
Genomic Characterisation of the Relationship and Causal Links Between Vascular Calcification, Alzheimer’s Disease, and Cognitive Traits

Abstract
1. Introduction
2. Materials and Methods
2.1. Overview of Study Design
2.2. Data Sources
2.3. Cross-Trait Genome-Wide Genetic Correlation Analyses
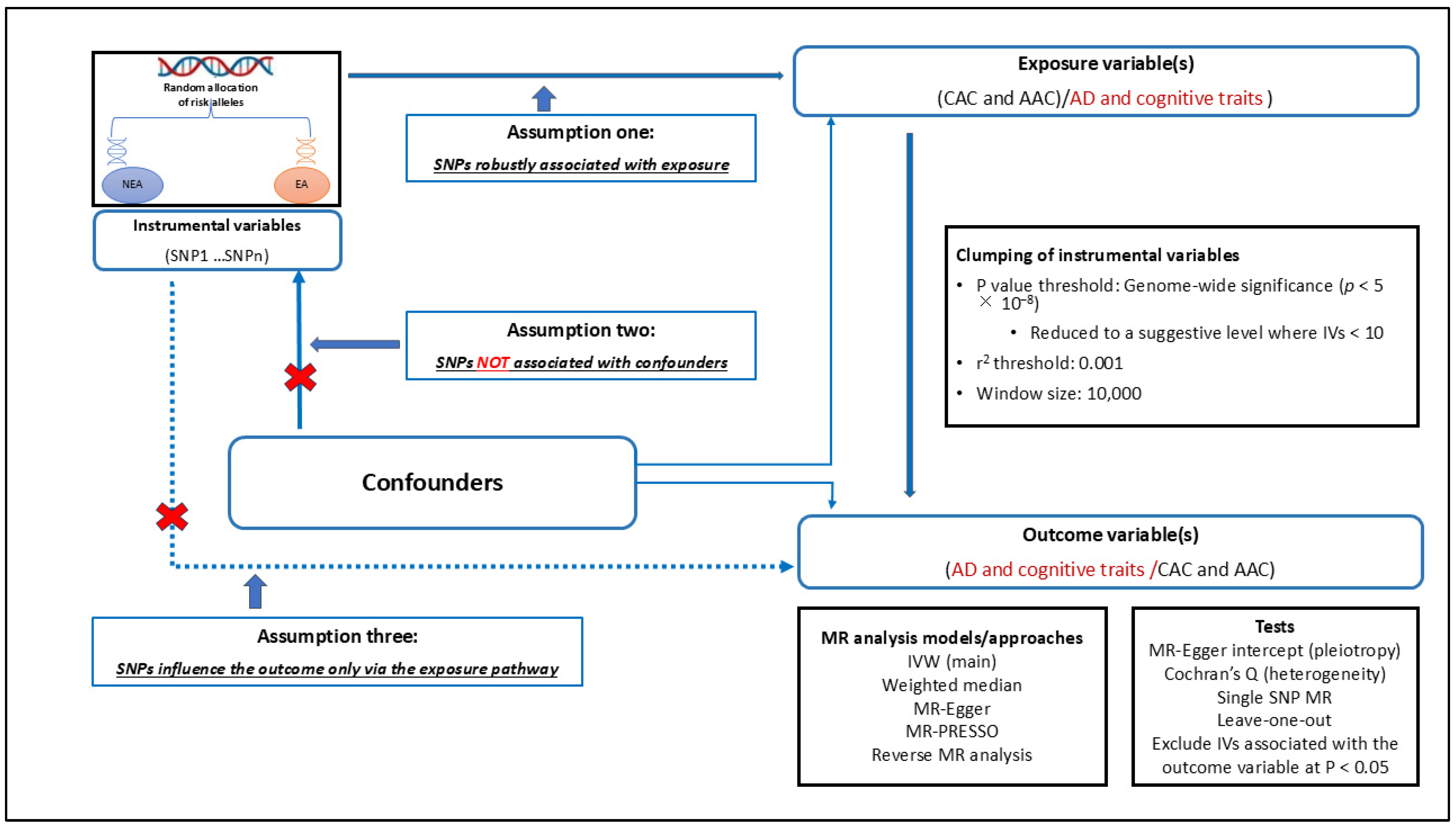
2.4. Causal Relationship Assessment Using Mendelian Randomisation
2.4.1. Selection of Instrumental Variables for MR Analysis
2.4.2. Performing MR Analyses
2.4.3. MR Sensitivity Analyses
2.5. Assessing Shared Genetic Risk Loci: The Pairwise Gwas and Gene-Based Approach
3. Results
3.1. Results of Genome-Wide Genetic Correlation Analyses
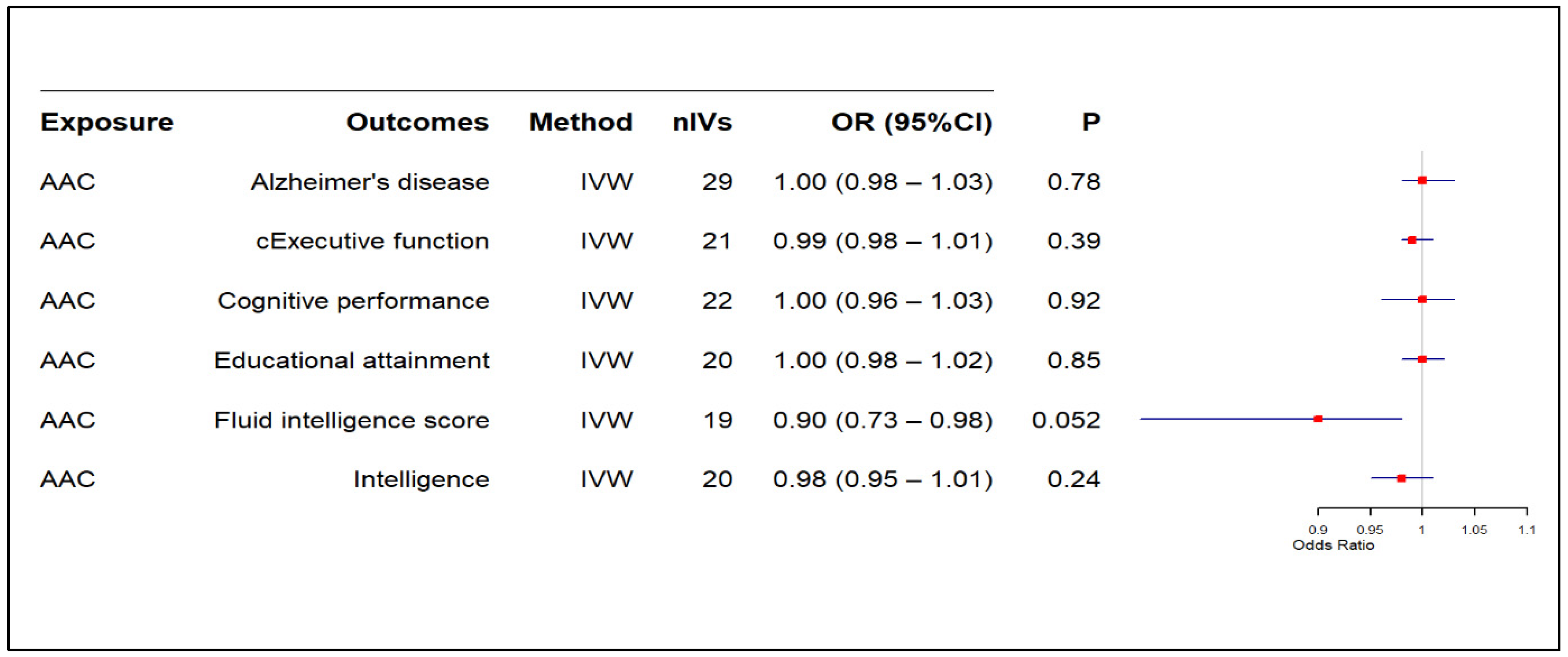
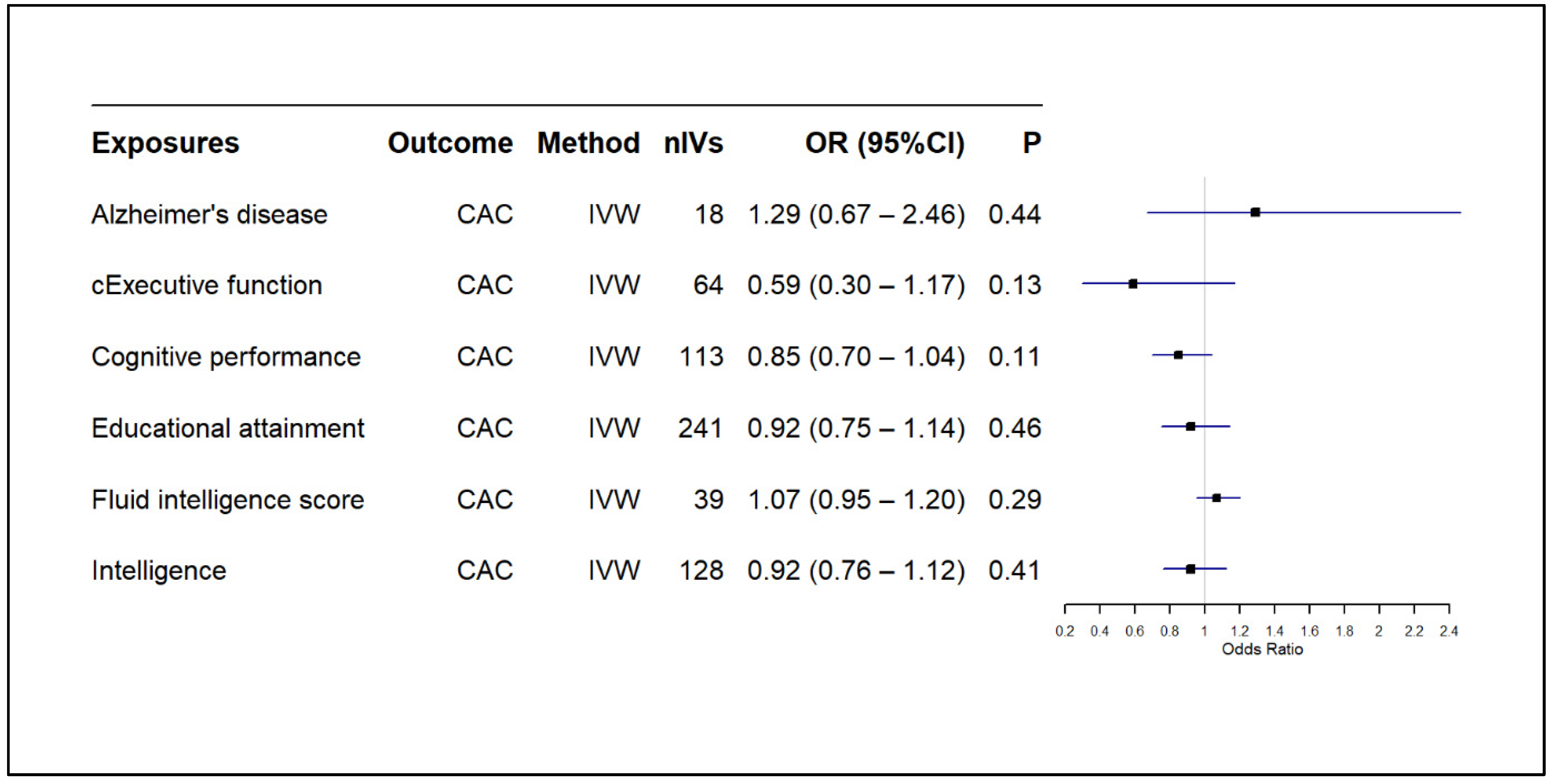
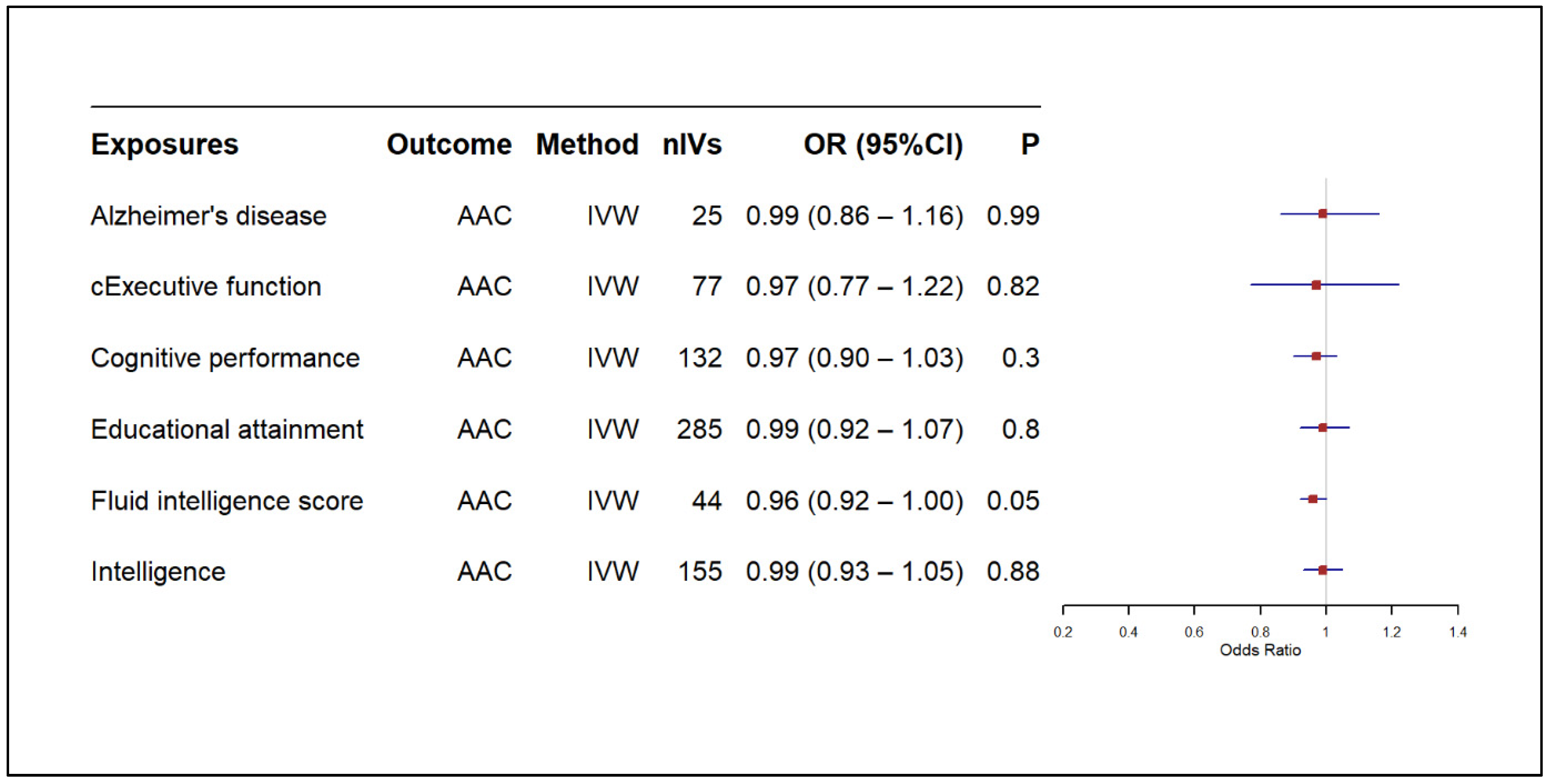
3.2. Results of MR-Based Causal Association Assessment
Addressing Horizontal Pleiotropy in Our MR Analysis
3.3. Shared Genomic Loci Between CAC or AAC and AD or Cognitive Traits
4. Discussion
Strengths and Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AAC | Abdominal aortic calcification |
| AD | Alzheimer’s disease |
| CAC | Coronary artery calcification |
| cEF | Common executive function |
| CI | Confidence interval |
| CT | Cognitive traits |
| EA | Effect allele |
| GWAS | Genome-wide association studies |
| GWAS-PW | Pair-wise GWAS |
| GWS | Genome-wide significant |
| IV | Instrumental variable |
| IVW | Inverse variance weighted |
| LD | Linkage disequilibrium |
| LDSC | Linkage disequilibrium score regression |
| MAF | Minor allele frequency |
| MR | Mendelian randomisation |
| NEA | Non-effect allele |
| PPA | Posterior probability of association |
| SNP | Single nucleotide polymorphism |
| 2SMR | Two-sample Mendelian randomisation |
References
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Nandi, A.; Counts, N.; Bröker, J.; Malik, S.; Chen, S.; Han, R.; Klusty, J.; Seligman, B.; Tortorice, D.; Vigo, D.; et al. Cost of care for Alzheimer’s disease and related dementias in the United States: 2016 to 2060. Npj Aging 2024, 10, 13. [Google Scholar] [CrossRef] [PubMed]
- Nandi, A.; Counts, N.; Chen, S.; Seligman, B.; Tortorice, D.; Vigo, D.; Bloom, D.E. Global and regional projections of the economic burden of Alzheimer’s disease and related dementias from 2019 to 2050: A value of statistical life approach. EClinicalMedicine 2022, 51, 101580. [Google Scholar] [CrossRef]
- Alzheimer’s Disease International. Dementia Statistics. Available online: https://www.alzint.org/about/dementia-facts-figures/dementia-statistics/ (accessed on 23 August 2023).
- Eisenmenger, L.B.; Peret, A.; Famakin, B.M.; Spahic, A.; Roberts, G.S.; Bockholt, J.H.; Johnson, K.M.; Paulsen, J.S. Vascular contributions to Alzheimer’s disease. Transl. Res. 2023, 254, 41–53. [Google Scholar] [CrossRef]
- Xie, B.; Shi, X.; Xing, Y.; Tang, Y. Association between atherosclerosis and Alzheimer’s disease: A systematic review and meta-analysis. Brain Behav. 2020, 10, e01601. [Google Scholar] [CrossRef]
- Frentz, I.; van Arendonk, J.; Leeuwis, A.E.; Vernooij, M.W.; van der Flier, W.M.; Bos, D.; De Deyn, P.P.; Wolters, F.J.; Ikram, M.A. Interaction Between Arteriosclerosis and Amyloid-β on Cognitive Function. J. Alzheimer’s Dis. 2024, 97, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Bos, D.; Vernooij, M.W.; de Bruijn, R.F.A.G.; Koudstaal, P.J.; Hofman, A.; Franco, O.H.; van der Lugt, A.; Ikram, M.A. Atherosclerotic calcification is related to a higher risk of dementia and cognitive decline. Alzheimer’s Dement. 2015, 11, 639–647.e1. [Google Scholar] [CrossRef] [PubMed]
