1. Introduction
MicroRNAs are small non-coding RNAs that are approximately 22 nucleotides in length. By binding to the 3′-untranslated regions (UTRs) of target messenger RNAs (mRNAs) via imperfect base-paring, miRNAs regulate gene expression [
1,
2,
3,
4,
5]. MicroRNAs play two different roles; while they suppress protein translation by interfering with gene expression, they act as positive regulators [
6]. Since the discovery of the first miRNA lin-4, many other miRNAs have been identified using high-throughput techniques [
7,
8]. Several studies have reported the important roles of miRNAs in various biological processes such as aging [
9], apoptosis [
10], development [
11], and proliferation [
12]. Therefore, identifying disease-related miRNAs is of great significance from a molecular biology perspective and for diagnosing complex human diseases. Considering the resources and time required for biological experiments, numerous studies have attempted to develop computational frameworks for detecting disease-related miRNAs.
To date, various studies have assumed that functionally similar miRNAs are highly related to phenotypically similar diseases to predict miRNA–disease associations (MDAs) [
13,
14].
Ha et al. employed a probabilistic matrix factorization model that integrates miRNA expression as implicit feedback to predict novel MDAs [
15]. Additionally, they incorporated disease similarity information into the matrix factorization model, achieving improved performance in identifying MDAs [
16]. Furthermore, the authors proposed a groundbreaking model by integrating neural collaborative filtering, based on a deep learning architecture, to infer relationships between miRNAs and diseases [
17]. Jiang et al. introduced a novel framework that applies a hypergeometric distribution. To detect disease-related miRNAs, this model utilizes various heterogeneous networks, including miRNAs and diseases [
18]. Shi et al. designed a random walk framework to detect disease-related miRNAs by using a bipartite network [
19]. Mørk et al. reported an efficient model that utilizes protein associations between miRNAs and diseases [
20]. This model gathers protein–miRNA and protein–disease associations via text mining. Xu et al. ranked disease-related miRNAs using disease genes and miRNA–target interaction data [
