Abstract
Tissue-resident memory T (TRM) cells persist long-term in non-lymphoid tissues and provide rapid local immune protection, yet emerging evidence shows they also act as key drivers of chronic inflammation and relapse in rheumatoid immune diseases such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), systemic sclerosis (SSc), and primary Sjögren’s syndrome (pSS). A systematic search of PubMed, Web of Science, and Google Scholar (through October 2025) identified studies on TRM cell biology, pathogenic roles, and therapeutic modulation in autoimmune diseases. This review summarizes the fundamental features of TRM cells, including their TGF-β and IL-15 dependent development, tissue-specific heterogeneity, and unique metabolic programs. It highlights disease-specific pathogenic mechanisms: promotion of osteoclastogenesis and chronic synovial inflammation via Granulocyte-macrophage colony stimulating factor (GM-CSF) and the IL-23/IL-17 axis in RA; amplification of type I interferon responses and autoantibody production in SLE; potential contribution to fibrosis through TGF-β secretion in SSc; and mediation of glandular injury through cytotoxicity in pSS. Therapeutic strategies targeting TRM cells—such as JAK inhibitors, IL-17/IL-23 blockade, disruption of residency signals, metabolic interventions, and microenvironmental remodeling via nanotechnology—are critically evaluated. Challenges remain in achieving tissue-specific targeting without compromising systemic immune memory. Future directions include spatial transcriptomics, organoid models, and artificial intelligence to support precision medicine. Targeting TRM cells presents a promising novel avenue for achieving long-term remission and potentially even a cure for rheumatoid immune diseases.
1. Introduction
Rheumatic and autoimmune diseases, such as rheumatoid arthritis (RA), psoriatic arthritis, and systemic lupus erythematosus (SLE), are characterized by immune-mediated dysregulation of stromal tissues, chronic inflammation, joint destruction, and aberrant immune mechanisms [1]. Therapeutic approaches have traditionally relied on non-steroidal anti-inflammatory drugs, glucocorticoids, and conventional disease-modifying antirheumatic drugs (DMARDs). In recent years, several innovative therapies have emerged, including biologics such as IL-6 inhibitors, small-molecule targeted agents like JAK inhibitors, cellular therapies including mesenchymal stem cells and CAR-T cells, as well as genome-editing technologies such as CRISPR-Cas9 [2,3,4,5,6]. Although these therapies provide meaningful clinical benefits by alleviating symptoms and modulating inflammatory pathways, frequent disease relapse and pathological rebound suggest that conventional approaches do not adequately control the underlying immune memory mechanisms [7,8]. Hence, overcoming this therapeutic limitation requires novel interventions targeting the fundamental logic of immune memory.
Tissue-resident memory T (TRM) cells are a subset of long-lived memory T cells that reside in non-lymphoid barrier tissues, exhibiting unique localization, functional, and metabolic adaptations [9,10]. Recent studies have shed new light on the mechanisms driving disease relapse in rheumatic autoimmune disorders. TRM cells, which reside predominantly in barrier tissues such as the skin, lungs, gut, liver, and brain, persist long-term and are poised for rapid response to local stimuli. They secrete potent inflammatory mediators and play critical roles in both anti-tumor immunity and autoreactive immune responses [11,12,13,14]. Typical surface markers of TRM cells include CD69, CD103, and CD49a, which collectively mediate tissue retention and functional activity [10,13,15]. CD69 suppresses S1P1-dependent tissue egress, CD103 binds epithelial E-cadherin to promote adhesion, and CD49a interacts with collagen to support basement-membrane localization and cytotoxic function [16,17].
The pathogenic role of TRM cells in rheumatic autoimmune diseases is increasingly recognized. In psoriatic arthritis, CD69+CD103+CD8+ TRM cells are enriched in synovial fluid and secrete high levels of IL-17A, indicating their involvement in local inflammation and disease relapse [18]; Similarly, in autoimmune hepatitis, hepatic CD8+ TRM cell abundance correlates positively with disease severity [19]. In murine arthritis models and human RA synovium, CD8+ TRM cells persist in previously inflamed sites during remission and recruit circulating effector cells via the chemokine (C-C motif) ligand 5 (CCL5) axis, triggering arthritis flare-ups—a phenomenon termed “lesion memory” [20]. Spatial transcriptomics and multiplex immunofluorescence analyses of RA synovial tissue have revealed dense clusters of CD8+CD69+CD103+ TRM cells co-localized with high levels of IL-6 and TNF-α. These findings suggest that TRM cells sustain persistent local inflammation through a “lesion memory”–driven mechanism [21]. Collectively, these findings highlight TRM cells as central mediators of “lesion memory,” sustaining disease activity and relapse in rheumatic autoimmune disorders.
Current therapies predominantly target circulating immune cells and exert limited effects on tissue-resident TRM cells. Consequently, developing strategies that specifically modulate TRM cells—such as disrupting key survival pathways like IL-15 or TGF-β signaling, or blocking TRM-associated surface molecules—is essential for achieving durable remission and potentially curing rheumatic autoimmune diseases [22,23].
Based on this background, the present study aims to elucidate the pathogenic mechanisms of TRM cells in the relapse of rheumatic autoimmune diseases and to explore their potential as therapeutic targets. By systematically analyzing TRM cell phenotypes, distribution, and functions, this work seeks to provide a theoretical foundation and translational direction for the development of precision therapies targeting rheumatic autoimmune disorders.
2. TRM Cell Biology in Rheumatoid Immune Diseases
2.1. Development and Maintenance Mechanisms of TRM Cells
2.1.1. Key Regulatory Signals
TRM cell differentiation is influenced by both intrinsic transcriptional programs and extrinsic factors like local cytokines, adhesion molecules, and metabolic adaptations [24].
Local cytokines, as essential components of the tissue microenvironment, are crucial for both the differentiation and maintenance of TRM cells [25]. Transforming growth factor-β (TGF-β) is a central inducer of the TRM phenotype. Together with T cell receptor (TCR) signaling, TGF-β promotes the expression of integrin αE (CD103), thereby guiding CD8+ T cells toward a tissue-resident state [26]. Moreover, TGF-β fine-tunes downstream gene transcription through SMAD-dependent pathways, thereby ensuring that TRM cells acquire and sustain long-term residency [27,28]. In coordination with other local cytokines, TGF-β suppresses the expression of circulation-associated genes, such as S1PR1 and KLF2, to further promote tissue retention [22]. Interleukin-15 (IL-15) is another indispensable cytokine for TRM cell maintenance. Through activation of the JAK–STAT pathway, IL-15 upregulates anti-apoptotic molecules including Bcl-2 and enhances metabolic activity, thereby supporting TRM cell survival, proliferation, and effector function [29]. IL-15 also sustains mitochondrial fatty acid oxidation (FAO), enabling TRM cells to preserve energy homeostasis and resist apoptosis over prolonged periods [30,31]. Additionally, in certain tissues, interleukin-7 (IL-7) signals through CD127 and downstream JAK–STAT5 to further promote TRM cell longevity and homeostasis [30,31]. Additionally, in certain tissues, IL-7 engages its receptor CD127 to activate downstream JAK–STAT5 signaling, further promoting TRM cells longevity and homeostasis [32,33]. Collectively, TGF-β, IL-15, and IL-7 reshape transcriptional and signaling networks by repressing egress-associated genes while activating survival and metabolic programs, ultimately orchestrating TRM cell differentiation, tissue residency, and persistence [25].
Another crucial determinant of long-term tissue residency in TRM cells is their high expression of adhesion molecules [29]. Among these, CD103 is the most characteristic, mediating physical anchoring of TRM cells to epithelial E-cadherin [34,35]. TGF-β signaling induces CD103 expression in CD8+ T cells, conferring the hallmark tissue-resident phenotype [28]. In addition, CD103 can interact with local extracellular matrix (ECM) components, such as collagen, to further stabilize TRM cell retention in collagen-rich tissues [34]. Collagen-binding receptors also contribute indispensably to TRM cell positioning and stability, helping cells maintain spatial organization and interact dynamically with their microenvironment [9]. These adhesion molecules collectively form a structural framework that enables rapid responses to local antigen re-exposure while sustaining long-term immune surveillance [13]. Moreover, adhesion molecules participate in intracellular signaling cascades that modulate metabolism and effector function, establishing positive feedback loops that reinforce TRM cell differentiation and persistence [36].
2.1.2. Metabolic Adaptation Mechanisms
To persist long-term in tissues, TRM cells must adapt to environmental challenges such as nutrient scarcity, hypoxia, and metabolic stress. They achieve this through specialized metabolic adaptation mechanisms that optimize energy utilization and survival [37]. Among these, FAO plays a central role. Multiple studies have shown that under nutrient-deprived conditions where amino acids and glucose are limited, TRM cells rely on Carnitine Palmitoyltransferase 1A (CPT1a)-mediated transport of fatty acids into mitochondria for β-oxidation [24,38]. Since TRM cells often inhabit nutrient-poor tissue niches, lipid metabolism becomes a key energy source to sustain their long-term functionality [39].
Hypoxia is another prominent feature of tissue environments, profoundly influencing TRM cell development and maintenance [40]. Under low oxygen tension, hypoxia-inducible factor-1α (HIF-1α) stabilizes and becomes transcriptionally active, initiating adaptive gene expression programs [41]. HIF-1α not only upregulates glycolytic enzymes but also modulates FAO and mitochondrial function, coordinating metabolic reprogramming and energy homeostasis within TRM cells [36,42,43]. By increasing glucose uptake and lactate production, HIF-1α helps partially compensate for hypoxia-induced energy deficits, while its cooperative regulation with FAO supports TRM cell survival in oxygen- and nutrient-limited environments [41]. Moreover, HIF-1α regulates ECM remodeling and adhesion molecule expression, providing structural and metabolic support that enables TRM cells to remain stably resident under hypoxic conditions [25,44]. During this adaptive process, TRM cells must simultaneously manage hypoxia-induced metabolic stress and integrate regulatory signals from local cytokines and adhesion molecules. Consequently, TRM cells establish a highly integrated regulatory network that coordinates metabolism, signaling, and survival, ensuring stable residency and robust immune responsiveness within tissues [45].
The development and maintenance of TRM cells depend on the synergistic regulation of local cytokines, adhesion molecules, and metabolic adaptation mechanisms, which together form a tightly interconnected immunoregulatory network [28,46]. Specifically, TGF-β functions as a central regulator—inducing CD103 expression to strengthen epithelial adhesion while reshaping cellular metabolism by activating FAO, thus enabling TRM cells to sustain energy homeostasis and survival in hypoxic, nutrient-deprived environments [25,47]. IL-15 and IL-7 complement this process by activating anti-apoptotic pathways such as PI3K–AKT, further extending TRM cell lifespan [48]. Meanwhile, adhesion molecules such as integrin αEβ7 not only provide structural anchorage but also mediate mechanotransducive signaling that coordinates the expression of metabolic genes, thereby promoting metabolic reprogramming [49,50]. Through these multidimensional interactions, TRM cells acquire unique tissue-residency properties that enable them to function as a potent immune barrier within epithelial and barrier tissues, mounting rapid and durable responses against local pathogen invasion or tumor dissemination and achieving precise tissue-specific immune protection [51].
2.2. Heterogeneity and Disease Specificity of TRM Cells
2.2.1. Tissue-Distribution Heterogeneity
TRM cells display marked differences in phenotypic markers, transcriptional programs, and functions across organs. For example, TRM cells in the skin, lung, and intestine commonly express CD69, whereas CD103 expression varies substantially: CD8+ TRM cells in the skin and intestinal epithelium often express high levels of CD103, while lung TRM cell populations are predominantly CD103-negative or low-expressing [52,53]. This heterogeneity reflects tissue-specific shaping of cell fate by local microenvironments; cell–cell interactions, cytokine milieus, and extracellular-matrix composition collectively drive tissue-tailored transcriptional maturation and phenotypes [37,54].
