflammation is regulated by PILRα via modulation of integrin activation. Nat. Immunol. 2013, 14, 34–40. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Pontén, F.; Jirström, K.; Uhlen, M. The Human Protein Atlas—A tool for pathology. J. Pathol. 2008, 216, 387–393. [Google Scholar] [CrossRef]
- Kohyama, M.; Matsuoka, S.; Shida, K.; Sugihara, F.; Aoshi, T.; Kishida, K.; Ishii, K.J.; Arase, H. Monocyte infiltration into obese and fibrilized tissues is regulated by PILRα. Eur. J. Immunol. 2016, 46, 1214–1223. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Huang, K.-l.; Marcora, E.; Pimenova, A.A.; Di Narzo, A.F.; Kapoor, M.; Jin, S.C.; Harari, O.; Bertelsen, S.; Fairfax, B.P.; Czajkowski, J.; et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat. Neurosci. 2017, 20, 1052–1061. [Google Scholar] [CrossRef]
- Li, Y.; Laws, S.M.; Miles, L.A.; Wiley, J.S.; Huang, X.; Masters, C.L.; Gu, B.J. Genomics of Alzheimer’s disease implicates the innate and adaptive immune systems. Cell Mol. Life Sci. 2021, 78, 7397–7426. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.; Brookes, K.J.; Turton, J.; Chaudhury, S.; Guetta-Baranes, T.; Guerreiro, R.; Bras, J.; Hernandez, D.; Singleton, A.; Francis, P.T.; et al. Whole-exome sequencing of the BDR cohort: Evidence to support the role of the PILRA gene in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2018, 44, 506–521. [Google Scholar] [CrossRef] [PubMed]
- Rathore, N.; Ramani, S.R.; Pantua, H.; Payandeh, J.; Bhangale, T.; Wuster, A.; Kapoor, M.; Sun, Y.; Kapadia, S.B.; Gonzalez, L.; et al. Paired Immunoglobulin-like Type 2 Receptor Alpha G78R variant alters ligand binding and confers protection to Alzheimer’s disease. PLoS Genet. 2018, 14, e1007427. [Google Scholar] [CrossRef] [PubMed]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef]
- Harrison, P.W.; Amode, M.R.; Austine-Orimoloye, O.; Azov, A.G.; Barba, M.; Barnes, I.; Becker, A.; Bennett, R.; Berry, A.; Bhai, J. Ensembl 2024. Nucleic Acids Res. 2024, 52, D891–D899. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef]