- Porter, T.; Sim, M.; Prince, R.L.; Schousboe, J.T.; Bondonno, C.; Lim, W.H.; Zhu, K.; Kiel, D.P.; Hodgson, J.M.; Laws, S.M.; et al. Abdominal aortic calcification on lateral spine images captured during bone density testing and late-life dementia risk in older women: A prospective cohort study. Lancet Reg. Health-West. Pac. 2022, 26, 100502. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.N.; Xie, W.X.; Hou, Z.H.; An, Y.Q.; Ren, X.S.; Ma, Y.J.; Li, C.L.; Wu, Y.F.; Lu, B. Association between coronary artery calcification and cognitive function in a Chinese community-based population. J. Geriatr. Cardiol. 2021, 18, 514–522. [Google Scholar] [CrossRef]
- Lee, S.J.; Lee, I.-K.; Jeon, J.-H. Vascular Calcification—New Insights into Its Mechanism. Int. J. Mol. Sci. 2020, 21, 2685. [Google Scholar] [CrossRef]
- Sethi, A.; Taylor, D.L.; Ruby, J.G.; Venkataraman, J.; Sorokin, E.; Cule, M.; Melamud, E. Calcification of the abdominal aorta is an under-appreciated cardiovascular disease risk factor in the general population. Front. Cardiovasc. Med. 2022, 9, 1003246. [Google Scholar] [CrossRef] [PubMed]
- Johansen, M.C.; Blaha, M.J. Coronary Artery Calcium: A Risk Factor for Brain Aging? Circ. Cardiovasc. Imaging 2021, 14, e013062. [Google Scholar] [CrossRef]
- Orringer, C.E.; Blaha, M.J.; Blankstein, R.; Budoff, M.J.; Goldberg, R.B.; Gill, E.A.; Maki, K.C.; Mehta, L.; Jacobson, T.A. The National Lipid Association scientific statement on coronary artery calcium scoring to guide preventive strategies for ASCVD risk reduction. J. Clin. Lipidol. 2021, 15, 33–60. [Google Scholar] [CrossRef]
- Vasan, R.S.; Pan, S.; Larson, M.G.; Mitchell, G.F.; Xanthakis, V. Arteriosclerosis, atherosclerosis, and cardiovascular health: Joint relations to the incidence of cardiovascular disease. Hypertension 2021, 78, 1232–1240. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, G.; Zhang, X.; Wang, S.; Wang, Y. A two-sample Mendelian randomization study of atherosclerosis and dementia. iScience 2023, 26, 108325. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Vonder, M.; Sidorenkov, G.; Ma, R.; Oudkerk, M.; Harst, P.v.d.; Deyn, P.P.D.; Vliegenthart, R. Coronary Artery Calcium and Cognitive Function in Dutch Adults: Cross-Sectional Results of the Population-Based ImaLife Study. J. Am. Heart Assoc. 2021, 10, e018172. [Google Scholar] [CrossRef] [PubMed]
- Muhire, G.; Iulita, M.F.; Vallerand, D.; Youwakim, J.; Gratuze, M.; Petry, F.R.; Planel, E.; Ferland, G.; Girouard, H. Arterial stiffness due to carotid calcification disrupts cerebral blood flow regulation and leads to cognitive deficits. J. Am. Heart Assoc. 2019, 8, e011630. [Google Scholar] [CrossRef]
- Cheng, H.-M.; Wang, J.-J.; Chen, C.-H. The Role of Vascular Calcification in Heart Failure and Cognitive Decline. Pulse 2017, 5, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Valderas, J.M.; Starfield, B.; Sibbald, B.; Salisbury, C.; Roland, M. Defining comorbidity: Implications for understanding health and health services. Ann. Fam. Med. 2009, 7, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Vonder, M.; Sidorenkov, G.; Oudkerk, M.; de Groot, J.C.; van der Harst, P.; de Bock, G.H.; De Deyn, P.P.; Vliegenthart, R. The relationship of coronary artery calcium and clinical coronary artery disease with cognitive function: A systematic review and meta-analysis. J. Atheroscler. Thromb. 2020, 27, 934–958. [Google Scholar] [CrossRef] [PubMed]
- Ngamdu, K.S.; Kalra, D.K. Risk of Stroke, Dementia, and Cognitive Decline with Coronary and Arterial Calcification. J. Clin. Med. 2024, 13, 4263. [Google Scholar] [CrossRef] [PubMed]