Even within the same organ, distinct TRM cell subpopulations are evident. In the skin, for instance, TRM cells can be subdivided into epidermal and dermal compartments: epidermal TRM cells typically exhibit high CD103 expression and stronger cytotoxic potential, whereas dermal TRM cells tend to display more active cytokine production and proliferative capacity [52,55]. In addition, TRM cells in the lung can also be divided into subpopulations that are retained stably in the lung interstitium and are continuously replenished by circulating T cells, which play different roles in antigen reencounter and continuous immune surveillance [9,56].
2.2.2. Functional Heterogeneity
Beyond spatial diversity, TRM cells also differ functionally. One subset is characterized by robust production of IFN-γ and TNF-α and exhibits potent pro-inflammatory and cytotoxic activities [57,58]; Another subset expresses immunoregulatory molecules such as IL-10 and Programmed death receptor 1 (PD-1) and contributes to local protection and immune modulation [59,60,61,62]. In addition, upon antigenic stimulation, a fraction of TRM cells can exit their tissue of residence and enter the circulation as so-called “ex- TRM cells,” which may display heightened proliferative capacity and multipotency [63,64].
Pro-Inflammatory TRM Cells: Drivers of Tissue Damage
Pro-inflammatory TRM cells serve as frontline responders in tissues. Upon antigenic or inflammatory cues, these cells rapidly secrete high levels of IFN-γ and TNF-α, promoting recruitment/activation of effector leukocytes and triggering inflammatory cell death and matrix degradation in resident cells, thereby exacerbating pathology [20,65,66]. In barrier sites such as skin, gut, and liver, abundant CD8+CD69+CD103+ TRM cells are positioned within the epithelium or mucosal basal zones, poised to respond to reinfection or tissue injury and to promptly release IFN-γ and TNF-α [16,21]. Following influenza infection, a CD49a+CD103+CD8+ TRM cell subset in the lung produces higher levels of IFN-γ than other T cells, contributing to viral clearance but also to local epithelial damage [67]. In psoriatic arthritis, synovial fluid is enriched for CD69+CD103+CD8+ TRM cells that secrete IFN-γ and TNF-α and, together with IL-17A, induce cartilage-degrading enzymes, thereby directly promoting cartilage destruction [68].
Regulatory TRM Cells: Guardians of Local Protection
TRM cells not only exert potent pro-inflammatory effects but can also adopt regulatory or inhibitory phenotypes that limit excessive inflammation and promote tissue repair. Regulatory TRM cells express IL-10, PD-1, and other inhibitory molecules, playing a key role in maintaining tissue balance and preventing immune-related damage. IL-10–producing TRM cells rapidly suppress overactivation of macrophages and other effector cells, thereby mitigating tissue injury [69]. PD-1/PD-L1 signaling negatively regulates TRM cell activity, curbing excessive IFN-γ and TNF-α production and facilitating tissue repair [21]. In secondary influenza challenge models, PD-1+ TRM cells help balance antiviral immunity with pulmonary immunopathology; blockade of PD-1 causes over-expansion of TRM cells and worsened tissue injury [70]. In a non-human primate model of SARS-CoV-2 infection, IL-10 signaling both promotes TRM cell formation and limits excessive T-cell expansion, thereby balancing antiviral responses with tissue protection [60]. In the pancreas, TRM cells maintain local immune homeostasis via the PD-1/PD-L1 axis, preventing autoimmune pancreatitis [71]. IL-10+ TRM cells limit T-cell–mediated damage in cutaneous wound and intestinal inflammation models, promoting wound healing and mucosal repair [72]. Thus, therapeutically enhancing IL-10+/PD-1+ TRM cells—or mimicking their functions—may offer new strategies for autoimmune diseases such as psoriatic arthritis and rheumatoid arthritis.
2.3. Disease-Specific TRM Cells Revealed by Single-Cell Sequencing
Recent single-cell and histological studies have identified disease-specific TRM cell subsets across rheumatic autoimmune conditions, showing that distinct chemokine-receptor repertoires and spatial localization underpin disease-driving programs.
In RA, single-cell sequencing of synovial tissue has revealed a phenotypically unique subset of PD-1+GPR56+ tissue-resident T cells co-expressing classical TRM cell markers such as CXCR6, CD69, and LAG3. These cells show significant clonal expansion and are enriched in ACPA-positive patients, indicating antigen-driven persistence [73]. Earlier work indicated that synovial CXCR6+ TRM cells can persist long-term within the joint microenvironment, continuously producing pro-inflammatory and regulatory mediators under inflammatory conditions, thereby sustaining chemokine/cytokine networks and, via crosstalk with B cells and other leukocytes, promoting chronicity and relapse of synovitis [74]. Integrated single-cell transcriptomics and TCR sequencing further indicate functional plasticity: these TRM cells support B-cell autoantibody production and can also contribute directly to cartilage and bone damage via cytotoxic pathways and amplification of local inflammation [75]. These insights nominate CXCR6-mediated retention or effector programs as potential precision-targeting strategies to control RA synovitis and joint destruction.
In SLE, single-cell analyses of skin lesions have identified a population of TRM cells expressing the skin-homing receptor CCR10. These CCR10+ TRM cells localize to the epidermis and peri-follicular niches, where CCR10-dependent homing and retention enable long-term persistence and production of IFN-γ, IL-17, and other inflammatory mediators that exacerbate chronic cutaneous inflammation [76]. Single-cell data suggest interactions between CCR10+ TRM cells and dendritic or B cells. These interactions may enhance antigen presentation, sustain T-cell activation, and contribute to autoimmunity [77]. While no clinical trials directly targeting CCR10+ TRM cells have yet been registered, current single-cell evidence supports their importance in SLE skin pathology and points to CCR10-mediated skin-homing pathways as candidate therapeutic targets.
SSc is a systemic autoimmune disease characterized by fibrosis of the skin and internal organs [78]. While no clinical trials directly targeting CCR10+ TRM cells have yet been registered, current single-cell evidence supports their importance in SLE skin pathology and points to CCR10-mediated skin-homing pathways as candidate therapeutic targets [79]. More recent large-scale single-cell work confirmed immune-cell compositional abnormalities and identified immune subsets associated with clinical heterogeneity, including T cells with resident/homing phenotypes [80]. In SSc-associated interstitial lung disease (SSc-ILD), studies further report enrichment of a CD8+ T-cell subset with type-II interferon features in both blood and lung tissues, whose migratory behavior may contribute to ILD progression [81]. These findings imply that TRM-like populations may participate in local immune dysregulation within SSc target organs.
In pSS, multiple single-cell sequencing studies have shown that the distribution and functional status of immune cells in the salivary glands and other exocrine glands play a key role in the destruction of glandular structure and loss of secretory function. Notably, CD8+ TRM cells are markedly expanded in salivary glands, expressing canonical residency markers (CD69, CD103) along with high activation signatures (e.g., HLA-DR) and cytotoxic mediators (e.g., IFN-γ, granzyme B), thereby damaging glandular epithelium through direct cytotoxicity and pro-inflammatory cytokines [82,83]. These locally expanded TRM cells also interact closely with dendritic cells and B cells, amplifying inflammatory cytokine and chemokine networks and culminating in glandular dysfunction and reduced secretion [82]. Single-cell transcriptomics further show that TRM cells can persist locally without continuous peripheral input; their sustained inflammatory state promotes epithelial apoptosis/dysfunction, clinically manifesting as xerostomia and xerophthalmia [83].
In summary, single-cell technology reveals the disease-specificity and pathological role of TRM cells in different autoimmune diseases. In RA, synovial CXCR6+ TRM cells sustain local inflammation and drive joint damage via chemokine and cytokine outputs; in cutaneous SLE, CCR10-high TRM cells persist in skin and exacerbate local responses through inflammatory mediators; in SSc, perivascular TRM cells may contribute to fibroblast activation and fibrosis (e.g., via TGF-β–linked pathways); and in pSS, glandular CD8+ TRM cells inflict epithelial injury through cytotoxic and inflammatory mechanisms, leading to secretory failure. Integrating single-cell with spatial omics will further clarify these mechanisms and accelerate the development of TRM cell-targeted immunotherapies tailored to disease context (As shown in Table 1).

Table 1.
Characteristics and functions of TRM cells in different rheumatic immune diseases.
3. TRM Cell-Driven Pathogenic Mechanisms
In recent years, numerous studies have shown that TRM cells not only protect the host by preventing pathogen re-encounter but also play pivotal roles in the pathogenesis of autoimmune diseases. In disorders such as RA [84,85] and SLE [86], TRM cells within tissues secrete inflammatory mediators, cytokines, and chemokines that drive uncontrolled local inflammation, culminating in tissue destruction, fibrosis, and organ dysfunction (As shown in Figure 1).
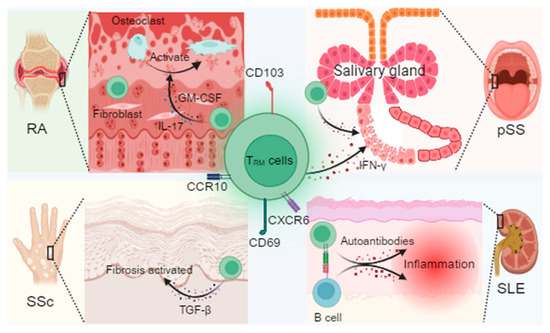
Figure 1.
Mechanistic overview of TRM cells and their pathogenic roles across autoimmune diseases. TRM cell markers include CD69, CD103, CXCR6, and CCR10, maintained by cytokine signaling through transforming growth factor-β (TGF-β), interleukin-15 (IL-15), and interleukin-7 (IL-7). In rheumatoid arthritis (RA), synovial TRM cells interact with fibroblasts, secrete granulocyte–macrophage colony-stimulating factor (GM-CSF), and promote osteoclast activation, contributing to joint erosion. In primary Sjögren’s syndrome (pSS), CD8+ TRM cells in the salivary glands release cytotoxic mediators that damage glandular epithelium. In systemic sclerosis (SSc), TRM-derived TGF-β drives fibroblast activation and tissue fibrosis, accompanied by epigenetic reprogramming of local immune cells. In systemic lupus erythematosus (SLE), TRM cells in the skin and kidney produce type I interferons and form immune synapses with B cells, facilitating autoantibody production. Together, these findings highlight TRM cells as critical mediators of tissue inflammation, fibrotic remodeling, and disease-specific “inflammatory memory”. Created in BioRender. Min, D. (2025) https://BioRender.com/lxe8sih (accessed on 29 October 2025).
3.1. TRM Cell-Mediated Pathogenic Mechanisms in Rheumatoid Arthritis (RA)
3.1.1. Synovial TRM Cells–Fibroblast Crosstalk and Promotion of Osteoclastogenesis
Within RA synovium, TRM cells are locally enriched in inflamed joints and display distinct residency phenotypes; these cells engage in close cell–cell interactions with synovial fibroblasts [87]. Synovial TRM cells exhibit pronounced clonal expansion and, upon recognition of autoantigens in situ, rapidly secrete multiple pro-inflammatory cytokines, including GM-CSF [88]. GM-CSF is a key inflammatory mediator that stimulates synovial fibroblast proliferation and activation and directly acts on macrophages and osteoclast precursors to induce their differentiation into mature osteoclasts, thereby promoting bone erosion within the joint [88,89]. Thus, immune–stromal interactions driven by TRM cell-derived inflammatory factors are central to RA joint damage. In addition, TRM cell-derived IL-17 and other cytokines form a positive feedback loop with synovial fibroblasts, which, in turn, produce IL-6, CXCL12, and RANKL. As a critical signal for osteoclast differentiation, excessive RANKL expression directly accelerates bone resorption and joint destruction [75,90]. This cytokine cascade amplifies into a self-reinforcing pathological circuit, sustaining inflammation and ultimately leading to irreversible tissue injury [90].