- Dong, S.; Zhao, N.; Spragins, E.; Kagda, M.S.; Li, M.; Assis, P.; Jolanki, O.; Luo, Y.; Cherry, J.M.; Boyle, A.P. Annotating and prioritizing human non-coding variants with RegulomeDB v. 2. Nat. Genet. 2023, 55, 724–726. [Google Scholar] [CrossRef]
- Consortium, G. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef]
- Noguchi, S.; Arakawa, T.; Fukuda, S.; Furuno, M.; Hasegawa, A.; Hori, F.; Ishikawa-Kato, S.; Kaida, K.; Kaiho, A.; Kanamori-Katayama, M. FANTOM5 CAGE profiles of human and mouse samples. Sci. Data 2017, 4, 1–10. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Ghaemmaghami, S.; Newman, J.R.; Weissman, J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 2009, 324, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.; Zheng-Bradley, X.; Smith, R.; Kulesha, E.; Xiao, C.; Toneva, I.; Vaughan, B.; Preuss, D.; Leinonen, R.; Shumway, M.; et al. The 1000 Genomes Project: Data management and community access. Nat. Methods 2012, 9, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Auer, P.L.; Reiner, A.P.; Wang, G.; Kang, H.M.; Abecasis, G.R.; Altshuler, D.; Bamshad, M.J.; Nickerson, D.A.; Tracy, R.P.; Rich, S.S.; et al. Guidelines for Large-Scale Sequence-Based Complex Trait Association Studies: Lessons Learned from the NHLBI Exome Sequencing Project. Am. J. Hum. Genet. 2016, 99, 791–801. [Google Scholar] [CrossRef]
- Chen, S.; Francioli, L.C.; Goodrich, J.K.; Collins, R.L.; Kanai, M.; Wang, Q.; Alföldi, J.; Watts, N.A.; Vittal, C.; Gauthier, L.D.; et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature 2024, 625, 92–100. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Miller, J.B.; McKinnon, L.M.; Whiting, M.F.; Kauwe, J.S.K.; Ridge, P.G. Codon Pairs are Phylogenetically Conserved: A comprehensive analysis of codon pairing conservation across the Tree of Life. PLoS ONE 2020, 15, e0232260. [Google Scholar] [CrossRef]
- Miller, J.B.; McKinnon, L.M.; Whiting, M.F.; Ridge, P.G. Codon use and aversion is largely phylogenetically conserved across the tree of life. Mol. Phylogenet. Evol. 2020, 144, 106697. [Google Scholar] [CrossRef]