21]. Xuan et al. reported a prediction model for HDMP by exploiting the k-nearest neighbors in a constructed network [
22]. The HDMP assumes that miRNAs belonging to the same cluster are strongly associated with the same disease. Chen et al. introduced a prediction method to identify novel MDA (RWRMDA). In the RWRMDA, a random walk algorithm is implemented by constructing a global miRNA functional similarity network [
23].
As machine learning is effectively applied in various scientific domains, various models have adopted it to perform research tasks in bioinformatics [
24,
25,
26]. Chen et al. introduced an approach that utilized k-nearest neighbors to infer miRNA–disease associations [
27]. This model integrates heterogeneous biological datasets and ranks disease-related miRNAs according to scores assigned by the support vector machine. Chen et al. applied hierarchical agglomerative clustering by considering miRNA–disease bias ratings [
28]. This research group proposed a detection method called RLSMDA [
29]. RLSMDA uses a semi-supervised classifier to predict miRNAs that are not associated with known diseases. Xiao et al. designed a prediction method that applied non-negative matrix factorization (MF) based on various heterogeneous omics data (GRNMF) [
30]. When the recommender algorithm was applied, GRNMF performed well for both miRNAs with no known disease associations and diseases with no known miRNA associations. Li et al. introduced a matrix factorization framework, MCMDA, for disease-related miRNA identification [
31]. MCMDA efficiently predicts disease-related miRNAs by updating the adjacency matrix of MDA. Chen et al. developed a method for potential miRNA–disease associations (HGIMDAs), in which various similarity values are used for comprehensive networks [
32]. Ha et al. reported a prediction method for PMAMCA that exploits matrix factorization to infer disease-related miRNAs [
33]. The authors adopted MF, a machine-learning algorithm that is actively applied in recommendation systems. Using MF, PMAMCA efficiently captured disease-related miRNAs by assigning miRNA expression values to the model. Chen et al. integrated the comprehensive similarity values of miRNA functional similarity, disease semantic similarity, and Gaussian interaction profile kernel similarity to efficiently infer disease-related miRNAs [