- Hess, A.S.; Abd-Elsayed, A. Observational Studies: Uses and Limitations. In Pain: A Review Guide; Abd-Elsayed, A., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 123–125. [Google Scholar]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2018, 47, D1005–D1012. [Google Scholar] [CrossRef] [PubMed]
- Adewuyi, E.O.; O’Brien, E.K.; Nyholt, D.R.; Porter, T.; Laws, S.M. A large-scale genome-wide cross-trait analysis reveals shared genetic architecture between Alzheimer’s disease and gastrointestinal tract disorders. Commun. Biol. 2022, 5, 691. [Google Scholar] [CrossRef] [PubMed]
- Adewuyi, E.O.; Porter, T.; O’Brien, E.K.; Olaniru, O.; Verdile, G.; Laws, S.M. Genome-wide cross-disease analyses highlight causality and shared biological pathways of type 2 diabetes with gastrointestinal disorders. Commun. Biol. 2024, 7, 643. [Google Scholar] [CrossRef]
- Davies, N.M.; Holmes, M.V.; Davey Smith, G. Reading Mendelian randomisation studies: A guide, glossary, and checklist for clinicians. BMJ 2018, 362, k601. [Google Scholar] [CrossRef] [PubMed]
- Sekula, P.; Fabiola Del Greco, M.; Pattaro, C.; Köttgen, A. Mendelian Randomization as an Approach to Assess Causality Using Observational Data. J. Am. Soc. Nephrol. 2016, 27, 3253–3265. [Google Scholar] [CrossRef] [PubMed]
- Richmond, R.C.; Davey Smith, G. Mendelian Randomization: Concepts and Scope. Cold Spring Harb. Perspect. Med. 2022, 12, a040501. [Google Scholar] [CrossRef]
- Skrivankova, V.W.; Richmond, R.C.; Woolf, B.A.; Yarmolinsky, J.; Davies, N.M.; Swanson, S.A.; VanderWeele, T.J.; Higgins, J.P.; Timpson, N.J.; Dimou, N. Strengthening the reporting of observational studies in epidemiology using Mendelian randomization: The STROBE-MR statement. Jama 2021, 326, 1614–1621. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, A.; Zhu, Z.; Vinkhuyzen, A.A.E.; Hill, W.D.; McRae, A.F.; Visscher, P.M.; Yang, J. Fast set-based association analysis using summary data from GWAS identifies novel gene loci for human complex traits. Sci. Rep. 2016, 6, 32894. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Liu, S.; Bakshi, A.; Jiang, L.; Chen, W.; Zheng, Z.; Sullivan, P.F.; Visscher, P.M.; Wray, N.R.; Yang, J.; et al. mBAT-combo: A more powerful test to detect gene-trait associations from GWAS data. Am. J. Hum. Genet. 2023, 110, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, J.K.; Berisa, T.; Liu, J.Z.; Ségurel, L.; Tung, J.Y.; Hinds, D.A. Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet. 2016, 48, 709–717. [Google Scholar] [CrossRef]
- Kavousi, M.; Bos, M.M.; Barnes, H.J.; Lino Cardenas, C.L.; Wong, D.; Lu, H.; Hodonsky, C.J.; Landsmeer, L.P.L.; Turner, A.W.; Kho, M.; et al. Multi-ancestry genome-wide study identifies effector genes and druggable pathways for coronary artery calcification. Nat. Genet. 2023, 55, 1651–1664. [Google Scholar] [CrossRef] [PubMed]
- Kauppila, L.I.; Polak, J.F.; Cupples, L.A.; Hannan, M.T.; Kiel, D.P.; Wilson, P.W. New indices to classify location, severity and progression of calcific lesions in the abdominal aorta: A 25-year follow-up study. Atherosclerosis 1997, 132, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef]
- Hatoum, A.S.; Morrison, C.L.; Mitchell, E.C.; Lam, M.; Benca-Bachman, C.E.; Reineberg, A.E.; Palmer, R.H.C.; Evans, L.M.; Keller, M.C.; Friedman, N.P. Genome-wide Association Study Shows That Executive Functioning Is Influenced by GABAergic Processes and Is a Neurocognitive Genetic Correlate of Psychiatric Disorders. Biol. Psychiatry 2023, 93, 59–70. [Google Scholar] [CrossRef]
