
Differential Gene Expression Analysis in a Lumbar Spinal Stenosis Rat Model via RNA Sequencing: Identification of Key Molecular Pathways and Therapeutic Insights

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Surgical Procedure for LSS Induction
2.2. RNA Extraction and Quality Assessment
2.3. mRNA Library Preparation and Sequencing
2.4. Sequencing Data Processing and Normalization
2.5. DEG Analysis
2.6. GO and KEGG Pathway Enrichment Analysis
3. Results
3.1. Correlation and Heatmap Analysis of Gene Expression in LSS and Sham Groups
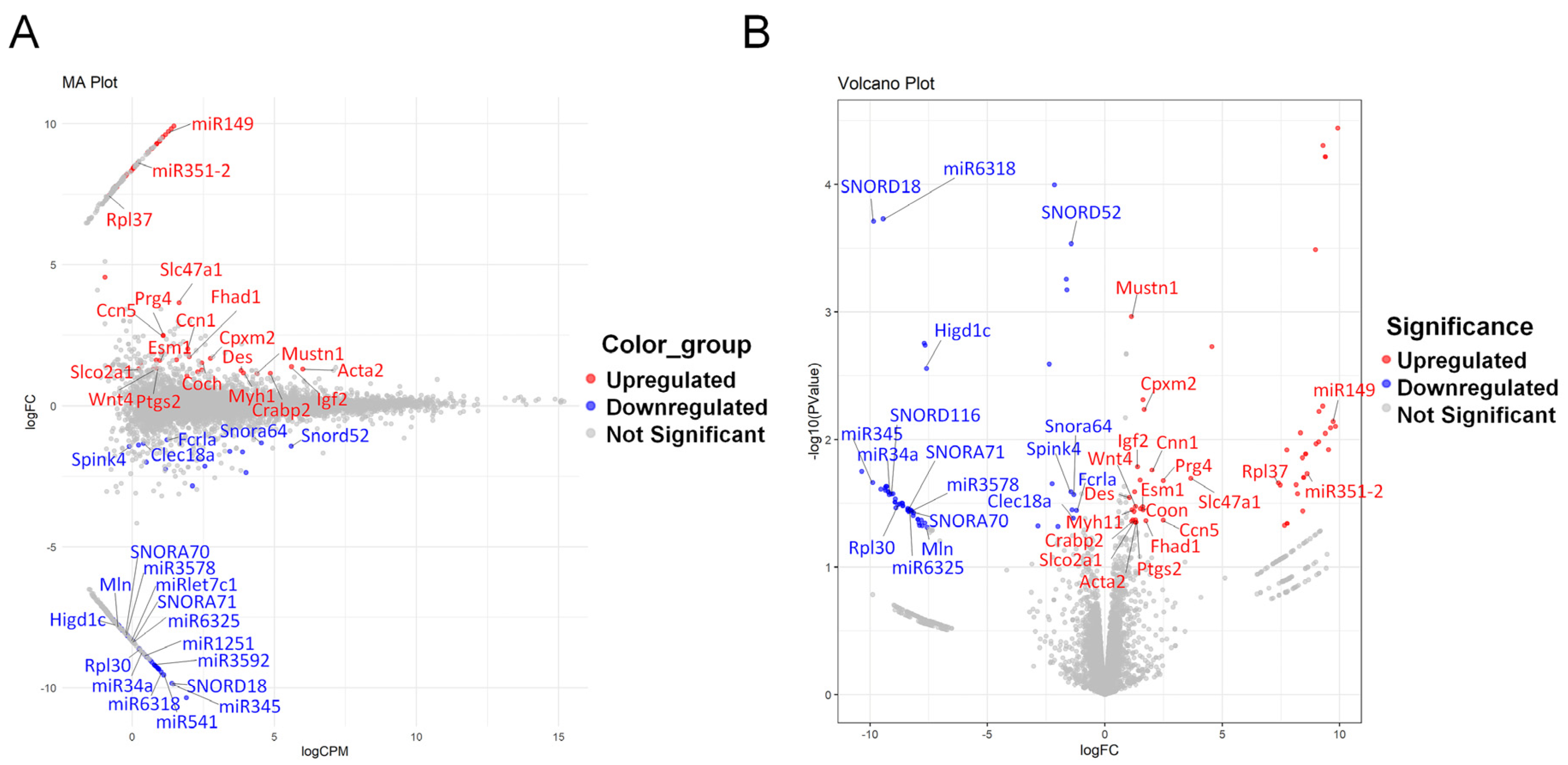
3.2. DEG Analysis and Identification of Key Genes in the LSS Model
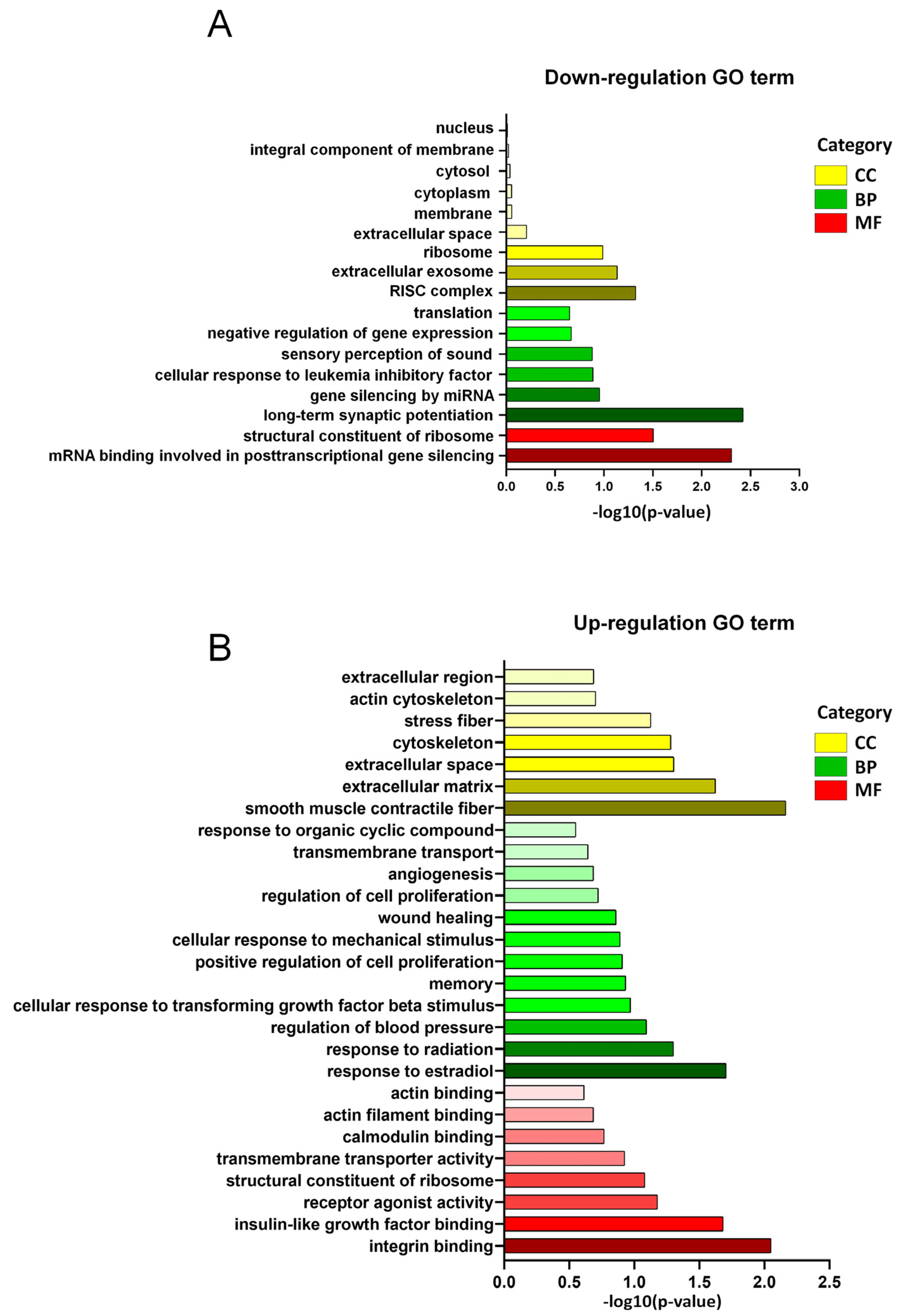
3.3. GO Enrichment of DEGs in LSS Model
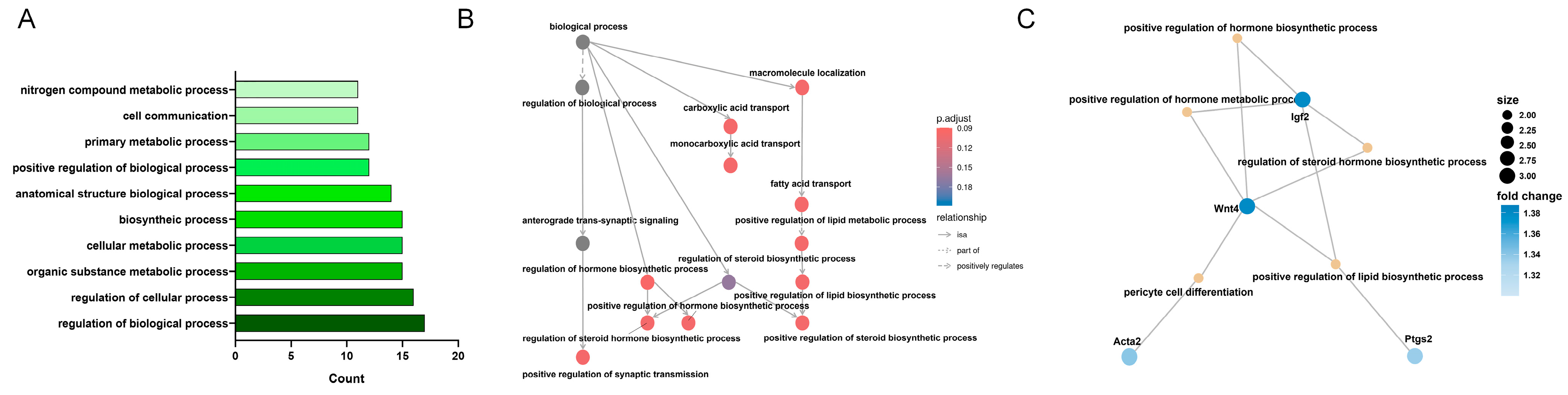
3.4. KEGG Enrichment Analysis of MF-Related Gene Expression Changes in the LSS Model
3.5. KEGG Enrichment Analysis of BP-Related Gene Expression Changes in the LSS Model
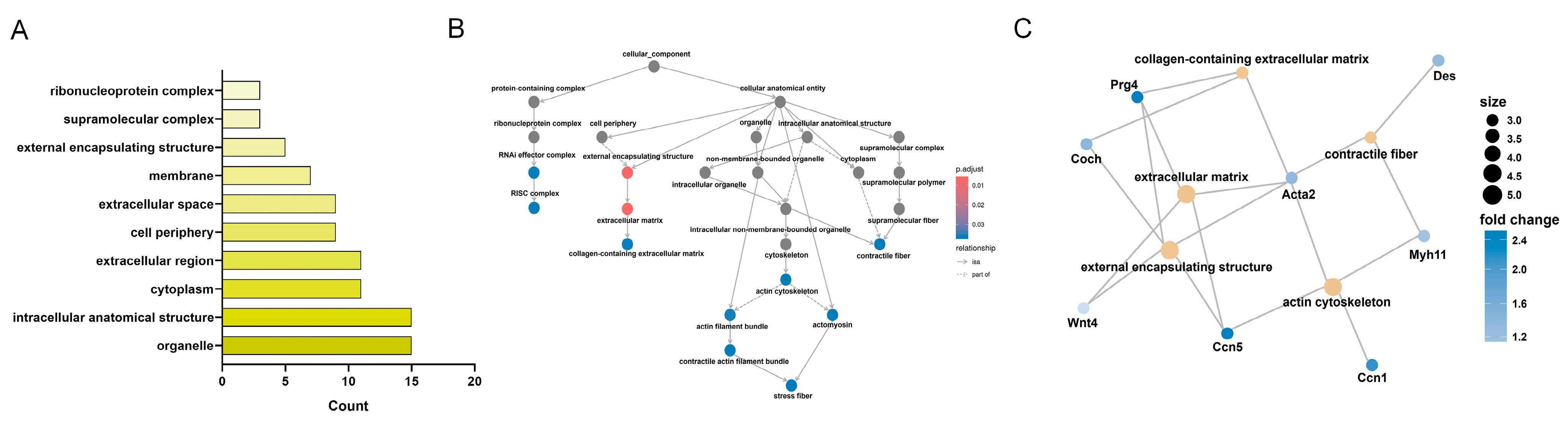
3.6. KEGG Enrichment Analysis of CC-Related Gene Expression Changes in the LSS Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, B.H.; Moon, S.H.; Suk, K.S.; Kim, H.S.; Yang, J.H.; Lee, H.M. Lumbar Spinal Stenosis: Pathophysiology and Treatment Principle: A Narrative Review. Asian Spine J. 2020, 14, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Munakomi, S.; Cruz, R. Lumbar Spinal Stenosis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Munakomi, S.; Foris, L.A.; Varacallo, M. Spinal Stenosis and Neurogenic Claudication. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Lurie, J.; Tomkins-Lane, C. Management of lumbar spinal stenosis. BMJ 2016, 352, h6234. [Google Scholar] [CrossRef] [PubMed]
- Byron, S.A.; Van Keuren-Jensen, K.R.; Engelthaler, D.M.; Carpten, J.D.; Craig, D.W. Translating RNA sequencing into clinical diagnostics: Opportunities and challenges. Nat. Rev. Genet. 2016, 17, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Kukurba, K.R.; Montgomery, S.B. RNA Sequencing and Analysis. Cold Spring Harb. Protoc. 2015, 2015, 951–969. [Google Scholar] [CrossRef]