3.1.2. Role of the IL-23/IL-17 Positive Feedback Loop in Maintaining Chronic Inflammation
Beyond GM-CSF–mediated osteoclast activation, TRM cells and local antigen-presenting cells (APCs) establish a positive feedback circuit centered on IL-23 and IL-17, which maintains and exacerbates synovial inflammation [87,91]. In this loop, dendritic cells and macrophages secrete IL-23, which effectively induces TRM cells and Th17 cells to produce IL-17. IL-17 not only directly activates synovial fibroblasts to release additional inflammatory mediators and RANKL but also promotes local GM-CSF production, thereby closing the inflammatory loop within the joint microenvironment [92,93]. Consequently, even when systemic inflammatory mediators decline, the intra-articular cytokine network can self-sustain a highly inflamed state, driving chronicity and progressive tissue destruction [20].
3.1.3. Extra-Articular Manifestations
The role of pulmonary TRM cells in RA-associated interstitial lung disease RA is not only limited to joint inflammation, but its extra-articular manifestations have also attracted much attention, among which RA-associated interstitial lung disease (RA-ILD) is one of the important manifestations of significant pathogenicity [94]. Emerging evidence suggests that pulmonary TRM cells also mediate local immune responses and may act as “ignition switches” in fibrotic lesions. Upon environmental or autoantigenic stimulation, lung TRM cells are activated to secrete inflammatory factors such as IL-17 and GM-CSF, which induce profibrotic mediator release from interstitial cells and activate fibroblasts [95,96]. This local immune activation partially mirrors intra-articular mechanisms, implying that pulmonary TRM cells may promote interstitial inflammation and fibrosis through cytokine networks analogous to those in synovium, ultimately impairing lung function [95]. Targeting TRM cells and their associated cytokine networks could, therefore, provide new therapeutic entry points for RA and its extra-articular complications [94].
3.2. TRM Cell Pathogenesis in Systemic Lupus Erythematosus (SLE)
3.2.1. Long-Term Retention of Skin TRM Cells and Production of Type I Interferon
In SLE—especially cutaneous manifestations such as cutaneous lupus erythematosus and discoid lupus—TRM cells have garnered increasing attention. Cutaneous TRM cells, characterized by CD69 and CD103 expression, exhibit long-term residency that supports sustained local immune activation [86,97]. Following triggers such as ultraviolet irradiation, these persistent TRM cells can be reactivated to produce large amounts of type I interferons (e.g., IFN-α). Type I interferons enhance antigen presentation and inflammation and activate neighboring keratinocytes to produce additional inflammatory mediators, thereby establishing a self-sustaining pro-inflammatory network in the skin [98]. Consequently, persistent activation of cutaneous TRM cells is a key driver of chronic inflammation, lesion persistence, and frequent relapse in SLE skin disease.
3.2.2. The Formation of TRM Cells and B Cell Immune Synapses in the Kidney Promotes the Production of Autoantibodies
Beyond the skin, lupus nephritis represents one of the most severe SLE manifestations. Recent studies indicate that renal TRM cells participate in local immune regulation and directly promote autoantibody production through interactions with B cells [99]. In the kidney, TRM cells can form immunological synapses with B cells via CD40/CD40L interactions, delivering potent co-stimulatory signals that drive B-cell proliferation and differentiation into antibody-secreting plasma cells [100,101]. Concurrently, TRM cell-derived IL-21 further activates B-cell secretory pathways, enhancing immunoglobulin production and class switching, thereby exacerbating pathogenic autoantibody generation [100]. This TRM cell–B-cell crosstalk is a critical component of SLE immunopathology, fostering immune-complex deposition and correlating with the severity of organ damage (e.g., glomerular injury).
3.3. TRM Cell-Mediated Pathogenic Mechanisms in Systemic Sclerosis (SSc)
3.3.1. TRM Cell-Derived TGF-β as a Driver of Cutaneous and Vascular Fibrosis
SSc is clinically defined by widespread fibrosis, presenting as skin thickening, internal-organ fibrosis, and vasculopathy. Recent studies suggest that aberrantly activated TRM cells may directly contribute to fibrogenesis in SSc through secretion of profibrotic cytokines such as TGF-β [102]. TGF-β is a central mediator of fibrosis, promoting fibroblast proliferation and differentiation into myofibroblasts and inducing extracellular matrix deposition (e.g., collagen), which culminates in tissue stiffening and functional impairment [103]. Although direct human evidence that TRM cells produce TGF-β in SSc remains limited, the multifaceted secretory capacity of TRM cells in other autoimmune settings, together with the highly inflamed SSc lesions, supports the hypothesis that aberrant TRM cell activation may constitute an important local source of TGF-β that accelerates fibrosis [79].
3.3.2. Epigenetic Reprogramming Underlies Aberrant TRM Cell Activation
In addition to profibrotic cytokine production, epigenetic dysregulation is thought to contribute to abnormal TRM cell function in SSc. T cells from patients with SSc exhibit pronounced DNA-methylation abnormalities and histone-modification defects, leading to dysregulated expression of immune regulatory genes and a persistently activated TRM cell-like state [104,105]. Such epigenetic reprogramming disrupts immune tolerance and predisposes TRM cells to produce pro-inflammatory and profibrotic cytokines—including TGF-β—thereby aggravating local fibrosis and endothelial pathology [106]. Thus, in SSc, aberrant TRM cell activation appears to be co-driven by exogenous inflammatory cues and intrinsic epigenetic factors, forming a self-sustaining fibrotic circuit that severely compromises the function of skin and internal organs [107,108].
3.4. TRM Cell-Mediated Pathogenic Mechanisms in Primary Sjögren’s Syndrome (pSS)
In pSS, pathological changes primarily affect salivary and lacrimal glands, leading to acinar destruction and loss of secretory function, clinically manifesting as xerostomia and xerophthalmia. Recent studies highlight TRM cells residing in salivary glands as key mediators of this process [109]. These glandular TRM cells persist long-term and, upon activation, can secrete lymphotoxin-α (LTα)—a TNF-family cytokine with direct tissue-damaging potential [110]. LTα establishes an early pro-inflammatory milieu within salivary glands; even in the absence of overt lymphocytic infiltration, glandular secretion can be impaired due to LTα-mediated immune injury [111,112]. As disease progresses, sustained TRM cell activity disrupts glandular architecture, damaging acini and ducts and ultimately causing irreversible functional loss, consistent with clinical xerostomia [83,113].
4. Therapeutic Targeting of TRM Cells in Rheumatoid Immune Diseases
4.1. Drug Repurposing
4.1.1. JAK Inhibitors
The survival and proliferation of TRM cells depend substantially on IL-15 signaling, which primarily activates downstream effectors via the JAK–STAT pathway—especially STAT5—to support TRM cell maintenance and function [114]. Current studies indicate that JAK inhibitors effectively block IL-15 signal transduction, thereby reducing TRM cell survival and the secretion of pro-inflammatory cytokines, with notable therapeutic benefits in inflammatory skin diseases such as vitiligo and psoriasis [115]. In addition, JAK inhibition attenuates antigen-triggered local inflammatory responses, providing a preliminary basis for subsequent combination strategies [116,117].
4.1.2. Anti-IL-17/23 Antibodies
IL-17 and IL-23 are pivotal in regulating TRM cell functions and sustaining an inflammatory microenvironment [118]. In psoriasis, tissue-resident TRM cells represent a key source of IL-17, whereas IL-23 provides essential survival and functional support; the IL-23/IL-17 axis relies on a local “inflammatory memory pool” formed by TRM cells to drive relapse [119]. Clinical studies have shown that anti-IL-23 antibodies (e.g., risankizumab) and anti-IL-17 antibodies (e.g., secukinumab) achieve significant efficacy in psoriasis and other Th17-mediated diseases. Their durable remission partly reflects indirect modulation of TRM cells, interrupting cytokine feedback circuits and dampening TRM cell-driven pro-inflammatory functions [120,121].
4.1.3. Low-Dose Radiotherapy (LDRT)
As a local modality, LDRT shows potential for the selective reduction of TRM cells. Traditionally used for anti-inflammatory and anti-proliferative effects, recent evidence suggests that precise dose control may locally “debulk” or decrease radio-tolerant TRM cell populations while sparing other immune cells, thereby alleviating local inflammation and improving clinical status [122]. This approach is particularly applicable to focal inflammatory conditions, such as RA [123], and psoriasis [122], and may complement systemic therapies.
4.2. Emerging Targeted Strategies
Although repurposed agents can partially restrain TRM cell-mediated inflammation, complete eradication remains challenging due to the tissue-retentive nature of TRM cells. Consequently, new strategies are being developed to disrupt residency programs and to intervene in key metabolic pathways (As shown in Table 2).
Table 2.
Existing and potential TRM cell-targeted therapy strategies.
4.2.1. Disrupting Residency Signals
TRM cell localization partly depends on responsiveness to sphingosine-1-phosphate (S1P). S1PR1 is critical for regulating lymphocyte egress and tissue trafficking. Pharmacologic activation of S1PR1 (e.g., fingolimod) can promote TRM cells egress from niches, reduce the density of local inflammatory cells, and ameliorate focal immune inflammation. By altering intra-tissue distribution, this strategy offers a new point of intervention that can be combined with other immunomodulators [55]. Although most data derive from inflammatory bowel disease and related contexts, the underlying mechanisms extend to other rheumatic diseases characterized by recurrent focal inflammation [131].
Integrin αEβ7 (linked to CD103 expression) is indispensable for TRM cell adhesion to epithelial cells. Inhibiting this integrin can disrupt TRM cell–tissue interactions, facilitating egress or reducing residency stability and thereby diminishing chronic local inflammation [132]. Preclinical data suggest that integrin inhibitors may become useful tools against inflammatory skin or rheumatic diseases [114]. In SSc, TGF-β is a key fibrogenic cytokine, and multiple αv integrins (e.g., αvβ1, αvβ3, αvβ5, αvβ6, αvβ8) participate in its activation. Thus, targeting αv-integrin–mediated latent TGF-β activation has emerged as a more prominent antifibrotic strategy than αEβ7 blockade and represents a promising approach in SSc [133].
4.2.2. Metabolic Interventions
TRM cells exhibit heightened energy demands to sustain long-term survival and effector competence, with a particular reliance on fatty-acid oxidation (FAO). In gastric adenocarcinoma, CD8+CD103+ TRM cells undergo metabolic reprogramming and depend on FAO, with CPT1a acting as a rate-limiting enzyme; rewiring metabolism prolongs TRM cell lifespan and enhances anti-tumor immunity [134]. Similarly, CPT1a-mediated FAO is critical in other tissue-resident immune populations. A 2024 study in lupus nephritis models reported reduced CPT1a expression in renal and circulating macrophages, impairing efferocytosis and exacerbating renal inflammation [124]. These data suggest that immune cell–specific metabolic targeting (e.g., modulating CPT1a) offers a promising avenue in autoimmunity. By analogy, targeting CPT1a in TRM cells may provide a metabolic handle for intervention and could be combined with other immunoregulatory therapies.
Metformin, a widely used antihyperglycemic agent, activates AMPK and inhibits mTORC1, downregulating anabolic and proliferative programs and potentially affecting the survival and function of metabolically dependent immune subsets [135]. Although direct evidence for TRM cell-specific effects is lacking, metformin’s immunomodulatory impact has attracted attention. Preclinical studies indicate modulation of T-cell subset balance (e.g., Th17/Treg) in multiple rheumatic models [125], and clinical data in SLE show reduced relapse risk and steroid-sparing trends with metformin add-on therapy, alongside improvements in patient-reported outcomes [126]. These findings support the hypothesis that systemic immune-metabolic tuning may indirectly influence TRM cell-involved chronic inflammation and justify further exploration in rheumatic disease.
4.3. Microenvironment-Remodeling Strategies
Long-term TRM cell persistence relies not only on intrinsic survival programs but also on extrinsic support from the local microenvironment—including ECM, chemokines, and neighboring immune cells. Remodeling this niche to weaken trophic support for TRM cells represents an innovative and promising therapeutic direction.