- Lee, J.J.; Wedow, R.; Okbay, A.; Kong, E.; Maghzian, O.; Zacher, M.; Nguyen-Viet, T.A.; Bowers, P.; Sidorenko, J.; Karlsson Linnér, R.; et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet. 2018, 50, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Savage, J.E.; Jansen, P.R.; Stringer, S.; Watanabe, K.; Bryois, J.; de Leeuw, C.A.; Nagel, M.; Awasthi, S.; Barr, P.B.; Coleman, J.R.I.; et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat. Genet. 2018, 50, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Stringer, S.; Frei, O.; Umićević Mirkov, M.; de Leeuw, C.; Polderman, T.J.C.; van der Sluis, S.; Andreassen, O.A.; Neale, B.M.; Posthuma, D. A global overview of pleiotropy and genetic architecture in complex traits. Nat. Genet. 2019, 51, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Bulik-Sullivan, B.K.; Loh, P.-R.; Finucane, H.K.; Ripke, S.; Yang, J.; Schizophrenia Working Group of the Psychiatric Genomics Consortium; Patterson, N.; Daly, M.J.; Price, A.L.; Neale, B.M. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015, 47, 291–295. [Google Scholar] [CrossRef]
- Adewuyi, E.O.; Mehta, D.; Sapkota, Y.; Auta, A.; Yoshihara, K.; Nyegaard, M.; Griffiths, L.R.; Montgomery, G.W. Genetic analysis of endometriosis and depression identifies shared loci and implicates causal links with gastric mucosa abnormality. Hum. Genet. 2021, 140, 529–552. [Google Scholar] [CrossRef] [PubMed]
- Adewuyi, E.O.; Sapkota, Y.; International Endogene Consortium (IEC); 23andMe Research Team; International Headache Genetics Consortium (IHGC); Auta, A.; Yoshihara, K.; Nyegaard, M.; Griffiths, L.R.; Montgomery, G.W. Shared molecular genetic mechanisms underlie endometriosis and migraine comorbidity. Genes 2020, 11, 268. [Google Scholar] [CrossRef]
- Tasnim, S.; Wilson, S.G.; Walsh, J.P.; Nyholt, D.R. Cross-Trait Genetic Analyses Indicate Pleiotropy and Complex Causal Relationships between Headache and Thyroid Function Traits. Genes 2022, 14, 16. [Google Scholar] [CrossRef]
- Carter, A.R.; Anderson, E.L. Correct illustration of assumptions in Mendelian randomization. Int. J. Epidemiol. 2024, 53, dyae050. [Google Scholar] [CrossRef] [PubMed]
- Davey Smith, G.; Hemani, G. Mendelian randomization: Genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 2014, 23, R89–R98. [Google Scholar] [CrossRef]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef]
- Verbanck, M.; Chen, C.-Y.; Neale, B.; Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018, 50, 693–698. [Google Scholar] [CrossRef]
- Adewuyi, E.O.; Mehta, D.; Nyholt, D. Genetic overlap analysis of endometriosis and asthma identifies shared loci implicating sex hormones and thyroid signalling pathways. Hum. Reprod. 2022, 37, 366–383. [Google Scholar] [CrossRef] [PubMed]
- Akosile, W.; Adewuyi, E. Genetic correlation and causality assessment between post-traumatic stress disorder and coronary artery disease-related traits. Gene 2022, 842, 146802. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.R.; Nyholt, D.R.; International Headache Genetics Consortium (IHGC). Cross-trait analyses identify shared genetics between migraine, headache, and glycemic traits, and a causal relationship with fasting proinsulin. Hum. Genet. 2023, 142, 1149–1172. [Google Scholar] [CrossRef] [PubMed]
- Tasnim, S.; Wilson, S.G.; Walsh, J.P.; Nyholt, D.R. Shared genetics and causal relationships between migraine and thyroid function traits. Cephalalgia 2023, 43, 03331024221139253. [Google Scholar] [CrossRef]
- Adewuyi, E.O.; O’Brien, E.K.; Porter, T.; Laws, S.M. Relationship of Cognition and Alzheimer’s Disease with Gastrointestinal Tract Disorders: A Large-Scale Genetic Overlap and Mendelian Randomisation Analysis. Int. J. Mol. Sci. 2022, 23, 16199. [Google Scholar] [CrossRef]