- Atrooz, F.; Alkadhi, K.A.; Salim, S. Understanding stress: Insights from rodent models. Curr. Res. Neurobiol. 2021, 2, 100013. [Google Scholar] [CrossRef]
- Hosszu, A.; Kaucsar, T.; Seeliger, E.; Fekete, A. Animal Models of Renal Pathophysiology and Disease. Methods Mol. Biol. 2021, 2216, 27–44. [Google Scholar] [CrossRef]
- Szpirer, C. Rat models of human diseases and related phenotypes: A systematic inventory of the causative genes. J. Biomed. Sci. 2020, 27, 84. [Google Scholar] [CrossRef]
- Kim, H.; Hong, J.Y.; Jeon, W.J.; Lee, J.; Ha, I.H. Evaluation of the effects of differences in silicone hardness on rat model of lumbar spinal stenosis. PLoS ONE 2021, 16, e0251464. [Google Scholar] [CrossRef]
- Kasprzak, A. Insulin-Like Growth Factor 1 (IGF-1) Signaling in Glucose Metabolism in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 6434. [Google Scholar] [CrossRef]
- Moradali, M.F.; Rehm, B.H.A. Bacterial biopolymers: From pathogenesis to advanced materials. Nat. Rev. Microbiol. 2020, 18, 195–210. [Google Scholar] [CrossRef]
- Casillas-Espinosa, P.M.; Powell, K.L.; O’Brien, T.J. Regulators of synaptic transmission: Roles in the pathogenesis and treatment of epilepsy. Epilepsia 2012, 53 (Suppl. S9), 41–58. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Cheng, Z. Hormonal regulation of metabolism-recent lessons learned from insulin and estrogen. Clin. Sci. 2023, 137, 415–434. [Google Scholar] [CrossRef] [PubMed]
- Schmoller, K.M.; Skotheim, J.M. The Biosynthetic Basis of Cell Size Control. Trends Cell Biol. 2015, 25, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Stillwell, W. Chapter 20—Bioactive Lipids. In An Introduction to Biological Membranes, 2nd ed.; Stillwell, W., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 453–478. [Google Scholar]