- Miller, J.B.; McKinnon, L.M.; Whiting, M.F.; Ridge, P.G. CAM: An alignment-free method to recover phylogenies using codon aversion motifs. PeerJ 2019, 7, e6984. [Google Scholar] [CrossRef]
- Miller, J.B.; Hippen, A.A.; Belyeu, J.R.; Whiting, M.F.; Ridge, P.G. Missing something? Codon aversion as a new character system in phylogenetics. Cladistics 2017, 33, 545–556. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Ishigaki, K.; Suzuki, A.; Tsuchida, Y.; Tsuchiya, H.; Sumitomo, S.; Nagafuchi, Y.; Miya, F.; Tsunoda, T.; Shoda, H.; et al. Splicing QTL analysis focusing on coding sequences reveals mechanisms for disease susceptibility loci. Nat. Commun. 2022, 13, 4659. [Google Scholar] [CrossRef]
- Kudla, G.; Lipinski, L.; Caffin, F.; Helwak, A.; Zylicz, M. High guanine and cytosine content increases mRNA levels in mammalian cells. PLoS Biol. 2006, 4, e180. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Wright, G.; Emrich, S.; Clark, P.L. %MinMax: A versatile tool for calculating and comparing synonymous codon usage and its impact on protein folding. Protein Sci. 2018, 27, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Cannarozzi, G.; Schraudolph, N.N.; Faty, M.; von Rohr, P.; Friberg, M.T.; Roth, A.C.; Gonnet, P.; Gonnet, G.; Barral, Y. A role for codon order in translation dynamics. Cell 2010, 141, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Brandman, O.; Hegde, R.S. Ribosome-associated protein quality control. Nat. Struct. Mol. Biol. 2016, 23, 7–15. [Google Scholar] [CrossRef]
- Zhou, Z.; Sun, B.; Yu, D.; Bian, M. Roles of tRNA metabolism in aging and lifespan. Cell Death Dis. 2021, 12, 548. [Google Scholar] [CrossRef]
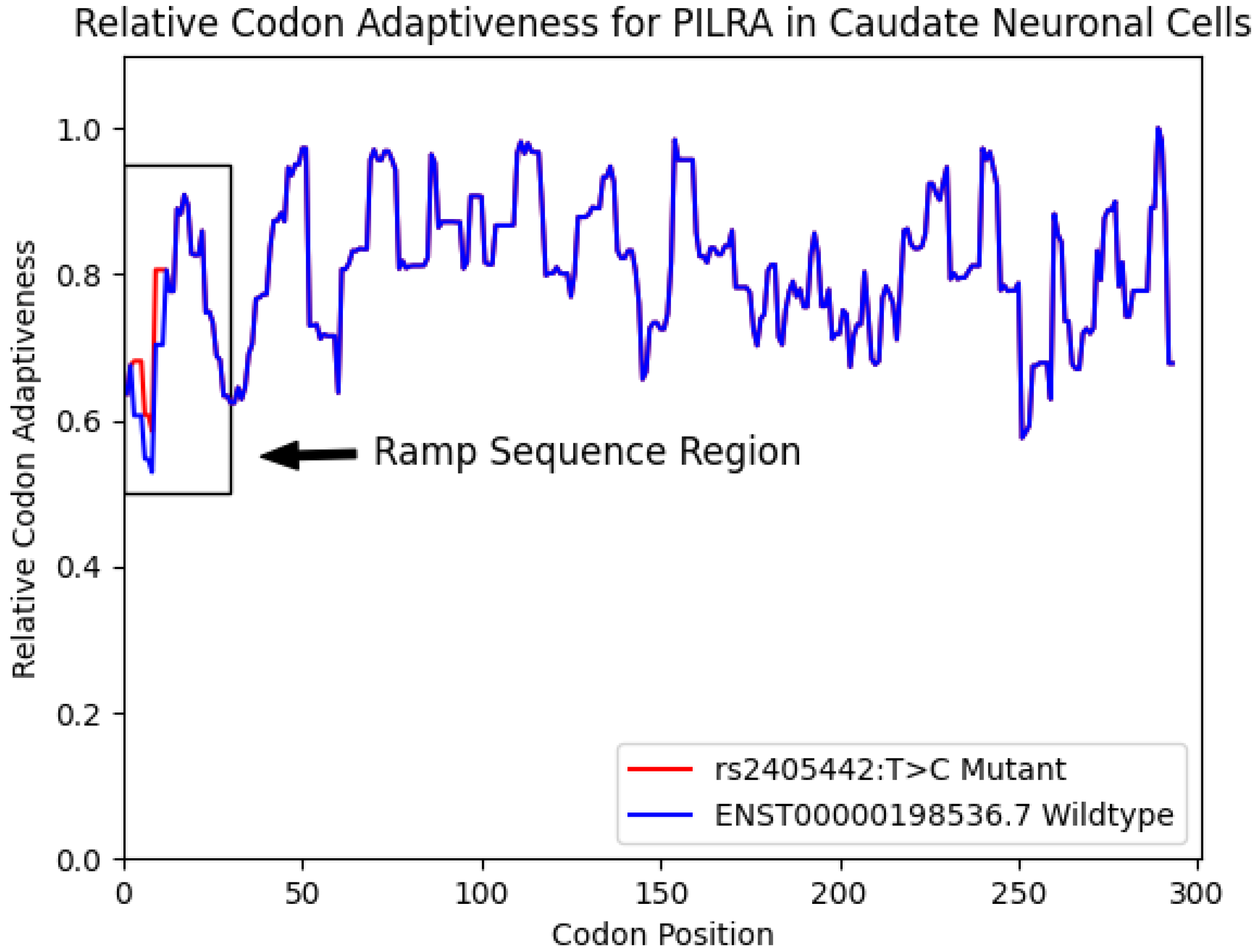
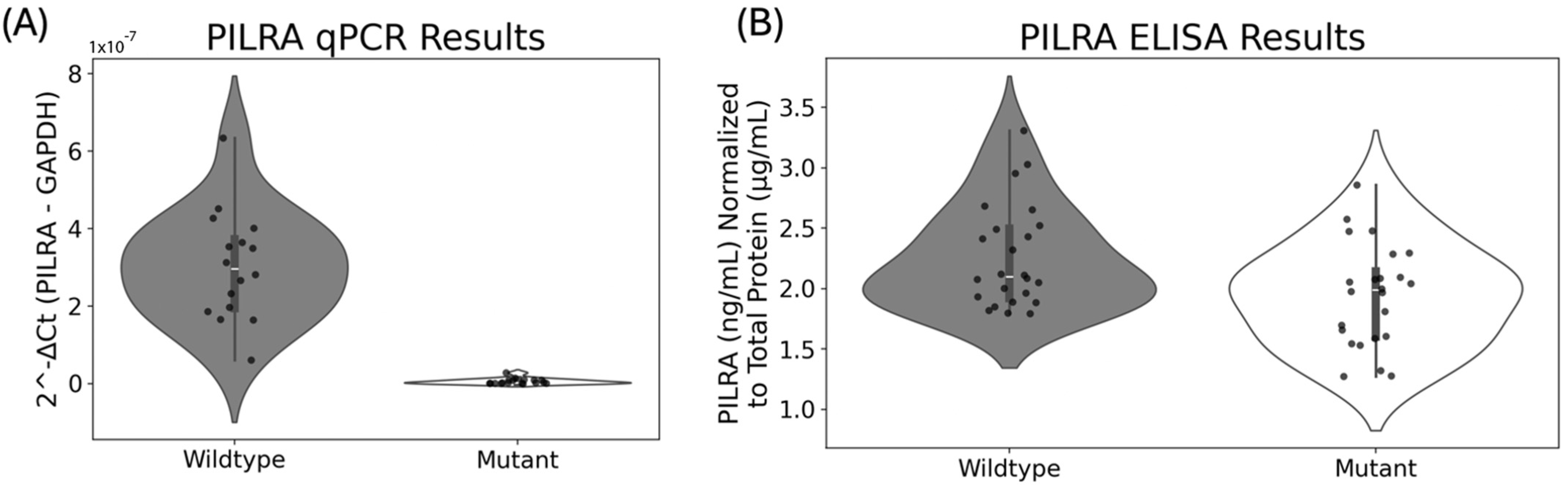

{kind=link}
{kind=link}
| SNP | Chromosome/Position | Nearest Gene | Transcripts with Ramp Sequence | Most Severe Variant Effect | Highest MAF | CADD Score | GERP Score | RegulomeDB Score | Most Severe Ramp Effect |