- Chu, Z.; Cheng, L.; Tong, Q. Carotid artery calcification score and its association with cognitive impairment. Clin. Interv. Aging 2019, 14, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Haycock, P.C.; Burgess, S.; Wade, K.H.; Bowden, J.; Relton, C.; Davey Smith, G. Best (but oft-forgotten) practices: The design, analysis, and interpretation of Mendelian randomization studies1. Am. J. Clin. Nutr. 2016, 103, 965–978. [Google Scholar] [CrossRef] [PubMed]
- Tin, A.; Fohner, A.E.; Yang, Q.; Brody, J.A.; Davies, G.; Yao, J.; Liu, D.; Caro, I.; Lindbohm, J.V.; Duggan, M.R.; et al. Identification of circulating proteins associated with general cognitive function among middle-aged and older adults. Commun. Biol. 2023, 6, 1117. [Google Scholar] [CrossRef]
- Raulin, A.-C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.-C. ApoE in Alzheimer’s disease: Pathophysiology and therapeutic strategies. Mol. Neurodegener. 2022, 17, 72. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Ma, T. The impact of apolipoprotein E, type∊ 4 allele on Alzheimer’s disease pathological biomarkers: A comprehensive post-mortem pilot-analysis. PLoS ONE 2025, 20, e0303486. [Google Scholar] [CrossRef]
- Li, Z.; Shue, F.; Zhao, N.; Shinohara, M.; Bu, G. APOE2: Protective mechanism and therapeutic implications for Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 63. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W. Apolipoprotein E: From cardiovascular disease to neurodegenerative disorders. J. Mol. Med. 2016, 94, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Arboleda-Velasquez, J.F.; Quiroz, Y.T.; Huentelman, M.J.; Beach, T.G.; Caselli, R.J.; Chen, Y.; Su, Y.; Myers, A.J.; Hardy, J.; et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat. Commun. 2020, 11, 667. [Google Scholar] [CrossRef]
- Sutton, N.R.; Malhotra, R.; St. Hilaire, C.; Aikawa, E.; Blumenthal, R.S.; Gackenbach, G.; Goyal, P.; Johnson, A.; Nigwekar, S.U.; Shanahan, C.M.; et al. Molecular Mechanisms of Vascular Health: Insights From Vascular Aging and Calcification. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. Cerebrovascular Effects of Apolipoprotein E: Implications for Alzheimer Disease. JAMA Neurol. 2013, 70, 440–444. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait 1 | Trait 2 | rg | se | p |
|---|---|---|---|---|
| CAC | AD GWAS [37] | 0.1 | 0.05 | 3.14 × 10−2 |
| AD GWAS [37] excluding APOE region | 0.1 | 0.05 | 5.33 × 10−2 | |
| AD GWAS [38] | 0.01 | 0.04 | 8.06 × 10−1 | |
| AD GWAS [38] excluding APOE region | 0 | 0.05 | 9.17 × 10−1 | |
| Cognitive performance | −0.11 | 0.02 | 1.59 × 10−6 | |
| Fluid intelligence scores | −0.11 | 0.03 | 1.87 × 10−5 | |
| Intelligence | −0.11 | 0.02 | 5.14 × 10−6 | |
| Common executive function | −0.04 | 0.02 | 7.55 × 10−2 | |
| Educational attainment | −0.1 | 0.02 | 1.08 × 10−6 | |
| Abdominal aortic calcification | 0.95 * | 0.34 | 4.70 × 10−3 | |
| AAC | AD GWAS [37] | 0.08 | 0.07 | 2.32 × 10−1 |
| AD GWAS [37] excluding APOE region | 0.07 | 0.07 | 3.49 × 10−1 | |
| AD GWAS [38] | 0.01 | 0.07 | 9.28 × 10−1 | |
| AD GWAS [38] excluding APOE region | −0.01 | 0.07 | 9.19 × 10−1 | |
| Cognitive performance | −0.1 | 0.03 | 5.49 × 10−3 | |
| Fluid intelligence scores | −0.07 | 0.03 | 5.24 × 10−2 | |
| Intelligence | −0.06 | 0.04 | 1.12 × 10−1 | |
| Common executive function | −0.04 | 0.04 | 2.53 × 10−1 | |
| Educational attainment | −0.13 | 0.03 | 6.39 × 10−5 |
| Exposure | Outcome | Heterogeneity Tests | Horizontal Pleiotropy Tests | |||