- Motohashi, H.; Inui, K. Multidrug and toxin extrusion family SLC47: Physiological, pharmacokinetic and toxicokinetic importance of MATE1 and MATE2-K. Mol. Asp. Med. 2013, 34, 661–668. [Google Scholar] [CrossRef]
- Batara, D.C.; Park, S.W.; Kim, H.J.; Choi, S.Y.; Ohn, T.; Choi, M.C.; Park, S.I.; Kim, S.H. Targeting the multidrug and toxin extrusion 1 gene (SLC47A1) sensitizes glioma stem cells to temozolomide. Am. J. Cancer Res. 2023, 13, 4021–4038. [Google Scholar]
- Qiao, H.; Yin, H.; Feng, Y.; Tang, H. Pan-cancer analysis reveals the relationship between RCSD1 immune infiltration and clinical prognosis in human tumors. Front. Immunol. 2022, 13, 1008778. [Google Scholar] [CrossRef]
- Yao, X.; Watkins, N.H.; Brown-Harding, H.; Bierbach, U. A membrane transporter determines the spectrum of activity of a potent platinum-acridine hybrid anticancer agent. Sci. Rep. 2020, 10, 15201. [Google Scholar] [CrossRef]
- Lin, Z.; Liu, J.; Long, F.; Kang, R.; Kroemer, G.; Tang, D.; Yang, M. The lipid flippase SLC47A1 blocks metabolic vulnerability to ferroptosis. Nat. Commun. 2022, 13, 7965. [Google Scholar] [CrossRef]
- Iqbal, S.M.; Leonard, C.; Regmi, S.C.; De Rantere, D.; Tailor, P.; Ren, G.; Ishida, H.; Hsu, C.; Abubacker, S.; Pang, D.S.; et al. Lubricin/Proteoglycan 4 binds to and regulates the activity of Toll-Like Receptors In Vitro. Sci. Rep. 2016, 6, 18910. [Google Scholar] [CrossRef]
- Krawetz, R.J.; Abubacker, S.; Leonard, C.; Masson, A.O.; Shah, S.; Narendran, N.; Tailor, P.; Regmi, S.C.; Labit, E.; Ninkovic, N.; et al. Proteoglycan 4 (PRG4) treatment enhances wound closure and tissue regeneration. NPJ Regen. Med. 2022, 7, 32. [Google Scholar] [CrossRef]
- Qadri, M.; Jay, G.D.; Zhang, L.X.; Richendrfer, H.; Schmidt, T.A.; Elsaid, K.A. Proteoglycan-4 regulates fibroblast to myofibroblast transition and expression of fibrotic genes in the synovium. Arthritis Res. Ther. 2020, 22, 113. [Google Scholar] [CrossRef] [PubMed]