4.3.1. Smart Nanoparticle Delivery of siRNA
This approach employs functionalized nanocarriers to deliver small interfering RNAs (siRNAs) to targeted cells within lesions, silencing pathogenic genes to perturb TRM cell survival and function. Multiple smarts, stimulus-responsive nanoparticles have been developed. For example, macrophage-targeted polymeric polymersomes can sequentially deliver TNF-α siRNA (siTNFα) followed by dexamethasone. In the collagen-induced arthritis (CIA) model, this “temporal nanotherapy” reverses glucocorticoid resistance via TNF-α silencing and then achieves synergistic anti-inflammatory and joint-protective effects with co-administration [127]. Another microenvironment-driven, deformable self-assembling nanoplatform responds to elevated MMP-2 and acidic pH in RA joints. Upon arrival, the platform deforms to form a physical barrier limiting inflammatory cell infiltration and precisely releases payloads (e.g., triptolide, metformin), thereby remodeling the vascular–immune–inflammatory milieu. Preclinical studies demonstrate specificity and safety of nanoparticle-mediated siRNA delivery for local gene modulation, supporting future clinical translation [128].
4.3.2. Synthetic-Biology Engineering of Fibroblasts
An additional strategy seeks to reprogram local fibroblasts using synthetic-biology tools to create in situ “drug factories” that continuously secrete therapeutic proteins. In rheumatic diseases—particularly RA—engineering fibroblasts to reconfigure the joint microenvironment is an active frontier [129]. In RA, aberrant activation of fibroblast-like synoviocytes (FLS) is central to synovial inflammation and bone destruction [130]; synthetic-biology interventions aim for precise control of these cells. In the future, such local microenvironmental control can be combined with systemic therapies (small molecules or biologics) to achieve complementary mechanisms and synergistic efficacy, offering a more comprehensive therapeutic paradigm for rheumatic autoimmune diseases [136].
5. Challenges and Future Perspectives
TRM cells, a subset of T cells that permanently reside in barrier tissues and non-lymphoid organs, play key roles in maintaining chronic inflammation and driving relapse in rheumatic autoimmune diseases such as RA, psoriatic arthritis, and SLE. Although targeting TRM cells offers new promise for durable disease control, clinical translation faces multiple challenges and will depend on advances in technology and personalized treatment strategies.
5.1. Challenges for Clinical Translation
5.1.1. Difficulties in Tissue-Specific Targeting
Marked heterogeneity of TRM cells across tissues complicates precise targeting. Studies indicate substantial functional differences across organs; for example, skin TRM cells may depend on antigen-specific deletion to sustain protective immunity, whereas synovial TRM cells appear less dependent on this mechanism. This raises the prospect of tissue-differentiated interventions but also underscores the difficulty of selectively modulating TRM cells across distinct tissues [55,137]. In inflammatory arthritis, synovial TRM cells can persist in remission and establish “joint-specific memory,” thereby mediating disease flares. Given that indiscriminate TRM cell depletion may compromise local immune homeostasis—and that TRM cell heterogeneity spans functions and regulatory programs—no broadly applicable strategy yet balances effective suppression of pathogenic inflammation with preservation of protective immunity. This constitutes a central complexity in current clinical translation [137].
5.1.2. Potential Impacts of TRM Cell Depletion on Immune Memory
Potential erosion of immune memory is a key concern in therapeutic TRM cell depletion. As sentinels of barrier immunity, TRM cells form the first defensive layer against local pathogen entry. Non-selective depletion may weaken local defenses and increase opportunistic infections [138]. Even putatively selective strategies risk diminished anti-infective memory and suboptimal vaccine responses if specificity is insufficient, leading to heightened infection risk and reduced vaccine efficacy [137]. In tissues such as joints and skin, TRM cells combine pro-inflammatory functions with immune surveillance; thus, intervention requires careful balancing of inflammatory control with baseline protection [137]. Achieving precise control of inflammation-related TRM cells while maintaining local and systemic immune homeostasis has therefore become a central translational question.
5.2. Key Technological Directions
5.2.1. Spatial Transcriptomics: Mapping TRM Cells–Stroma Interactions
Spatial transcriptomics has emerged as a powerful tool to resolve cellular positioning and interactions in situ, enabling precise mapping of TRM cell niches and their crosstalk with neighboring cells. In lupus nephritis (LN), spatial profiling revealed a distinct inflammatory–injury niche in the renal cortex enriched for VCAM1+ proximal tubule cells, myofibroblasts, and multiple immune lineages. VCAM1+ tubule cells actively recruit immune cells via chemokines (e.g., CXCL12) and signal with surrounding cells through TGF-β pathways, collectively shaping a pro-inflammatory, pro-fibrotic microenvironment [139]. In RA synovium, spatial analyses identified a key interaction between CD8+ T cells and endothelial cells via the CCL5–ACKR1 ligand–receptor axis, with spatial co-localization that facilitates transendothelial leukocyte trafficking and exacerbates synovitis [140].
5.2.2. Organoid Models: Modeling Long-Term TRM Cell Persistence and Optimizing Drug Discovery
Organoid systems recapitulate tissue architecture and function and are increasingly used to model immune–epithelial interfaces. A 2024 study established human intestinal immune organoids (IIOs) containing autologous TRM cells. TRM cells actively invaded and integrated into the epithelial barrier at an approximate 1:16 ratio—mirroring healthy gut distributions—and remained viable for ≥14 days under low-cytokine conditions [141]. In rheumatic disease research [142,143], patient-derived 3D organoids have reproduced fibroblast-like synoviocyte (FLS) hyperproliferation and cytokine cascades (e.g., TNF-α, IL-6). Single-cell sequencing confirms preservation of patient-specific transcriptional signatures, enabling mechanistic dissection and pharmacologic testing.
5.2.3. AI-Guided Epigenetic Target Discovery for Precision Therapy
Artificial intelligence (AI) is increasingly applied to identify epigenetic targets. DNA methylation and other epigenetic marks play pivotal roles in autoimmune diseases, particularly RA and SLE. Pro-inflammatory cytokines and microenvironmental stress can reprogram methylation patterns, which may predict disease course, subtypes, and therapeutic response [144,145].
5.3. The Future of Individualized Therapy
5.3.1. TRM Cell Biomarker Assessment
Given their persistence and rapid activation in local inflammation, TRM cell abundance and functional state correlate with disease activity. Assessment typically relies on canonical surface markers such as CD69 and CD103. In cutaneous lupus, lesional skin exhibits significantly increased CD69+ and/or CD103+ TRM cells compared with healthy skin, directly indicating local accumulation in autoimmune lesions [97]. In psoriatic arthritis, a pro-inflammatory CD161 + CCR6+ type-17–like TRM cell subset produces IL-17A, TNF-α, and IFN-γ, suggesting potential biomarker utility for distinguishing arthritis subtypes [68].
CD39+ TRM cells represent a phenotypic subset whose expression levels reflect local inflammatory burden and dysregulated immune control [55]. In inflammatory bowel disease and RA, changes in CD39 expression correlate with clinical fluctuations, supporting CD39+ TRM cell load as a candidate indicator of severity and prognosis [146]. High-resolution platforms—including flow cytometry, immunofluorescence, and spatial transcriptomics—now enable quantitative and phenotypic profiling of TRM cell subsets.
5.3.2. Combination Therapies
Onotherapy with immunosuppressants may be insufficient to fully eliminate or reprogram pathogenic TRM cells. Therapies that selectively remove aberrant resident cells—such as engineered fusion antibodies, toxin conjugates, or cellular approaches—offer potential precision routes to target TRM cells [55,147]. Recent work has used targeted lipid nanoparticles (tLNPs) to deliver mRNA and reprogram CD8+ T cells in samples from healthy donors and autoimmune patients, opening in vivo therapeutic avenues for cancer and autoimmunity [148]. However, cell removal alone rarely prevents relapse; microenvironmental repair is equally critical. Mesenchymal stem cell (MSC)-based immunomodulation and advanced biomaterials are gaining traction. In multiple sclerosis, MSC infusion induced T-cell anergy, reduced Th17 cells, and promoted antigen-specific Treg polarization, reversing Th17/Treg imbalance, dampening inflammation and demyelination, and restoring tolerance [149].
Emerging biomaterials—such as smart nanoparticles and instructive scaffolds—enable localized, sustained delivery of anti-inflammatory agents, promote matrix repair, and support tissue regeneration. A 2025 study engineered an immunomodulatory nanomedicine (MP@NEs/CT) co-delivering multi-epitope citrullinated autoantigens and triptolide (TPL), achieving effective RA control and inducing antigen-specific tolerance [150].
Although the direct “clearance + repair” combination strategy is still in the exploration stage in the field of TRM cells and rheumatology, some cutting-edge studies have shown a trend of synergistic integration of therapies with different mechanisms of action, which provides ideas for real combined strategies in the future. The 2025 study mentioned above is a good example. The nanodrug (MP@NEs/CT) designed in this study carries two functions at the same time: on the one hand, it carries a low-dose triptolidine (TPL) that can eliminate abnormally activated immune cells (such as Th1 and Th17 cells) through immunosuppression; On the other hand, the multiepitope citrullinated autoantigen it carries can induce antigen-specific immune tolerance and promote the proliferation of regulatory T cells (Treg) and B cells (Breg) with protective effects, thereby restoring immune balance [150]. This idea of integrating “clearance” and “regulation” functions in the same delivery system is very close to the synergistic concept of “removing disease-causing cells” and “repairing the microenvironment”.
Current research on rheumatic immune diseases is rapidly moving towards precise and individualized treatment. In the key direction of technological innovation, spatial transcriptomics technology has provided a powerful tool for elucidating the spatial distribution and interaction between TRM cells and stromal cells in the inflammatory region, and laid a foundation for revealing the cellular communication network in the lesion microenvironment. By reproducing the complex local microenvironment in vivo, organoid models can not only simulate the mechanism of long-term survival of TRM cells, but also provide a more reliable in vitro testing platform for new drug screening and efficacy optimization. At the same time, in the future of personalized therapy, through the fine evaluation of biomarkers such as CD39+ TRM cells, it can directly reflect the severity of the disease and the inflammatory burden, so as to achieve dynamic monitoring and treatment plan adjustment. In the future, the deep integration of multiple technologies and clinical translational research will further promote the leap from basic mechanism research to precision treatment strategies, ultimately enabling doctors to formulate personalized and efficient treatment plans based on the unique immune microenvironment, epigenetic characteristics and cell distribution status of each rheumatic immune disease patient, thereby reducing the risk of recurrence and improving the quality of life of patients.
6. Conclusions
TRM cells have firmly established themselves as central drivers of chronicity and relapse in rheumatic autoimmune diseases. This review has synthesized evidence illustrating how these long-lived tissue guardians, crucial for peripheral immunity, become key effectors of pathology in diseases like RA, SLE, SSc, and pSS. Their unique biology—governed by cytokines (TGF-β, IL-15), adhesion molecules (CD103), and metabolic adaptations (fatty acid oxidation)—enables their long-term persistence in tissues, forming stable “lesional memories” that sustain inflammation even during systemic remission.