|---|---|---|---|---|---|---|
| Method | Cochran’s Q p Value | Method | Intercept | p Value | ||
| Coronary artery calcification vs. AD and cognitive traits | ||||||
| CAC | Alzheimer’s disease | IVW | 0.72 | Egger intercept | −0.0013 | 0.72 |
| cExecutive function | 0.94 | −0.00042 | 0.59 | |||
| Cognitive performance | 0.5 | 0.0013 | 0.43 | |||
| Educational attainment | 0.81 | 0.00041 | 0.61 | |||
| Fluid intelligence scores | 0.65 | −0.0026 | 0.63 | |||
| Intelligence | 0.39 | 0.00032 | 0.86 | |||
| Abdominal aortic calcification vs. AD and cognitive traits | ||||||
| AAC | Alzheimer’s disease | IVW | 0.88 | Egger intercept | −0.0014 | 0.55 |
| cExecutive function | 0.71 | −0.0019 | 0.11 | |||
| Cognitive performance | 0.19 | −0.0069 | 0.024 | |||
| Educational attainment | 0.56 | 0.0013 | 0.47 | |||
| Fluid intelligence scores | 0.69 | −0.0057 | 0.5 | |||
| Intelligence | 0.72 | −0.0027 | 0.35 | |||
| AD and cognitive traits vs. coronary artery calcification | ||||||
| Alzheimer’s disease | CAC | IVW | 0.26 | Egger intercept | −0.021 | 0.11 |
| cExecutive function | 0.96 | 0.018 | 0.21 | |||
| Cognitive performance | 0.95 | 0.0097 | 0.89 | |||
| Educational attainment | 1 | 0.0022 | 0.7 | |||
| Fluid intelligence scores | 0.96 | −0.019 | 0.45 | |||
| Intelligence | 0.86 | −0.0014 | 0.88 | |||
| AD and cognitive traits vs. abdominal aortic calcification | ||||||
| Alzheimer’s disease | AAC | IVW | 0.99 | Egger intercept | −0.00019 | 0.95 |
| cExecutive function | 0.99 | 0.0047 | 0.55 | |||
| Cognitive performance | 0.95 | 0.0036 | 0.26 | |||
| Educational attainment | 1 | −0.00096 | 0.63 | |||
| Fluid intelligence scores | 0.78 | 0.0065 | 0.88 | |||
| Intelligence | 0.98 | −0.00017 | 0.95 | |||
| CAC/ AAC | AD | Chr: BP | PPA4 | Shared Genes Gene | CAC/AAC | AD | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene Pgene * | Top SNP | Top SNP p | Gene Pgene * | Top SNP | Top SNP p | |||||
| CAC | AD | 19: 44,744,370– 46,102,289 | 1.00 | BCAM | 7.08 × 10−9 | rs118147862 | 1.40 × 10−10 | 0 | rs41289512 | 1.5 × 10−278 |
| TOMM40 | 3.15 × 10−8 | rs41290120 | 1.57 × 10−11 | 0 | rs12972156 | 0 | ||||
| NECTIN2 | 4.29 × 10−8 | rs41290120 | 1.57 × 10−11 | 0 | rs12972156 | 0 | ||||
| APOE | 2.69 × 10−7 | rs41290120 | 1.57 × 10−11 | 0 | rs12972156 | 0 | ||||
| APOC1 | 8.13 × 10−7 | rs41290120 | 1.57 × 10−11 | 0 | rs12972156 | 0 | ||||
| CBLC | 1.01 × 10−5 | rs118147862 | 1.40 × 10−10 | 3.67 × 10−264 | rs41289512 | 1.5 × 10−278 | ||||
| APOC4 | 1.99 × 10−5 | rs7412 | 4.61 × 10−10 | 0 | rs2075650 | 0 | ||||
| APOC2 | 2.16 × 10−5 | rs7412 | 4.61 × 10−10 | 0 | rs10119 | 0 | ||||
| APOC4-APOC2 | 2.97 × 10−5 | rs7412 | 4.61 × 10−10 | 0 | rs2075650 | 0 | ||||
| EXOC3L2 | 4.56 × 10−5 | rs12461144 | 7.03 × 10−5 | 1.95 × 10−66 | rs10415850 | 2.17 × 10−33 | ||||
| TRAPPC6A | 5.08 × 10−5 | rs12461144 | 7.03 × 10−5 | 4.30 × 10−74 | rs28469095 | 1.07 × 10−38 | ||||
| BLOC1S3 | 8.06 × 10−5 | rs12461144 | 7.03 × 10−5 | 1.17 × 10−65 | rs28469095 | 1.07 × 10−38 | ||||
| NKPD1 | 3.92 × 10−4 | rs10421247 | 1.04 × 10−4 | 1.06 × 10−79 | rs28469095 | 1.07 × 10−38 | ||||
| CLPTM1 | 6.14 × 10−4 | rs7412 | 4.61 × 10−10 | 0 | rs769449 | 0 | ||||
| PPP1R37 | 1.59 × 10−3 | rs10421247 | 1.04 × 10−4 | 5.47 × 10−84 | rs28469095 | 1.07 × 10−38 | ||||
| BCL3 | 3.23 × 10−3 | rs148933445 | 1.26 × 10−7 | 7.93 × 10−127 | rs2965169 | 9.24 × 10−58 | ||||
| MARK4 | 5.66 × 10−3 | rs12461144 | 7.03 × 10−5 | 3.44 × 10−98 | rs28469095 | 1.07 × 10−38 | ||||
| CEACAM16 | 8.13 × 10−3 | rs62117204 | 1.58 × 10−6 | 7.75 × 10−121 | rs2965169 | 9.24 × 10−58 | ||||