- Qadri, M.M. Targeting CD44 Receptor Pathways in Degenerative Joint Diseases: Involvement of Proteoglycan-4 (PRG4). Pharmaceuticals 2023, 16, 1425. [Google Scholar] [CrossRef]
- Risbud, M.V.; Schoepflin, Z.R.; Mwale, F.; Kandel, R.A.; Grad, S.; Iatridis, J.C.; Sakai, D.; Hoyland, J.A. Defining the phenotype of young healthy nucleus pulposus cells: Recommendations of the Spine Research Interest Group at the 2014 annual ORS meeting. J. Orthop. Res. 2015, 33, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Teeple, E.; Aslani, K.; Shalvoy, M.R.; Medrano, J.E.; Zhang, L.; Machan, J.T.; Fleming, B.C.; Jay, G.D. Lubricin deficiency in the murine lumbar intervertebral disc results in elevated torsional apparent modulus. J. Biomech. 2015, 48, 2210–2213. [Google Scholar] [CrossRef] [PubMed]
- Sao, K.; Risbud, M.V. Proteoglycan Dysfunction: A Common Link Between Intervertebral Disc Degeneration and Skeletal Dysplasia. Neurospine 2024, 21, 162–178. [Google Scholar] [CrossRef]
- Zhu, J.Y.; Chen, M.; Mu, W.J.; Luo, H.Y.; Guo, L. The functional role of Higd1a in mitochondrial homeostasis and in multiple disease processes. Genes Dis. 2023, 10, 1833–1845. [Google Scholar] [CrossRef]
- An, H.J.; Cho, G.; Lee, J.O.; Paik, S.G.; Kim, Y.S.; Lee, H. Higd-1a interacts with Opa1 and is required for the morphological and functional integrity of mitochondria. Proc. Natl. Acad. Sci. USA 2013, 110, 13014–13019. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, S.; Li, X.; Li, X.; Zhao, S.; Guo, J.; Wang, S.; Wang, R.; Zhang, M.; Qiu, W. HIGD1B, as a novel prognostic biomarker, is involved in regulating the tumor microenvironment and immune cell infiltration; its overexpression leads to poor prognosis in gastric cancer patients. Front. Immunol. 2024, 15, 1415148. [Google Scholar] [CrossRef]
- Zhu, J.Y.; Chen, M.; Mu, W.J.; Luo, H.Y.; Guo, L. Higd1a facilitates exercise-mediated alleviation of fatty liver in diet-induced obese mice. Metabolism 2022, 134, 155241. [Google Scholar] [CrossRef]