The pathogenic roles of TRM cells are both diverse and disease-specific. Single-cell technologies have unveiled distinct subsets, such as synovial CXCR6+ TRM cells in RA that fuel osteoclastogenesis and chronic synovitis via GM-CSF and the IL-23/IL-17 axis, and CCR10+ TRM cells in SLE skin that act as reservoirs for type I interferon production. In addition, recent studies have shown that CD4+ TRM cells, particularly TH1 and TH17 subsets, also contribute significantly to RA pathology by driving synovial inflammation, enhancing cytokine secretion (e.g., IFN-γ, IL-17), and promoting tissue damage through immune interactions with fibroblasts and endothelial cells. Their persistence in the joint microenvironment is regulated by factors such as TGF-β, IL-15, and metabolic pathways like fatty acid oxidation, similar to their CD8+ counterparts [151]. In pSS, cytotoxic CD8+ TRM cells directly mediate glandular destruction, while in SSc, TRM cells are implicated in promoting fibrosis, potentially through TGF-β secretion. This detailed understanding underscores that TRM cells are not merely bystanders but active architects of tissue damage across different autoimmune contexts. Targeting TRM cells presents a paradigm shift from broadly immunosuppressive strategies towards precision medicine. Near-term opportunities lie in drug repurposing: JAK inhibitors disrupt critical IL-15 survival signals, and anti-IL-17/IL-23 antibodies break pathogenic cytokine feedback loops. However, the future lies in novel mechanisms designed to undermine their residency. Strategies in development include disrupting tissue retention via S1PR1 agonists (e.g., fingolimod) or integrin inhibitors, and targeting their core metabolism through CPT1a inhibition. The most innovative approaches involve microenvironmental remodeling, such as smart nanoparticles for targeted siRNA delivery and synthetic biology to engineer reparative stromal cells, aiming to actively reset the diseased tissue niche rather than just suppress immunity.
While TRM-targeting strategies hold promise, challenges persist. The heterogeneity of TRM cells across tissues makes it difficult to develop targeted therapies. A key concern is the potential impairment of protective immunity, as non-selective depletion of TRM cells could compromise the defense against pathogens and weaken vaccine responses. Thus, the central challenge lies in selectively silencing pathogenic functions of TRM cells while preserving their protective roles. Future progress will depend on the integration of cutting-edge technologies, including spatial transcriptomics to map TRM cell interactions in lesions, patient-derived organoids for more relevant drug testing, and artificial intelligence to identify novel epigenetic and biomarker targets. The evaluation of TRM cell-associated biomarkers, such as CD39, will be crucial for patient stratification and monitoring, facilitating personalized treatment strategies.
In perspective, TRM cells represent a critical frontier in the quest for lasting disease control in rheumatic autoimmunity. While hurdles of specificity and safety exist, the converging advances in immunology and bioengineering are paving the way for a new generation of therapies. Targeting TRM cells moves beyond transient immunosuppression, offering a transformative strategy to fundamentally reset local tissue immunity and bring the goal of long-term remission and potential cures within closer reach.
Author Contributions
Conceptualization, D.X. and G.L.; writing—original draft, Y.T. and J.Z.; visualization, L.W.; investigation, F.Z. and C.Z.; writing—review and editing, D.X., Y.Y. and G.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Health Commission Capacity Building and Continuing Education Center (GWJJMB202510025060), National Natural Science Foundation of China (32500813), Postdoctoral Fellowship Program of CPSF (GZC20251503), Sichuan Science and Technology Program (2024ZYD0149), Special Research Foundation for the Postdoctoral Program of Sichuan Province (TB2024017), Key Research and Development Project of Deyang Science and Technology Bureau (2024SZY016, 2023SZZ009), and Special Fund for Incubation Projects of Deyang People’s Hospital (FRH202501, FHT202501).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study.
Acknowledgments
We would like to acknowledge BioRender (https://www.biorender.com/) for providing the platform and tools for creating the illustrations.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| TRM cells | Tissue-resident memory T cells |
| RA | Rheumatoid arthritis |
| SLE | Systemic lupus erythematosus |
| SSc | Systemic sclerosis |
| pSS | Primary Sjögren’s syndrome |
| TGF-β | Transforming growth factor-β |
| CCL5 | chemokine (C-C motif) ligand 5 |
| TCR | T cell receptor |
| IL-15 | Interleukin-15 |
| GM-CSF | Granulocyte-macrophage colony stimulating factor |
| FAO | Fatty acid oxidation |
| CPT1a | Carnitine Palmitoyltransferase 1A |
| IL-7 | Interleukin-7 |
| ECM | Extracellular matrix |
| HIF-1α | Hypoxia-inducible factor 1α |
| PD-1 | Programmed death receptor 1 |
| SSc-ILD | SSc-associated interstitial lung disease |
| RA-ILD | RA-associated interstitial lung disease |
| APCs | Antigen-presenting cells |
| LT-α | Lymphotoxin-α |
| LDRT | Low-dose radiotherapy |
| S1P | Sphingosine-1-phosphate |
| FLS | Fibroblast-like synoviocytes |
| AI | Artificial intelligence |
| TPL | Triptolide |
| TLNPs | Targeted lipid nanoparticles |
| Treg | Regulatory T cells |
| Breg | Regulatory B cells |
References
- Firestein, G.S.; McInnes, I.B. Immunopathogenesis of Rheumatoid Arthritis. Immunity 2017, 46, 183–196. [Google Scholar] [CrossRef]
- Kishimoto, T.; Kang, S. IL-6 Revisited: From Rheumatoid Arthritis to CAR T Cell Therapy and COVID-19. Annu. Rev. Immunol. 2022, 40, 323–348. [Google Scholar] [CrossRef]
- Harigai, M.; Honda, S. Selectivity of Janus Kinase Inhibitors in Rheumatoid Arthritis and Other Immune-Mediated Inflamma tory Diseases: Is Expectation the Root of All Headache? Drugs 2020, 80, 1183–1201. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Sun, Y.; Xu, W.; Chang, F.; Wang, Y.; Ding, J. Mesenchymal Stem Cells-Involved Strategies for Rheumatoid Arthritis Therapy. Adv. Sci. 2024, 11, e2305116. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Mackensen, A.; Mougiakakos, D. CAR T-cell therapy in autoimmune diseases. Lancet 2023, 402, 2034–2044. [Google Scholar] [CrossRef]
- Chen, J.; Tan, J.; Wang, N.; Li, H.; Cheng, W.; Li, J.; Wang, B.; Sedgwick, A.C.; Chen, Z.; Chen, G.; et al. Specific macrophage RhoA targeting CRISPR-Cas9 for mitigating osteoclastogenesis-induced joint damage in inflammatory arthritis. Cell Rep. Med. 2025, 6, 102046. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Crickx, E.; Weill, J.-C.; Reynaud, C.-A.; Mahévas, M. Anti-CD20-mediated B-cell depletion in autoimmune diseases: Successes, failures and future perspectives. Kidney Int. 2020, 97, 885–893. [Google Scholar] [CrossRef]
- Mueller, S.N.; Mackay, L.K. Tissue-resident memory T cells: Local specialists in immune defence. Nat. Rev. Immunol. 2016, 16, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Park, C.O.; Kupper, T.S. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat. Med. 2015, 21, 688–697. [Google Scholar] [CrossRef]
- Masopust, D.; Soerens, A.G. Tissue-Resident T Cells and Other Resident Leukocytes. Annu. Rev. Immunol. 2019, 37, 521–546. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, D.E.; Mohamed, A.; Huang, Y.H.; Turk, M.J. In the right place at the right time: Tissue-resident memory T cells in immunity to cancer. Curr. Opin. Immunol. 2023, 83, 102338. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Lu, G.; Mai, G.; Guo, Q.; Xu, G. Tissue-resident memory T cells in diseases and therapeutic strategies. MedComm 2025, 6, e70053. [Google Scholar] [CrossRef]
- Amsen, D.; van Gisbergen, K.P.J.M.; Hombrink, P.; van Lier, R.A.W. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat. Immunol. 2018, 19, 538–546. [Google Scholar] [CrossRef]
- Chen, L.; Shen, Z. Tissue-resident memory T cells and their biological characteristics in the recurrence of inflammatory skin disorders. Cell. Mol. Immunol. 2020, 17, 64–75. [Google Scholar] [CrossRef]
- Kumar, B.V.; Ma, W.; Miron, M.; Granot, T.; Guyer, R.S.; Carpenter, D.J.; Senda, T.; Sun, X.; Ho, S.-H.; Lerner, H.; et al. Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep. 2017, 20, 2921–2934. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhou, R.; Chen, Z. Tissue-Resident Memory T Cell: Ontogenetic Cellular Mechanism and Clinical Translation. Clin. Exp. Immunol. 2023, 214, 249–259. [Google Scholar] [CrossRef]
- Steel, K.J.A.; Srenathan, U.; Ridley, M.; Durham, L.E.; Wu, S.-Y.; Ryan, S.E.; Hughes, C.D.; Chan, E.; Kirkham, B.W.; Taams, L.S. Polyfunctional, Proinflammatory, Tissue-Resident Memory Phenotype and Function of Synovial Interleukin-17A+CD8+ T Cells in Psoriatic Arthritis. Arthritis Rheumatol. 2020, 72, 435–447. [Google Scholar] [CrossRef]
- Pallett, L.J.; Davies, J.; Colbeck, E.J.; Robertson, F.; Hansi, N.; Easom, N.J.W.; Burton, A.R.; Stegmann, K.A.; Schurich, A.; Swadling, L.; et al. IL-2high tissue-resident T cells in the human liver: Sentinels for hepatotropic infection. J. Exp. Med. 2017, 214, 1567–1580. [Google Scholar] [CrossRef]
- Chang, M.H.; Levescot, A.; Nelson-Maney, N.; Blaustein, R.B.; Winden, K.D.; Morris, A.; Wactor, A.; Balu, S.; Grieshaber-Bouyer, R.; Wei, K.; et al. Arthritis flares mediated by tissue-resident memory T cells in the joint. Cell Rep. 2021, 37, 109902. [Google Scholar] [CrossRef]
- Li, J.; Xiao, C.; Li, C.; He, J. Tissue-resident immune cells: From defining characteristics to roles in diseases. Signal Transduct. Target. Ther. 2025, 10, 12. [Google Scholar] [CrossRef]
- Milner, J.J.; Goldrath, A.W. Transcriptional programming of tissue-resident memory CD8+ T cells. Curr. Opin. Immunol. 2018, 51, 162–169. [Google Scholar] [CrossRef]