|---|---|---|---|---|---|---|---|---|---|
| rs2405442:T>C | 7:100373690 | PILRA | 4/4 (100%) | Synonymous | 0.50 (T) | 4.238 | −2.24 | 1f | Loss of Ramp |
| rs12453:T>C | 11:60178272 | MS4A6A | 8/14 (57%) | Synonymous | 0.50 (C) | 0.578 | −4.07 | 1f | Ramp Size |
| rs1859788:A>G | 7:100374211 | PILRA | 4/4 (100%) | Missense | 0.50 (A) | 12.85 | 1.01 | 1f | Ramp Size |
| rs12459419:C>T | 19:51225221 | CD33 | 2/6 (33%) | Missense | 0.48 (T) | 14.75 | 0.06 | 1f | Ramp Size |
| rs7982:A>G | 8:27604964 | CLU | 1/2 (50%) | Missense | 0.49 (A) | 0.920 | −3.07 | 1f | Gene with Ramp |
| rs2296160:A>G | 1:207621975 | CR1 | 5/5 (100%) | Missense | 0.35 (A) | 0.001 | −3.64 | 7 | Gene with Ramp |
| rs3752241:C>G | 19:1053525 | ABCA7 | 0/18 (0%) | Synonymous | 0.29 (G) | 3.833 | −4.46 | 1f | N/A |
| rs117618017:C>T | 15:63277703 | APH1B | 0/3 (0%) | Missense | 0.31 (T) | 16.39 | −1.84 | 1f | N/A |
| rs429358:T>C | 19:44908684 | APOE | 0/5 (0%) | Missense | 0.38 (C) | 16.65 | 2.01 | 1f | N/A |
| rs9268480:C>T | 6:32396067 | BTNL2 | 0/2 (0%) | Synonymous | 0.35 (T) | 3.813 | −1.07 | 1f | N/A |
| rs1135173:G>A | 2:233146227 | INPP5D | 0/2 (0%) | Synonymous | 0.49 (A) | 4.311 | −3.25 | 1f | N/A |
| rs157581:T>C | 19:44892457 | TOMM40 | 0/4 (0%) | Missense | 0.50 (C) | 14.60 | −1.14 | 1f | N/A |
| rs11556505:C>T | 19:44892887 | TOMM40 | 0/4 (0%) | Missense | 0.18 (T) | 6.035 | −6.99 | 5 | N/A |
| rs75932628:C>T | 6:41161514 | TREM2 | 0/2 (0%) | Missense | 0.02 (T) | 26.1 | NA | 2b | N/A |
| Tissues with PILRA Ramp Sequence | Cell Types with PILRA Ramp Sequence |
|---|---|
| Amygdala | Appendix lymphoid tissue |
| Cerebral cortex | Caudate glial |
| Colon | Caudate neuronal |
| Corpus callosum | Cerebellum Purkinje |
| Ductus deferens | Cervix uterine glandular |
| Duodenum | Dendritic cells |
| Esophagus | Hippocampus glial |
| Fallopian tube | Lung pneumocytes |
| Gallbladder | Lymph node nongerminal center |
| Heart muscle | Monocytes |
| Hippocampal formation | Oral mucosa squamous epithelial |
| Hypothalamus | Pancreas islets of Langerhans |