- Cooke, S.F.; Bliss, T.V. Plasticity in the human central nervous system. Brain 2006, 129, 1659–1673. [Google Scholar] [CrossRef]
- Gutierrez, B.A.; Limon, A. Synaptic Disruption by Soluble Oligomers in Patients with Alzheimer’s and Parkinson’s Disease. Biomedicines 2022, 10, 1743. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.L.; Yan, Q.J.; Zhu, F. Abnormal synaptic plasticity and impaired cognition in schizophrenia. World J. Psychiatry 2022, 12, 541–557. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, S.I.; Swaminathan, J.; Sumaroka, M.; Liebhaber, S.; Gewirtz, A.M. Effects of local mRNA structure on posttranscriptional gene silencing. Proc. Natl. Acad. Sci. USA 2008, 105, 13787–13792. [Google Scholar] [CrossRef] [PubMed]
- Stokes, T. Post-transcriptional gene silencing: Conservation and sequences. Trends Plant Sci. 2000, 5, 514. [Google Scholar] [CrossRef]
- Opron, K.; Burton, Z.F. Ribosome Structure, Function, and Early Evolution. Int. J. Mol. Sci. 2018, 20, 40. [Google Scholar] [CrossRef]
- Pratt, A.J.; MacRae, I.J. The RNA-induced silencing complex: A versatile gene-silencing machine. J. Biol. Chem. 2009, 284, 17897–17901. [Google Scholar] [CrossRef]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef]
- Chicurel, M.E.; Singer, R.H.; Meyer, C.J.; Ingber, D.E. Integrin binding and mechanical tension induce movement of mRNA and ribosomes to focal adhesions. Nature 1998, 392, 730–733. [Google Scholar] [CrossRef]
- Sinha, S.; Iyer, D.; Granata, A. Embryonic origins of human vascular smooth muscle cells: Implications for in vitro modeling and clinical application. Cell. Mol. Life Sci. 2014, 71, 2271–2288. [Google Scholar] [CrossRef]
- Hafen, B.B.; Burns, B. Physiology, Smooth Muscle. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Wang, J.H.; Thampatty, B.P.; Lin, J.S.; Im, H.J. Mechanoregulation of gene expression in fibroblasts. Gene 2007, 391, 1–15. [Google Scholar] [CrossRef]