34]. Chen et al. developed a disease-related miRNA detection method called MDHGI that integrates comprehensive similarity values [
35]. Chen et al. developed an NC-MCMDA model to identify novel disease-related miRNAs. NCMCMDA adopts a matrix completion algorithm by combining comprehensive similarity values to reflect similarity-based neighborhood constraints [
36]. However, a common drawback of the aforementioned computational models is their high dependence on known miRNA–disease information, and the performance of the models varies greatly depending on parameter selection. Ha et al. leveraged matrix factorization, a commonly employed machine learning approach in recommendation systems, to extract disease-associated miRNAs effectively by incorporating disease and miRNA similarity information [
37]. The authors also proposed a machine learning-based model for identifying disease-associated miRNAs by employing metric learning techniques [
38]. Ning et al. presented AMHMDA, a novel method integrating attention-aware multi-view similarity networks and hypergraph learning to predict miRNA–disease associations [
39]. Jin et al. introduced MAMFGAT, a novel model leveraging adaptive modality fusion and graph attention networks to predict miRNA–disease associations by integrating multiple similarity and association networks [
40]. Peng et al. proposed MHCLMDA, a novel method utilizing multiple hypergraph contrastive learning and variational auto-encoders to predict miRNA–disease associations by integrating consistent feature representations across multiple views [
41].
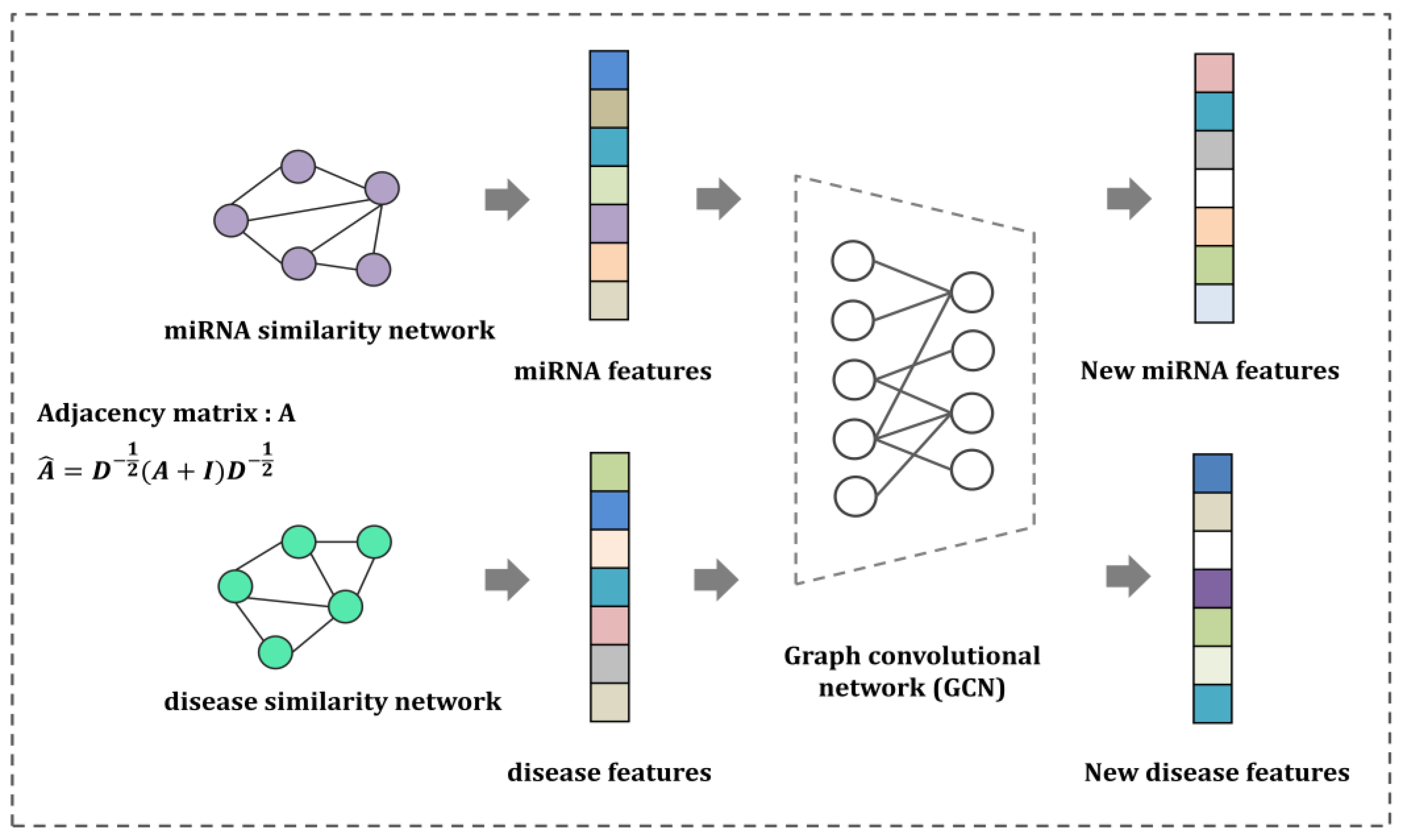
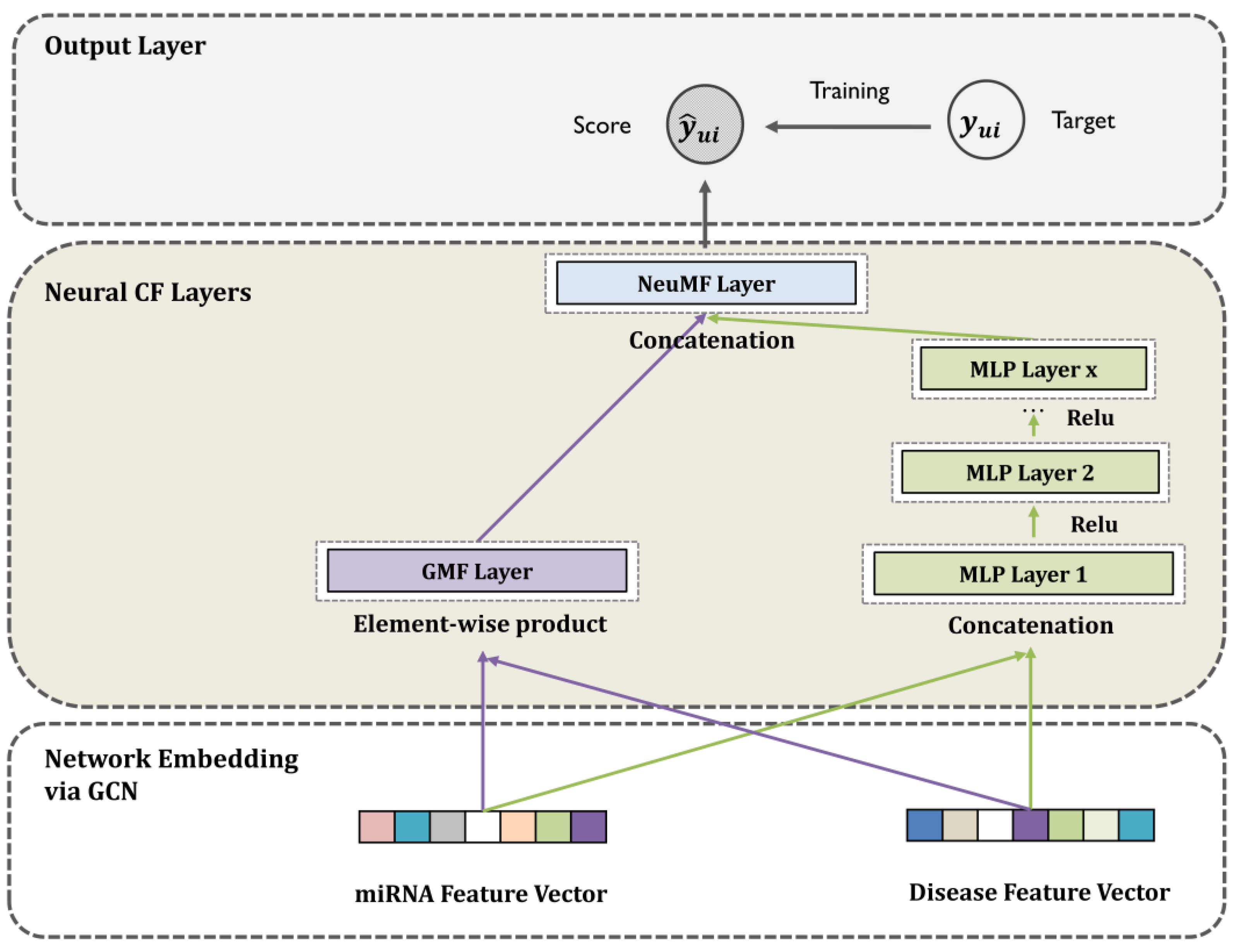
Here, we propose a novel and feasible machine learning framework using neural collaborative filtering (NCF) and a graph convolutional network (GCN) to infer miRNA–disease associations (GCNCF). This study aims to (1) overcome the limitations of the inner product score function, which violates the triangle inequality, and (2) build a feasible and effective machine