| Olfactory region | Prostate glandular |
| Pancreas | Seminal vesicle glandular |
| Retina | Skin1 fibroblasts |
| Salivary gland | Skin keratinocytes |
| Seminal vesicle | Skin melanocytes |
| Skeletal muscle | Soft tissue1 fibroblasts |
| Skin | Spleen cells in red pulp |
| Small intestine | Spleen cells in white pulp |
| Spleen | Thyroid gland glandular |
| Stomach | Tonsil nongerminal center |
| Tongue | Total PBMC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miller, J.B.; Brandon, J.A.; Harmon, L.M.; Sabra, H.W.; Lucido, C.C.; Murcia, J.D.G.; Nations, K.A.; Payne, S.H.; Ebbert, M.T.W.; Kauwe, J.S.K.; et al. Ramp Sequence May Explain Synonymous Variant Association with Alzheimer’s Disease in the Paired Immunoglobulin-like Type 2 Receptor Alpha (PILRA). Biomedicines 2025, 13, 739. https://doi.org/10.3390/biomedicines13030739
Miller JB, Brandon JA, Harmon LM, Sabra HW, Lucido CC, Murcia JDG, Nations KA, Payne SH, Ebbert MTW, Kauwe JSK, et al. Ramp Sequence May Explain Synonymous Variant Association with Alzheimer’s Disease in the Paired Immunoglobulin-like Type 2 Receptor Alpha (PILRA). Biomedicines. 2025; 13(3):739. https://doi.org/10.3390/biomedicines13030739
Chicago/Turabian StyleMiller, Justin B., J. Anthony Brandon, Lauren M. Harmon, Hady W. Sabra, Chloe C. Lucido, Josue D. Gonzalez Murcia, Kayla A. Nations, Samuel H. Payne, Mark T. W. Ebbert, John S. K. Kauwe, and et al. 2025. "Ramp Sequence May Explain Synonymous Variant Association with Alzheimer’s Disease in the Paired Immunoglobulin-like Type 2 Receptor Alpha (PILRA)" Biomedicines 13, no. 3: 739. https://doi.org/10.3390/biomedicines13030739
APA StyleMiller, J. B., Brandon, J. A., Harmon, L. M., Sabra, H. W., Lucido, C. C., Murcia, J. D. G., Nations, K. A., Payne, S. H., Ebbert, M. T. W., Kauwe, J. S. K., & Ridge, P. G. (2025). Ramp Sequence May Explain Synonymous Variant Association with Alzheimer’s Disease in the Paired Immunoglobulin-like Type 2 Receptor Alpha (PILRA). Biomedicines, 13(3), 739. https://doi.org/10.3390/biomedicines13030739