- Didangelos, A.; Yin, X.; Mandal, K.; Baumert, M.; Jahangiri, M.; Mayr, M. Proteomics characterization of extracellular space components in the human aorta. Mol. Cell. Proteom. MCP 2010, 9, 2048–2062. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, L.N.; Colborn, T.; Hayes, T.B.; Heindel, J.J.; Jacobs, D.R., Jr.; Lee, D.H.; Shioda, T.; Soto, A.M.; vom Saal, F.S.; Welshons, W.V.; et al. Hormones and endocrine-disrupting chemicals: Low-dose effects and nonmonotonic dose responses. Endocr. Rev. 2012, 33, 378–455. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, L.P.; Gomez de Cedron, M.; Ramirez de Molina, A. Alterations of Lipid Metabolism in Cancer: Implications in Prognosis and Treatment. Front. Oncol. 2020, 10, 577420. [Google Scholar] [CrossRef] [PubMed]
- Kadlecova, M.; Dobesova, Z.; Zicha, J.; Kunes, J. Abnormal Igf2 gene in Prague hereditary hypertriglyceridemic rats: Its relation to blood pressure and plasma lipids. Mol. Cell. Biochem. 2008, 314, 37–43. [Google Scholar] [CrossRef]
- Shen, L.; Li, C.; Wang, Z.; Zhang, R.; Shen, Y.; Miles, T.; Wei, J.; Zou, Z. Early-life exposure to severe famine is associated with higher methylation level in the IGF2 gene and higher total cholesterol in late adulthood: The Genomic Research of the Chinese Famine (GRECF) study. Clin. Epigenetics 2019, 11, 88. [Google Scholar] [CrossRef]
- Sottnik, J.L.; Shackleford, M.T.; Robinson, S.K.; Villagomez, F.R.; Bahnassy, S.; Oesterreich, S.; Hu, J.; Madak-Erdogan, Z.; Riggins, R.B.; Corr, B.R.; et al. WNT4 Regulates Cellular Metabolism via Intracellular Activity at the Mitochondria in Breast and Gynecologic Cancers. Cancer Res. Commun. 2024, 4, 134–151. [Google Scholar] [CrossRef]
- Belfiore, A.; Rapicavoli, R.V.; Le Moli, R.; Lappano, R.; Morrione, A.; De Francesco, E.M.; Vella, V. IGF2: A Role in Metastasis and Tumor Evasion from Immune Surveillance? Biomedicines 2023, 11, 229. [Google Scholar] [CrossRef]
- Foulquier, S.; Daskalopoulos, E.P.; Lluri, G.; Hermans, K.C.M.; Deb, A.; Blankesteijn, W.M. WNT Signaling in Cardiac and Vascular Disease. Pharmacol. Rev. 2018, 70, 68–141. [Google Scholar] [CrossRef]