- Mackay, L.K.; Minnich, M.; Kragten, N.A.M.; Liao, Y.; Nota, B.; Seillet, C.; Zaid, A.; Man, K.; Preston, S.; Freestone, D.; et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 2016, 352, 459–463. [Google Scholar] [CrossRef]
- Varanasi, S.K.; Kumar, S.V.; Rouse, B.T. Determinants of Tissue-Specific Metabolic Adaptation of T Cells. Cell Metab. 2020, 32, 908–919. [Google Scholar] [CrossRef]
- Hasan, F.; Chiu, Y.; Shaw, R.M.; Wang, J.; Yee, C. Hypoxia acts as an environmental cue for the human tissue-resident memory T cell differentiation program. JCI Insight 2021, 6, e138970. [Google Scholar] [CrossRef]
- Corgnac, S.; Boutet, M.; Kfoury, M.; Naltet, C.; Mami-Chouaib, F. The Emerging Role of CD8+ Tissue Resident Memory T (TRM) Cells in Antitumor Immunity: A Unique Functional Contribution of the CD103 Integrin. Front. Immunol. 2018, 9, 1904. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Bevan, M.J. Transforming growth factor-β signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity 2013, 39, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Chu, T.H.; Sheridan, B.S. TGF-β: Many Paths to CD103+ CD8 T Cell Residency. Cells 2021, 10, 989. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Deng, J.; Liu, X.; Zhou, Y.; Bi, T.; Chen, J.; Wang, J. Tissue-resident immune cells in cervical cancer: Emerging roles and therapeutic implications. Front. Immunol. 2025, 16, 1541950. [Google Scholar] [CrossRef]
- Jarjour, N.N.; Wanhainen, K.M.; Peng, C.; Gavil, N.V.; Maurice, N.J.; Borges da Silva, H.; Martinez, R.J.; Dalzell, T.S.; Huggins, M.A.; Masopust, D.; et al. Responsiveness to interleukin-15 therapy is shared between tissue-resident and circulating memory CD8+ T cell subsets. Proc. Natl. Acad. Sci. USA 2022, 119, e2209021119. [Google Scholar] [CrossRef]
- Xu, A.; Bhanumathy, K.K.; Wu, J.; Ye, Z.; Freywald, A.; Leary, S.C.; Li, R.; Xiang, J. IL-15 signaling promotes adoptive effector T-cell survival and memory formation in irradiation-induced lymphopenia. Cell Biosci. 2016, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Kim, Y.; Jung, Y.W. The Function of Memory CD8+ T Cells in Immunotherapy for Human Diseases. Immune Netw. 2023, 23, e10. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Tang, T.-X.; Deng, H.; Yang, X.-P.; Tang, Z.-H. Interleukin-7 Biology and Its Effects on Immune Cells: Mediator of Generation, Differentiation, Survival, and Homeostasis. Front. Immunol. 2021, 12, 747324. [Google Scholar] [CrossRef]
- Xu, W.; Bergsbaken, T.; Edelblum, K.L. The multifunctional nature of CD103 (αEβ7 integrin) signaling in tissue-resident lym phocytes. Am. J. Physiol. Cell Physiol. 2022, 323, C1161–C1167. [Google Scholar] [CrossRef]
- Topham, D.J.; Reilly, E.C. Tissue-Resident Memory CD8+ T Cells: From Phenotype to Function. Front. Immunol. 2018, 9, 515. [Google Scholar] [CrossRef]
- Salmond, R.J. Regulation of T Cell Activation and Metabolism by Transforming Growth Factor-Beta. Biology 2023, 12, 297. [Google Scholar] [CrossRef]
- van der Gracht, E.T.I.; Behr, F.M.; Arens, R. Functional Heterogeneity and Therapeutic Targeting of Tissue-Resident Memory T Cells. Cells 2021, 10, 164. [Google Scholar] [CrossRef]
- Pan, Y.; Tian, T.; Park, C.O.; Lofftus, S.Y.; Mei, S.; Liu, X.; Luo, C.; O’Malley, J.T.; Gehad, A.; Teague, J.E.; et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 2017, 543, 252–256. [Google Scholar] [CrossRef]
- Han, S.-J.; Glatman Zaretsky, A.; Andrade-Oliveira, V.; Collins, N.; Dzutsev, A.; Shaik, J.; Morais da Fonseca, D.; Harrison, O.J.; Tamoutounour, S.; Byrd, A.L.; et al. White Adipose Tissue Is a Reservoir for Memory T Cells and Promotes Protective Memory Responses to Infection. Immunity 2017, 47, 1154–1168.e6. [Google Scholar] [CrossRef]
- Liikanen, I.; Lauhan, C.; Quon, S.; Omilusik, K.; Phan, A.T.; Bartrolí, L.B.; Ferry, A.; Goulding, J.; Chen, J.; Scott-Browne, J.P.; et al. Hypoxia-inducible factor activity promotes antitumor effector function and tissue residency by CD8+ T cells. J. Clin. Investig. 2021, 131, e143729. [Google Scholar] [CrossRef] [PubMed]
- Yfantis, A.; Mylonis, I.; Chachami, G.; Nikolaidis, M.; Amoutzias, G.D.; Paraskeva, E.; Simos, G. Transcriptional Response to Hypoxia: The Role of HIF-1-Associated Co-Regulators. Cells 2023, 12, 798. [Google Scholar] [CrossRef]
- Shen, H.; Ojo, O.A.; Ding, H.; Mullen, L.J.; Xing, C.; Hossain, M.I.; Yassin, A.; Shi, V.Y.; Lewis, Z.; Podgorska, E.; et al. HIF1α-regulated glycolysis promotes activation-induced cell death and IFN-γ induction in hypoxic T cells. Nat. Commun. 2024, 15, 9394. [Google Scholar] [CrossRef]
- Huang, X.; Zhao, L.; Peng, R. Hypoxia-Inducible Factor 1 and Mitochondria: An Intimate Connection. Biomolecules 2022, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Hanumegowda, C.; Mohan, M.E. Hypoxia Inducible Factor-1α in Pulmonary Fibrosis: A Promising Therapeutic Target. Euro Pean J. Med. Health Sci. 2025, 7, 27–31. [Google Scholar] [CrossRef]
- Chen, Z.; Dudek, J.; Maack, C.; Hofmann, U. Pharmacological inhibition of GLUT1 as a new immunotherapeutic approach after myocardial infarction. Biochem. Pharmacol. 2021, 190, 114597. [Google Scholar] [CrossRef] [PubMed]
- Osman, M.; Park, S.L.; Mackay, L.K. Tissue-resident memory T (TRM) cells: Front-line workers of the immune system. Eur. J. Immunol. 2023, 53, e2250060. [Google Scholar] [CrossRef]
- Liu, H.; Chen, Y.-G. The Interplay Between TGF-β Signaling and Cell Metabolism. Front. Cell Dev. Biol. 2022, 10, 846723. [Google Scholar] [CrossRef] [PubMed]
- Dijkgraaf, F.E.; Kok, L.; Schumacher, T.N.M. Formation of Tissue-Resident CD8+ T-Cell Memory. Cold Spring Harb. Perspect. Biol. 2021, 13, a038117. [Google Scholar] [CrossRef]
- Harjunpää, H.; Somermäki, R.; Saldo Rubio, G.; Fusciello, M.; Feola, S.; Faisal, I.; Nieminen, A.I.; Wang, L.; Llort Asens, M.; Zhao, H.; et al. Loss of β2-integrin function results in metabolic reprogramming of dendritic cells, leading to increased dendritic cell functionality and anti-tumor responses. Oncoimmunology 2024, 13, 2369373. [Google Scholar] [CrossRef]
- Splitt, R.L.; DeMali, K.A. Metabolic reprogramming in response to cell mechanics. Biol. Cell 2023, 115, e202200108. [Google Scholar] [CrossRef]
- Barros, L.; Ferreira, C.; Veldhoen, M. The fellowship of regulatory and tissue-resident memory cells. Mucosal Immunol. 2022, 15, 64–73. [Google Scholar] [CrossRef]
- Christo, S.N.; Park, S.L.; Mueller, S.N.; Mackay, L.K. The Multifaceted Role of Tissue-Resident Memory T Cells. Annu. Rev. Immunol. 2024, 42, 317–345. [Google Scholar] [CrossRef]
- Zheng, M.Z.M.; Wakim, L.M. Tissue resident memory T cells in the respiratory tract. Mucosal Immunol. 2022, 15, 379–388. [Google Scholar] [CrossRef]
- van Gisbergen, K.P.J.M.; Zens, K.D.; Münz, C. T-cell memory in tissues. Eur. J. Immunol. 2021, 51, 1310–1324. [Google Scholar] [CrossRef]
- Samat, A.A.K.; van der Geest, J.; Vastert, S.J.; van Loosdregt, J.; van Wijk, F. Tissue-Resident Memory T Cells in Chronic In flammation-Local Cells with Systemic Effects? Cells 2021, 10, 409. [Google Scholar] [CrossRef] [PubMed]
- Low, J.S.; Farsakoglu, Y.; Amezcua Vesely, M.C.; Sefik, E.; Kelly, J.B.; Harman, C.C.D.; Jackson, R.; Shyer, J.A.; Jiang, X.; Cauley, L.S.; et al. Tissue-resident memory T cell reactivation by diverse antigen-presenting cells imparts distinct functional responses. J. Exp. Med. 2020, 217, e20192291. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.-H.; Lee, S.; Kwak, M.; Kim, B.-S.; Chung, Y. CD8 T-cell subsets: Heterogeneity, functions, and therapeutic potential. Exp. Mol. Med. 2023, 55, 2287–2299. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Wang, Z.; Li, S. Corrigendum: Heterogeneity and plasticity of tissue-resident memory T cells in skin diseases and homeostasis: A review. Front. Immunol. 2024, 15, 1460250. [Google Scholar] [CrossRef]
- Heeg, M.; Goldrath, A.W. Insights into phenotypic and functional CD8+ TRM heterogeneity. Immunol. Rev. 2023, 316, 8–22. [Google Scholar] [CrossRef]
- Nelson, C.E.; Foreman, T.W.; Fukutani, E.R.; Kauffman, K.D.; Sakai, S.; Fleegle, J.D.; Gomez, F.; Gould, S.T.; Le Nouën, C.; Liu, X.; et al. IL-10 suppresses T cell expansion while promoting tissue-resident memory cell formation during SARS-CoV-2 infection in rhesus macaques. PLoS Pathog. 2024, 20, e1012339. [Google Scholar] [CrossRef]
- Yenyuwadee, S.; Sanchez-Trincado Lopez, J.L.; Shah, R.; Rosato, P.C.; Boussiotis, V.A. The evolving role of tissue-resident memory T cells in infections and cancer. Sci. Adv. 2022, 8, eabo5871. [Google Scholar] [CrossRef]
- Musial, S.C.; Kleist, S.A.; Degefu, H.N.; Ford, M.A.; Chen, T.; Isaacs, J.F.; Boussiotis, V.A.; Skorput, A.G.J.; Rosato, P.C. Alarm Functions of PD-1+ Brain-Resident Memory T Cells. J. Immunol. 2024, 213, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Rodger, B.; Stagg, A.J.; Lindsay, J.O. The role of circulating T cells with a tissue resident phenotype (ex-TRM) in health and disease. Front. Immunol. 2024, 15, 1415914. [Google Scholar] [CrossRef]
- Pizzolla, A.; Nguyen, T.H.O.; Sant, S.; Jaffar, J.; Loudovaris, T.; Mannering, S.I.; Thomas, P.G.; Westall, G.P.; Kedzierska, K.; Wakim, L.M. Influenza-specific lung-resident memory T cells are proliferative and polyfunctional and maintain diverse TCR profiles. J. Clin. Investig. 2018, 128, 721–733. [Google Scholar] [CrossRef]
- Marchesini Tovar, G.; Gallen, C.; Bergsbaken, T. CD8+ Tissue-Resident Memory T Cells: Versatile Guardians of the Tissue. J. Immunol. 2024, 212, 361–368. [Google Scholar] [CrossRef]
- Iijima, N. The emerging role of effector functions exerted by tissue-resident memory T cells. Oxf. Open Immunol. 2024, 5, iqae006. [Google Scholar] [CrossRef]
- Reilly, E.C.; Sportiello, M.; Emo, K.L.; Amitrano, A.M.; Jha, R.; Kumar, A.B.R.; Laniewski, N.G.; Yang, H.; Kim, M.; Topham, D.J. CD49a Identifies Polyfunctional Memory CD8 T Cell Subsets that Persist in the Lungs After Influenza Infection. Front. Immunol. 2021, 12, 728669. [Google Scholar] [CrossRef]