learning approach to infer novel MDA while retaining the network structure and attributes. In this model, we first integrated heterogeneous similarity values such as miRNA functional similarity, disease semantic similarity, and Gaussian interaction profile kernel similarity to construct miRNA and disease similarity networks. Subsequently, a GCN is adopted to capture accurate network embeddings while retaining the structure and properties of the network. Finally, the NCF was applied to overcome the violation of the triangle inequality issue while enhancing the prediction accuracy. Consequently, GCNCF achieved reliable performance in detecting miRNA–disease associations, with AUC scores of 0.9273 and 0.9075 through global and local leave-one-out cross-validation (LOOCV). Moreover, extensive experiments based on evaluation measurements qualitatively confirmed the comparable GCNCF performance.
4. Discussion and Conclusions
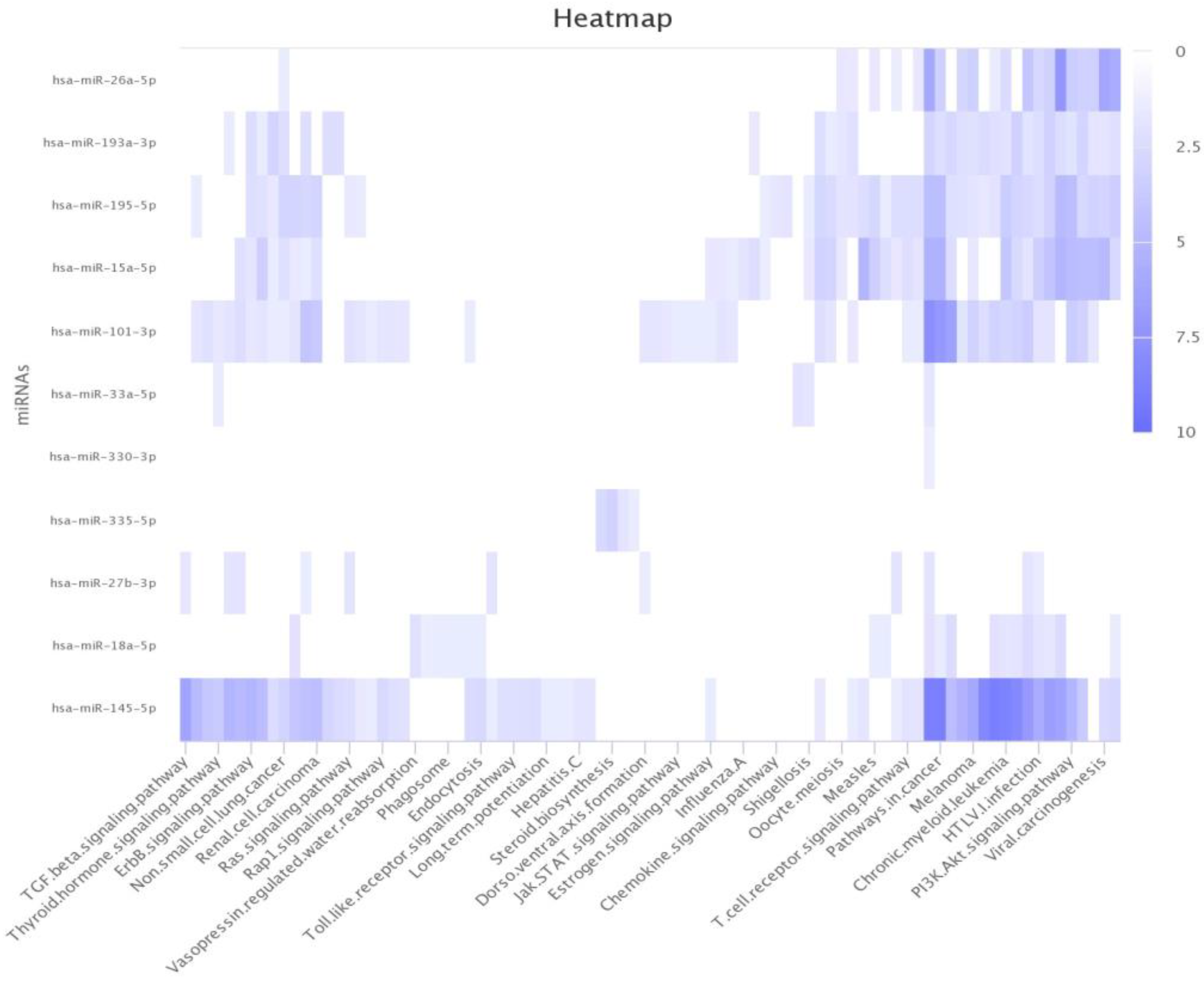
With the exponential growth in heterogeneous networks, the extraction of meaningful network embeddings via machine learning has become increasingly important. The crucial factors for network embedding are (1) learning low-dimensional representations for nodes in the network and (2) capturing the semantics behind the pairwise relationship (e.g., miRNA–disease association prediction). This study was motivated by the fact that the network implies node structural roles and attributes while learning the low-dimensional representations of nodes. Specifically, this study addresses the MDA prediction problem by formalizing neural collaborative filtering using GCNCF. GCNCF comprehensively integrates miRNA and disease similarities through miRNA functional similarity, disease semantic similarity, and a Gaussian interaction profile kernel to build miRNA and disease networks. We focused on modeling a network-embedding approach with a GCN that was suitable for learning low-dimensional vectors while aiming to capture the network structure and roles more accurately and realistically. Consequently, we systemically fused the linearities and non-linearities of miRNAs and disease feature vectors by designing neural collaborative filtering to predict novel miRNA–disease associations. The experimental results under various evaluation metrics validated that GCNCF outperformed the four previous models.