| AAC | AD | 19: 44,744,108– 46,102,684 | 1.00 | TOMM40 | 2.36 × 10−11 | rs1065853 | 3.07 × 10−13 | 0 | rs12972156 | 0 |
| NECTIN2 | 2.44 × 10−10 | rs1065853 | 3.07 × 10−13 | 0 | rs12972156 | 0 | ||||
| APOE | 2.55 × 10−9 | rs1065853 | 3.07 × 10−13 | 0 | rs12972156 | 0 | ||||
| APOC1 | 3.97 × 10−9 | rs1065853 | 3.07 × 10−13 | 0 | rs12972156 | 0 | ||||
| APOC2 | 6.12 × 10−9 | rs1065853 | 3.07 × 10−13 | 0 | rs10119 | 0 | ||||
| APOC4 | 1.43 × 10−8 | rs1065853 | 3.07 × 10−13 | 0 | rs2075650 | 0 | ||||
| APOC4-APOC2 | 1.55 × 10−8 | rs1065853 | 3.07 × 10−13 | 0 | rs2075650 | 0 | ||||
| CLPTM1 | 2.55 × 10−8 | rs1065853 | 3.07 × 10−13 | 0 | rs769449 | 0 | ||||
| BCAM | 1.06 × 10−5 | rs4803760 | 3.00 × 10−7 | 0 | rs41289512 | 1.46 × 10−278 | ||||
| CBLC | 4.26 × 10−4 | rs4803760 | 3.00 × 10−7 | 3.67 × 10−264 | rs41289512 | 1.46 × 10−278 | ||||
| CAC/AAC | CT | Chr: BP | PPA4 | Shared Genes | CAC/AAC | CT | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene p * | Top SNP | Top SNP p | Gene-p * | Top SNP | Top SNP p | |||||
| CAC | cEF | 19: 44,744,370–46,102,547 | 1.00 | ** PHLDB3 | 4.38 × 10−2 | rs62115754 | 2.28 × 10−3 | 3.88 × 10−2 | rs11668385 | 1.10 × 10−2 |
| CAC | EA | 19: 44,744,370–46,102,547 | 0.99 | TOMM40 | 3.15 × 10−8 | rs41290120 | 1.57 × 10−11 | 7.56 × 10−3 | rs405509 | 1.07 × 10−5 |
| NECTIN2 | 4.29 × 10−8 | rs41290120 | 1.57 × 10−11 | 2.05 × 10−2 | rs405509 | 1.07 × 10−5 | ||||
| APOE | 2.69 × 10−7 | rs41290120 | 1.57 × 10−11 | 7.88 × 10−3 | rs405509 | 1.07 × 10−5 | ||||
| APOC1 | 8.13 × 10−7 | rs41290120 | 1.57 × 10−11 | 6.22 × 10−3 | rs405509 | 1.07 × 10−5 | ||||
| APOC4 | 1.99 × 10−5 | rs7412 | 4.61 × 10−10 | 5.83 × 10−3 | rs405509 | 1.07 × 10−5 | ||||
| APOC2 | 2.16 × 10−5 | rs7412 | 4.61 × 10−10 | 1.01 × 10−3 | rs405509 | 1.07 × 10−5 | ||||
| APOC4-APOC2 | 2.97 × 10−5 | rs7412 | 4.61 × 10−10 | 5.72 × 10−3 | rs405509 | 1.07 × 10−5 | ||||
| EXOC3L2 | 4.56 × 10−5 | rs12461144 | 7.03 × 10−5 | 8.51 × 10−5 | rs386569 | 8.22 × 10−6 | ||||
| TRAPPC6A | 5.08 × 10−5 | rs12461144 | 7.03 × 10−5 | 3.77 × 10−2 | rs12974200 | 3.56 × 10−3 | ||||
| BLOC1S3 | 8.06 × 10−5 | rs12461144 | 7.03 × 10−5 | 1.50 × 10−3 | rs151165225 | 3.27 × 10−5 | ||||
| CLPTM1 | 6.14 × 10−4 | rs7412 | 4.61 × 10−10 | 4.07 × 10−2 | rs405509 | 1.07 × 10−5 | ||||
| PPP1R37 | 1.59 × 10−3 | rs10421247 | 1.04 × 10−4 | 1.94 × 10−2 | rs139290129 | 5.95 × 10−4 | ||||
| MARK4 | 5.66 × 10−3 | rs12461144 | 7.03 × 10−5 | 7.59 × 10−7 | rs10402747 | 1.71 × 10−8 | ||||
| AAC | cEF | 19: 44,744,147–46,101,600 | 1.00 | TOMM40 | 2.36 × 10−11 | rs1065853 | 3.07 × 10−13 | 1.57 × 10−15 | rs429358 | 9.52 × 10−20 |
| NECTIN2 | 2.44 × 10−10 | rs1065853 | 3.07 × 10−13 | 1.08 × 10−14 | rs429358 | 9.52 × 10−20 | ||||
| APOE | 2.55 × 10−9 | rs1065853 | 3.07 × 10−13 | 1.68 × 10−16 | rs429358 | 9.52 × 10−20 | ||||
| APOC1 | 3.97 × 10−9 | rs1065853 | 3.07 × 10−13 | 1.18 × 10−16 | rs429358 | 9.52 × 10−20 | ||||
| APOC2 | 6.12 × 10−9 | rs1065853 | 3.07 × 10−13 | 3.02 × 10−16 | rs429358 | 9.52 × 10−20 | ||||
| APOC4 | 1.43 × 10−8 | rs1065853 | 3.07 × 10−13 | 1.02 × 10−15 | rs429358 | 9.52 × 10−20 | ||||
| APOC4-APOC2 | 1.55 × 10−8 | rs1065853 | 3.07 × 10−13 | 7.48 × 10−16 | rs429358 | 9.52 × 10−20 | ||||
| CLPTM1 | 2.55 × 10−8 | rs1065853 | 3.07 × 10−13 | 2.71 × 10−15 | rs429358 | 9.52 × 10−20 | ||||
| BCAM | 1.06 × 10−5 | rs4803760 | 3.00 × 10−7 | 2.63 × 10−4 | rs4803764 | 4.24 × 10−4 | ||||
| CBLC | 4.26 × 10−4 | rs4803760 | 3.00 × 10−7 | 1.78 × 10−6 | rs12162222 | 6.16 × 10−4 | ||||
| AAC | EA | 19: 44,744,147–46,101,600 | 0.96 | TOMM40 | 2.36 × 10−11 | rs1065853 | 3.07 × 10−13 | 7.56 × 10−3 | rs405509 | 1.07 × 10−5 |