- Povoleri, G.A.M.; Durham, L.E.; Gray, E.H.; Lalnunhlimi, S.; Kannambath, S.; Pitcher, M.J.; Dhami, P.; Leeuw, T.; Ryan, S.E.; Steel, K.J.A.; et al. Psoriatic and rheumatoid arthritis joints differ in the composition of CD8+ tissue-resident memory T cell subsets. Cell Rep. 2023, 42, 112514. [Google Scholar] [CrossRef] [PubMed]
- Sul, C.; Nozik, E.; Malainou, C. A Tale of Two Cytokines: IL-10 Blocks IFN-γ in Influenza A Virus-Staphylococcus aureus Coinfection. Am. J. Respir. Cell Mol. Biol. 2024, 71, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, S.; Goplen, N.P.; Li, C.; Cheon, I.S.; Dai, Q.; Huang, S.; Shan, J.; Ma, C.; Ye, Z.; et al. PD-1hi CD8+ resident memory T cells balance immunity and fibrotic sequelae. Sci. Immunol. 2019, 4, eaaw1217. [Google Scholar] [CrossRef]
- Weisberg, S.P.; Carpenter, D.J.; Chait, M.; Dogra, P.; Gartrell-Corrado, R.D.; Chen, A.X.; Campbell, S.; Liu, W.; Saraf, P.; Snyder, M.E.; et al. Tissue-Resident Memory T Cells Mediate Immune Homeostasis in the Human Pancreas through the PD-1/PD-L1 Pathway. Cell Rep. 2019, 29, 3916–3932.e5. [Google Scholar] [CrossRef]
- Overacre-Delgoffe, A.E.; Hand, T.W. Regulation of tissue-resident memory T cells by the Microbiota. Mucosal Immunol. 2022, 15, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.; Wadsworth, M.H.; Lendvai, A.; Christensen, S.M.; Hensvold, A.H.; Gerstner, C.; van Vollenhoven, A.; Kravarik, K.; Winkler, A.; Malmström, V.; et al. Single cell sequencing identifies clonally expanded synovial CD4+ TPH cells expressing GPR56 in rheumatoid arthritis. Nat. Commun. 2022, 13, 4046. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wei, K.; Slowikowski, K.; Fonseka, C.Y.; Rao, D.A.; Kelly, S.; Goodman, S.M.; Tabechian, D.; Hughes, L.B.; Salomon-Escoto, K.; et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat. Immunol. 2019, 20, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A. Immunoprofiling the Inflamed Tissue in Rheumatic Diseases Using Single-Cell Technologies. Ph.D. Thesis, Karolinska Institutet, Stockholm, Sweden, 2025. [Google Scholar]
- Strobl, J.; Haniffa, M. Functional heterogeneity of human skin-resident memory T cells in health and disease. Immunol. Rev. 2023, 316, 104–119. [Google Scholar] [CrossRef]
- Dunlap, G.S.; Billi, A.C.; Xing, X.; Ma, F.; Maz, M.P.; Tsoi, L.C.; Wasikowski, R.; Hodgin, J.B.; Gudjonsson, J.E.; Kahlenberg, J.M.; et al. Single-cell transcriptomics reveals distinct effector profiles of infiltrating T cells in lupus skin and kidney. JCI Insight 2022, 7, e156341. [Google Scholar] [CrossRef]
- van den Hoogen, F.; Khanna, D.; Fransen, J.; Johnson, S.R.; Baron, M.; Tyndall, A.; Matucci-Cerinic, M.; Naden, R.P.; Medsger, T.A.; Carreira, P.E.; et al. 2013 classification criteria for systemic sclerosis: An American college of rheumatology/European league against rheumatism collaborative initiative. Ann. Rheum. Dis. 2013, 72, 1747–1755. [Google Scholar] [CrossRef]
- Gaydosik, A.M.; Tabib, T.; Domsic, R.; Khanna, D.; Lafyatis, R.; Fuschiotti, P. Single-cell transcriptome analysis identifies skin-specific T-cell responses in systemic sclerosis. Ann. Rheum. Dis. 2021, 80, 1453–1460. [Google Scholar] [CrossRef]
- Shimagami, H.; Nishimura, K.; Matsushita, H.; Metsugi, S.; Kato, Y.; Kawasaki, T.; Tsujimoto, K.; Edahiro, R.; Itotagawa, E.; Naito, M.; et al. Single-cell analysis reveals immune cell abnormalities underlying the clinical heterogeneity of patients with systemic sclerosis. Nat. Commun. 2025, 16, 4949. [Google Scholar] [CrossRef]
- Padilla, C.M.; Valenzi, E.; Tabib, T.; Nazari, B.; Sembrat, J.; Rojas, M.; Fuschiotti, P.; Lafyatis, R. Increased CD8+ tissue resident memory T cells, regulatory T cells and activated natural killer cells in systemic sclerosis lungs. Rheumatology 2024, 63, 837–845. [Google Scholar] [CrossRef]
- Xu, T.; Zhu, H.-X.; You, X.; Ma, J.-F.; Li, X.; Luo, P.-Y.; Li, Y.; Lian, Z.-X.; Gao, C.-Y. Single-cell profiling reveals pathogenic role and differentiation trajectory of granzyme K+CD8+ T cells in primary Sjögren’s syndrome. JCI Insight 2023, 8, e167490. [Google Scholar] [CrossRef] [PubMed]
- Mauro, D.; Lin, X.; Pontarini, E.; Wehr, P.; Guggino, G.; Tang, Y.; Deng, C.; Gandolfo, S.; Xiao, F.; Rui, K.; et al. CD8+ tissue-resident memory T cells are expanded in primary Sjögren’s disease and can be therapeutically targeted by CD103 blockade. Ann. Rheum. Dis. 2024, 83, 1345–1357. [Google Scholar] [CrossRef]
- Wu, H.; Liao, W.; Li, Q.; Long, H.; Yin, H.; Zhao, M.; Chan, V.; Lau, C.S.; Lu, Q. Pathogenic role of tissue-resident memory T cells in autoimmune diseases. Autoimmun. Rev. 2018, 17, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.; Zhao, W.; Wu, R.; Su, R.; Jin, R.; Luo, J.; Gao, C.; Li, X.; Wang, C. Tissue-resident memory T cells: The key frontier in local synovitis memory of rheumatoid arthritis. J. Autoimmun. 2022, 133, 102950. [Google Scholar] [CrossRef]
- Ryan, G.E.; Harris, J.E.; Richmond, J.M. Resident Memory T Cells in Autoimmune Skin Diseases. Front. Immunol. 2021, 12, 652191. [Google Scholar] [CrossRef]
- Alivernini, S.; Tolusso, B.; Ferraccioli, G.; Gremese, E.; Kurowska-Stolarska, M.; McInnes, I.B. Driving chronicity in rheumatoid arthritis: Perpetuating role of myeloid cells. Clin. Exp. Immunol. 2018, 193, 13–23. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet 2017, 389, 2328–2337. [Google Scholar] [CrossRef]
- Firestein, G.S. The immunopathogenesis of rheumatoid arthritis. Curr. Opin. Rheumatol. 1991, 3, 398–406. [Google Scholar] [CrossRef]
- Kondo, N.; Kuroda, T.; Kobayashi, D. Cytokine Networks in the Pathogenesis of Rheumatoid Arthritis. Int. J. Mol. Sci. 2021, 22, 10922. [Google Scholar] [CrossRef]
- Najm, A.; McInnes, I.B. IL-23 orchestrating immune cell activation in arthritis. Rheumatology 2021, 60, iv4–iv15. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.-Y.; Kim, J.-Y.; Kim, K.-W.; Park, M.-K.; Moon, Y.; Kim, W.-U.; Kim, H.-Y. IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-kappaB- and PI3-kinase/Akt-dependent pathways. Arthritis Res. Ther. 2004, 6, R120–R128. [Google Scholar] [CrossRef]
- Hirota, K.; Hashimoto, M.; Ito, Y.; Matsuura, M.; Ito, H.; Tanaka, M.; Watanabe, H.; Kondoh, G.; Tanaka, A.; Yasuda, K.; et al. Autoimmune Th17 Cells Induced Synovial Stromal and Innate Lymphoid Cell Secretion of the Cytokine GM-CSF to Initiate and Augment Autoimmune Arthritis. Immunity 2018, 48, 1220–1232.e5. [Google Scholar] [CrossRef]
- Lin, Y.-J.; Anzaghe, M.; Schülke, S. Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Ar thritis. Cells 2020, 9, 880. [Google Scholar] [CrossRef]
- Kadura, S.; Raghu, G. Rheumatoid arthritis-interstitial lung disease: Manifestations and current concepts in pathogenesis and management. Eur. Respir. Rev. 2021, 30, 210011. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, A.; Cassone, G.; Luppi, F.; Atienza-Mateo, B.; Cavazza, A.; Sverzellati, N.; González-Gay, M.A.; Salvarani, C.; Sebastiani, M. Rheumatoid arthritis related interstitial lung disease. Expert Rev. Clin. Immunol. 2021, 17, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.-J.; Song, S.; Roh, J.Y.; Jung, Y.; Kim, H.J. Expression pattern of tissue-resident memory T cells in cutaneous lupus ery thematosus. Lupus 2021, 30, 1427–1437. [Google Scholar] [CrossRef]
- Tian, J.; Shi, L.; Zhang, D.; Yao, X.; Zhao, M.; Kumari, S.; Lu, J.; Yu, D.; Lu, Q. Dysregulation in keratinocytes drives systemic lupus erythematosus onset. Cell. Mol. Immunol. 2025, 22, 83–96. [Google Scholar] [CrossRef]
- Vincent, F.B.; Figgett, W.A.; Hibbs, M.L. Hallmark of Systemic Lupus Erythematosus: Role of B Cell Hyperactivity. In Pathogenesis of Systemic Lupus Erythematosus: Insights from Translational Research; Hoi, A., Ed.; Springer International Publishing: Cham, Switzerland, 2021; pp. 9–36. [Google Scholar]
- Bocharnikov, A.V.; Keegan, J.; Wacleche, V.S.; Cao, Y.; Fonseka, C.Y.; Wang, G.; Muise, E.S.; Zhang, K.X.; Arazi, A.; Keras, G.; et al. PD-1hiCXCR5– T peripheral helper cells promote B cell responses in lupus via MAF and IL-21. JCI Insight 2019, 4, e130062. [Google Scholar] [CrossRef]
- Ramanujam, M.; Steffgen, J.; Visvanathan, S.; Mohan, C.; Fine, J.S.; Putterman, C. Phoenix from the flames: Rediscovering the role of the CD40-CD40L pathway in systemic lupus erythematosus and lupus nephritis. Autoimmun. Rev. 2020, 19, 102668. [Google Scholar] [CrossRef]
- Altorok, N.; Kahaleh, B. Epigenetics and systemic sclerosis. Semin. Immunopathol. 2015, 37, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N. Transforming growth factor-β in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Tang, R.; Ding, K. Epigenetic Modifications in the Pathogenesis of Systemic Sclerosis. Int. J. Gen. Med. 2022, 15, 3155–3166. [Google Scholar] [CrossRef]
- Li, T.; Ortiz-Fernández, L.; Andrés-León, E.; Ciudad, L.; Javierre, B.M.; López-Isac, E.; Guillén-Del-Castillo, A.; Simeón-Aznar, C.P.; Ballestar, E.; Martin, J. Epigenomics and transcriptomics of systemic sclerosis CD4+ T cells reveal long-range dysregulation of key inflammatory pathways mediated by disease-associated susceptibility loci. Genome Med. 2020, 12, 81. [Google Scholar] [CrossRef]
- Dees, C.; Pötter, S.; Zhang, Y.; Bergmann, C.; Zhou, X.; Luber, M.; Wohlfahrt, T.; Karouzakis, E.; Ramming, A.; Gelse, K.; et al. TGF-β-induced epigenetic deregulation of SOCS3 facilitates STAT3 signaling to promote fibrosis. J. Clin. Investig. 2020, 130, 2347–2363. [Google Scholar] [CrossRef]
- Volkmann, E.R.; Andréasson, K.; Smith, V. Systemic sclerosis. Lancet 2023, 401, 304–318. [Google Scholar] [CrossRef]
- Denton, C.P.; Khanna, D. Systemic sclerosis. Lancet 2017, 390, 1685–1699. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, I. Calcium signaling defects underlying salivary gland dysfunction. Biochim. Biophys. Acta (BBA)—Molec Ular Cell Res. 2018, 1865, 1771–1777. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Makarenkova, H.P. Innate Immunity and Biological Therapies for the Treatment of Sjögren’s Syndrome. Inter Natl. J. Mol. Sci. 2020, 21, 9172. [Google Scholar] [CrossRef]