GCNCF leverages graph convolutional networks (GCNs) to capture higher-order structural information from the input graph, effectively modeling complex relationships between entities. Unlike NCMD and MDHGI, which primarily rely on matrix decomposition and feature aggregation, GCNCF dynamically updates node representations through neighborhood information propagation. Additionally, the integration of neural collaborative filtering (NCF) enables the modeling of nonlinear and latent interactions often overlooked by traditional matrix factorization methods like GRNMF. This synergistic combination of GCN and NCF provides dual benefits: GCN establishes a robust foundation for representation learning, while NCF enhances predictive accuracy by capturing intricate interaction dynamics. These advancements are validated by the experimental results, where GCNCF consistently outperforms referenced models across multiple evaluation metrics, as detailed in the Results section. We believe these enhancements clearly highlight the novel contributions of GCNCF and its advantages over existing approaches.
However, our model has room for further improvement, which could be explored in future work. The model can be broadly divided into two stages: the feature representation stage, where diseases and miRNAs are mapped to low-dimensional vectors, and the prediction stage, where the relationships between miRNAs and diseases are inferred. To improve the first stage, employing more advanced machine learning models for feature representation could provide more accurate and meaningful representations of these objects, making them more comprehensible to the computer. In addition, applying more sophisticated machine learning techniques in the relationship prediction model could enhance performance and offer significant potential for model improvement. Also, generalizing the model to new, unseen diseases remains challenging. Exploring transfer learning or integrating domain-specific knowledge could help improve performance for diseases with limited data.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}