- Tang, Y.; Zhuo, D.; Yu, Y.; Pu, W.; Ma, Y.; Zhang, Y.; Huang, Y.; Zhang, Q.; Tang, K.; Meng, C.; et al. Single-cell RNA sequencing reveals the CRTAC1+ population actively contributes to the pathogenesis of spinal ligament degeneration by SPP1+ macrophage. Aging Cell 2024, 23, e14320. [Google Scholar] [CrossRef]
- Park, H.; Kim, J.; Yoo, S.; Yun, J.-M.; Song, K. Common Peroneal Nerve Entrapment Masked by Acute Lumbar Disc Herniation: Integrated Korean Medicine Treatment with Ultrasound-Guided Pharmacopuncture. Perspect. Integr. Med. 2024, 3, 177–183. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, J.Y.; Jeon, W.-J.; Kim, H.; Yeo, C.; Kim, H.; Lee, Y.J.; Ha, I.-H. Differential Gene Expression Analysis in a Lumbar Spinal Stenosis Rat Model via RNA Sequencing: Identification of Key Molecular Pathways and Therapeutic Insights. Biomedicines 2025, 13, 192. https://doi.org/10.3390/biomedicines13010192
Hong JY, Jeon W-J, Kim H, Yeo C, Kim H, Lee YJ, Ha I-H. Differential Gene Expression Analysis in a Lumbar Spinal Stenosis Rat Model via RNA Sequencing: Identification of Key Molecular Pathways and Therapeutic Insights. Biomedicines. 2025; 13(1):192. https://doi.org/10.3390/biomedicines13010192
Chicago/Turabian StyleHong, Jin Young, Wan-Jin Jeon, Hyunseong Kim, Changhwan Yeo, Hyun Kim, Yoon Jae Lee, and In-Hyuk Ha. 2025. "Differential Gene Expression Analysis in a Lumbar Spinal Stenosis Rat Model via RNA Sequencing: Identification of Key Molecular Pathways and Therapeutic Insights" Biomedicines 13, no. 1: 192. https://doi.org/10.3390/biomedicines13010192
APA StyleHong, J. Y., Jeon, W.-J., Kim, H., Yeo, C., Kim, H., Lee, Y. J., & Ha, I.-H. (2025). Differential Gene Expression Analysis in a Lumbar Spinal Stenosis Rat Model via RNA Sequencing: Identification of Key Molecular Pathways and Therapeutic Insights. Biomedicines, 13(1), 192. https://doi.org/10.3390/biomedicines13010192