- Shen, L.; Suresh, L.; Wu, J.; Xuan, J.; Li, H.; Zhang, C.; Pankewycz, O.; Ambrus, J.L. A role for lymphotoxin in primary Sjogren’s disease. J. Immunol. 2010, 185, 6355–6363. [Google Scholar] [CrossRef]
- Sharma, R.; Chaudhari, K.S.; Kurien, B.T.; Grundahl, K.; Radfar, L.; Lewis, D.M.; Lessard, C.J.; Li, H.; Rasmussen, A.; Sivils, K.L.; et al. Sjögren Syndrome without Focal Lymphocytic Infiltration of the Salivary Glands. J. Rheumatol. 2020, 47, 394–399. [Google Scholar] [CrossRef]
- Yura, Y.; Hamada, M. Outline of Salivary Gland Pathogenesis of Sjögren’s Syndrome and Current Therapeutic Approaches. Int. J. Mol. Sci. 2023, 24, 11179. [Google Scholar] [CrossRef]
- Blauvelt, A. Resident Memory T Cells in Psoriasis: Key to a Cure? J. Psoriasis Psoriatic Arthritis 2022, 7, 157–159. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Liu, F. Targeting the IL-15/CD122 signaling pathway: Reversing TRM cell-mediated immune memory in vitiligo. Front. Immunol. 2025, 16, 1639732. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, J.; Israelsson, E.; Björhall, K.; Yrlid, L.F.; Thörn, K.; Thorén, A.; Toledo, E.A.; Jinton, L.; Öberg, L.; Wingren, C.; et al. Selective Janus kinase 1 inhibition resolves inflammation and restores hair growth offering a viable treatment option for alopecia areata. Ski. Health Dis. 2023, 3, e209. [Google Scholar] [CrossRef]
- Yari, S.; Kikuta, J.; Shigyo, H.; Miyamoto, Y.; Okuzaki, D.; Furusawa, Y.; Minoshima, M.; Kikuchi, K.; Ishii, M. JAK inhibition ameliorates bone destruction by simultaneously targeting mature osteoclasts and their precursors. Inflamm. Regen. 2023, 43, 18. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li, S.; Ying, S.; Tang, S.; Ding, Y.; Li, Y.; Qiao, J.; Fang, H. The IL-23/IL-17 Pathway in Inflammatory Skin Diseases: From Bench to Bedside. Front. Immunol. 2020, 11, 594735. [Google Scholar] [CrossRef]
- Dong, C.; Lin, L.; Du, J. Characteristics and sources of tissue-resident memory T cells in psoriasis relapse. Curr. Res. Immunol. 2023, 4, 100067. [Google Scholar] [CrossRef]
- Huang, Y.-W.; Tsai, T.-F. A drug safety evaluation of risankizumab for psoriasis. Expert Opin. Drug Saf. 2020, 19, 395–402. [Google Scholar] [CrossRef]
- Çelik, M.S.; Aktaş, H. The effect of IL-17 and IL-23 ınhibitors on hematological ınflammatory parameters in patients with psoriasis vulgaris. Ir. J. Med. Sci. 2025, 194, 1329–1334. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.B.; Hernandez, R.; Carlson, P.; Grudzinski, J.; Bates, A.M.; Jagodinsky, J.C.; Erbe, A.; Marsh, I.R.; Arthur, I.; Aluicio-Sarduy, E.; et al. Low-dose targeted radionuclide therapy renders immunologically cold tumors responsive to immune checkpoint blockade. Sci. Transl. Med. 2021, 13, eabb3631. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-G.; Choi, H.S.; Choe, Y.-H.; Jeon, H.M.; Heo, J.Y.; Cheon, Y.-H.; Kang, K.M.; Lee, S.-I.; Jeong, B.K.; Kim, M. Low-Dose Radiotherapy Attenuates Experimental Autoimmune Arthritis by Inducing Apoptosis of Lymphocytes and Fibroblast-Like Synoviocytes. Immune Netw. 2024, 24, e32. [Google Scholar] [CrossRef] [PubMed]
- Játiva, S.; Torrico, S.; Calle, P.; Poch, E.; Muñoz, A.; García, M.; Larque, A.B.; Salido, M.T.T.; Hotter, G. The phagocytosis dys function in lupus nephritis is related to monocyte/macrophage CPT1a. Immunol. Lett. 2024, 266, 106841. [Google Scholar] [CrossRef] [PubMed]
- Son, H.-J.; Lee, J.; Lee, S.-Y.; Kim, E.-K.; Park, M.-J.; Kim, K.-W.; Park, S.-H.; Cho, M.-L. Metformin attenuates experimental autoimmune arthritis through reciprocal regulation of Th17/Treg balance and osteoclastogenesis. Mediat. Inflamm. 2014, 2014, 973986. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Moret, Y.A.; Lo, K.B.; Tan, I.J. Metformin in Systemic Lupus Erythematosus: Investigating Cardiovascular Impact and Nephroprotective Effects in Lupus Nephritis. ACR Open Rheumatol. 2024, 6, 497–503. [Google Scholar] [CrossRef]
- Sha, Y.; Yang, L.; Jiang, J.; Cao, J.; Sun, M.; Yin, L.; Zhong, Z.; Meng, F. Pre-silencing of TNF-α by targeted siRNA delivery mitigates glucocorticoid resistance of dexamethasone in rheumatoid arthritis. J. Control. Release 2025, 386, 114122. [Google Scholar] [CrossRef]
- Hu, C.; Ma, J.; Chen, X.; Chen, Y.; Song, Y.; Tang, Q.; He, X.; Wang, Y.; Gao, H.; Zhang, J. Microenvironment-driven transform able self-assembly nanoplatform enables spatiotemporal remodeling for rheumatoid arthritis therapy. Sci. Adv. 2025, 11, eadu5245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Y.; Liu, Y. Fibroblast activation and heterogeneity in fibrotic disease. Nat. Rev. Nephrol. 2025, 21, 613–632. [Google Scholar] [CrossRef]
- Diao, W.; Jiao, Y.; Deng, T.; Gu, J.; Wang, B.; Zhang, Q.; Wang, P.; Xu, N.; Xiao, C. Synovium-On-A-Chip: Simulating the Microenvironment of the Rheumatoid Arthritis Synovium via Multicell Interactions to Target Fibroblast-Like Synoviocytes. Adv. Sci. 2025, e11945. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.M.; Kim, W.-U. Targeted Immunotherapy for Autoimmune Disease. Immune Netw. 2022, 22, e9. [Google Scholar] [CrossRef]
- Chen, S.; Zheng, Y.; Ran, X.; Du, H.; Feng, H.; Yang, L.; Wen, Y.; Lin, C.; Wang, S.; Huang, M.; et al. Integrin αEβ7+ T cells direct intestinal stem cell fate decisions via adhesion signaling. Cell Res. 2021, 31, 1291–1307. [Google Scholar] [CrossRef] [PubMed]
- Leask, A.; Fadl, A.; Naik, A. A modest proposal: Targeting αv integrin-mediated activation of latent TGFbeta as a novel thera peutic approach to treat scleroderma fibrosis. Expert Opin. Investig. Drugs 2024, 33, 279–285. [Google Scholar] [CrossRef]
- Lin, R.; Zhang, H.; Yuan, Y.; He, Q.; Zhou, J.; Li, S.; Sun, Y.; Li, D.Y.; Qiu, H.-B.; Wang, W.; et al. Fatty Acid Oxidation Controls CD8+ Tissue-Resident Memory T-cell Survival in Gastric Adenocarcinoma. Cancer Immunol. Res. 2020, 8, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Shah, R.B.; Singhal, S.; Dutta, S.B.; Bansal, S.; Sinha, S.; Haque, M. Metformin: A Review of Potential Mechanism and Therapeutic Utility Beyond Diabetes. Drug Des. Devel. Ther. 2023, 17, 1907–1932. [Google Scholar] [CrossRef]
- Wang, X.; Liang, Q.; Luo, Y.; Ye, J.; Yu, Y.; Chen, F. Engineering the next generation of theranostic biomaterials with synthetic biology. Bioact. Mater. 2023, 32, 514–529. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.H.; Fuhlbrigge, R.C.; Nigrovic, P.A. Joint-specific memory, resident memory T cells and the rolling window of op portunity in arthritis. Nat. Rev. Rheumatol. 2024, 20, 258–271. [Google Scholar] [CrossRef]
- Abou-Daya, K.I.; Tieu, R.; Zhao, D.; Rammal, R.; Sacirbegovic, F.; Williams, A.L.; Shlomchik, W.D.; Oberbarnscheidt, M.H.; Lakkis, F.G. Resident memory T cells form during persistent antigen exposure leading to allograft rejection. Sci. Immunol. 2021, 6, eabc8122. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zheng, A.; Liu, N.; Tan, Z.; Shi, Y.; Ma, T.; Luo, S.; Zhu, L.; Zhou, Z.; Yuan, F.; et al. Integrative spatial multiomics analysis reveals regulatory mechanisms of VCAM1+ proximal tubule cells in lupus nephritis. Ann. Rheum. Dis. 2025, in press. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Gu, M.; Li, X.; Hu, X.; Chen, C.; Kang, Y.; Pan, B.; Chen, W.; Xian, G.; Wu, X.; et al. ITGA5+ synovial fibroblasts orchestrate proinflammatory niche formation by remodelling the local immune microenvironment in rheumatoid arthritis. Ann. Rheum. Dis. 2025, 84, 232–252. [Google Scholar] [CrossRef] [PubMed]
- Recaldin, T.; Steinacher, L.; Gjeta, B.; Harter, M.F.; Adam, L.; Kromer, K.; Mendes, M.P.; Bellavista, M.; Nikolaev, M.; Lazzaroni, G.; et al. Human organoids with an autologous tissue-resident immune compartment. Nature 2024, 633, 165–173. [Google Scholar] [CrossRef]
- Wang, X.; He, J.; Zhang, Q.; He, J.; Wang, Q. Constructing a 3D co-culture in vitro synovial tissue model for rheumatoid arthritis research. Mater. Today Bio 2025, 31, 101492. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Hahn, M.; Wauford, B.; Blaustein, R.; Wei, K.; Nigrovic, P. Generation of Human Resident Memory T Cells in 3D Synovial Organoid Model. Arthritis Rheumatol. 2023, 75 (Suppl. S4), 42–43. [Google Scholar]
- Guo, S.; Xu, L.; Chang, C.; Zhang, R.; Jin, Y.; He, D. Epigenetic Regulation Mediated by Methylation in the Pathogenesis and Precision Medicine of Rheumatoid Arthritis. Front. Genet. 2020, 11, 811. [Google Scholar] [CrossRef] [PubMed]
- Ballestar, E.; Sawalha, A.H.; Lu, Q. Clinical value of DNA methylation markers in autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 2020, 16, 514–524. [Google Scholar] [CrossRef]
- Murakami, M. Tissue-resident memory T cells: Decoding intra-organ diversity with a gut perspective. Inflamm. Re Gener. 2024, 44, 19. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.-F.; Zhao, N.; Hu, C.-H. Harnessing mesenchymal stem/stromal cells-based therapies for rheumatoid arthritis: Mecha nisms, clinical applications, and microenvironmental interactions. Stem Cell Res. Ther. 2025, 16, 379. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T.L.; Bao, Y.; Zhang, Y.; Matsuda, D.; Riener, R.; Wang, A.; Li, J.J.; Soldevila, F.; Chu, D.S.H.; Nguyen, D.P.; et al. In vivo CAR T cell generation to treat cancer and autoimmune disease. Science 2025, 388, 1311–1317. [Google Scholar] [CrossRef]
- Hu, H.; Li, H.; Li, R.; Liu, P.; Liu, H. Re-establishing immune tolerance in multiple sclerosis: Focusing on novel mechanisms of mesenchymal stem cell regulation of Th17/Treg balance. J. Transl. Med. 2024, 22, 663. [Google Scholar] [CrossRef]
- Zhong, X.; Deng, X.; Yang, Y.; Xie, X.; Li, B.; Peng, X. Immuno-engineered macrophage membrane-coated nanodrug to restore immune balance for rheumatoid arthritis treatment. Acta Biomater. 2025, 197, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Su, D.; Zhang, L.; Liu, T.; Wang, Q.; Yan, C.; Liu, M.; Ji, H.; Lei, J.; Zheng, M.; et al. Mitochondrial Control of Proteasomal Psmb5 Drives the Differentiation of Tissue-Resident Memory T Cells in Patients with Rheumatoid Arthritis. Arthritis Rheumatol. 2024, 76, 1743–1757. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).