| NECTIN2 | 2.44 × 10−10 | rs1065853 | 3.07 × 10−13 | 2.05 × 10−2 | rs405509 | 1.07 × 10−5 | ||||
| APOE | 2.55 × 10−9 | rs1065853 | 3.07 × 10−13 | 7.88 × 10−3 | rs405509 | 1.07 × 10−5 | ||||
| APOC1 | 3.97 × 10−9 | rs1065853 | 3.07 × 10−13 | 6.22 × 10−3 | rs405509 | 1.07 × 10−5 | ||||
| APOC2 | 6.12 × 10−9 | rs1065853 | 3.07 × 10−13 | 1.01 × 10−2 | rs405509 | 1.07 × 10−5 | ||||
| APOC4 | 1.43 × 10−8 | rs1065853 | 3.07 × 10−13 | 5.83 × 10−3 | rs405509 | 1.07 × 10−5 | ||||
| APOC4-APOC2 | 1.55 × 10−8 | rs1065853 | 3.07 × 10−13 | 5.72 × 10−3 | rs405509 | 1.07 × 10−5 | ||||
| CLPTM1 | 2.55 × 10−8 | rs1065853 | 3.07 × 10−13 | 4.07 × 10−2 | rs405509 | 1.07 × 10−5 | ||||
| AAC | FIS | 19: 44,744,147–46,101,600 | 0.96 | TOMM40 | 2.36 × 10−11 | rs1065853 | 3.07 × 10−13 | 1.54 × 10−3 | rs11668861 | 1.23 × 10−3 |
| NECTIN2 | 2.44 × 10−10 | rs1065853 | 3.07 × 10−13 | 2.19 × 10−3 | rs8113311 | 7.09 × 10−4 | ||||
| APOE | 2.55 × 10−9 | rs1065853 | 3.07 × 10−13 | 3.20 × 10−3 | rs11668861 | 1.23 × 10−3 | ||||
| APOC1 | 3.97 × 10−9 | rs1065853 | 3.07 × 10−13 | 6.93 × 10−3 | rs11668861 | 1.23 × 10−3 | ||||
| BCAM | 1.06 × 10−5 | rs4803760 | 3.00 × 10−7 | 3.39 × 10−3 | rs8113311 | 7.09 × 10−4 | ||||
| CBLC | 4.26 × 10−4 | rs4803760 | 3.00 × 10−7 | 2.41 × 10−2 | rs8113311 | 7.09 × 10−4 | ||||
| Locus (Chr:BP) | Key Genes | Associated Traits | Functional Annotation | Biological Implications |
|---|---|---|---|---|
| 19: 44,744,370–46,102,289 | APOE, TOMM40, NECTIN2, APOC1, APOC2, APOC4 | CAC, AAC, AD, Cognitive Traits | APOE is a major lipid transport protein linked to AD risk; TOMM40 is involved in mitochondrial protein transport; NECTIN2 plays a role in cell adhesion and immune signalling | Associated with neurodegeneration, vascular health, and AD |
| 19: 44,744,370–46,102,547 | PHLDB3 | CAC, Cognitive Traits (cEF) | Plays a role in cell signalling, potential involvement in neuronal function | May influence cognitive function and neurodevelopment |
| 19: 44,744,108–46,102,684 | BCAM, CBLC | AAC, AD | BCAM encodes a laminin-binding protein, implicated in cell adhesion; CBLC is involved in ubiquitin signalling and protein degradation | Suggests vascular contributions to AD risk through endothelial interactions |
| 19: 44,744,147–46,101,600 | TOMM40, APOE, APOC1, NECTIN2 | AAC, Cognitive Traits (cEF, EA, FIS) | Overlaps with well-established AD risk loci, involved in lipid metabolism, mitochondrial function, and immune response | Supports shared genetic architecture between vascular calcification and cognition |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adewuyi, E.O.; Laws, S.M. Genomic Characterisation of the Relationship and Causal Links Between Vascular Calcification, Alzheimer’s Disease, and Cognitive Traits. Biomedicines 2025, 13, 618. https://doi.org/10.3390/biomedicines13030618
Adewuyi EO, Laws SM. Genomic Characterisation of the Relationship and Causal Links Between Vascular Calcification, Alzheimer’s Disease, and Cognitive Traits. Biomedicines. 2025; 13(3):618. https://doi.org/10.3390/biomedicines13030618
Chicago/Turabian StyleAdewuyi, Emmanuel O., and Simon M. Laws. 2025. "Genomic Characterisation of the Relationship and Causal Links Between Vascular Calcification, Alzheimer’s Disease, and Cognitive Traits" Biomedicines 13, no. 3: 618. https://doi.org/10.3390/biomedicines13030618
APA StyleAdewuyi, E. O., & Laws, S. M. (2025). Genomic Characterisation of the Relationship and Causal Links Between Vascular Calcification, Alzheimer’s Disease, and Cognitive Traits. Biomedicines, 13(3), 618. https://doi.org/10.3390/biomedicines13030618