
Pathological Mechanisms and Novel Testing Methods in Thrombotic Thrombocytopenic Purpura


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Known Mechanisms, Laboratory Values, and Treatment
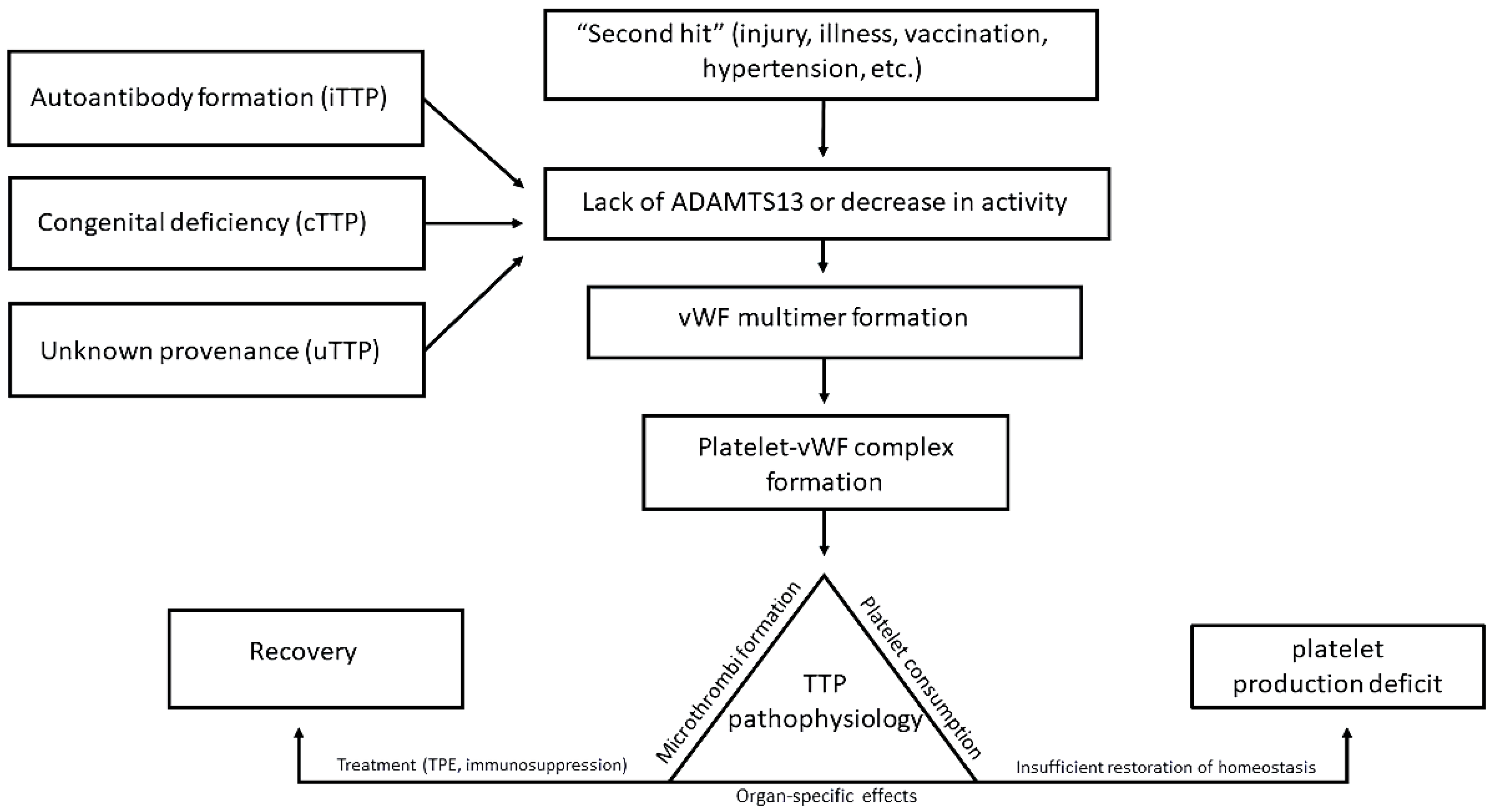
3. Potential New Pathologic Mechanisms in TTP
3.1. Mechanisms and Prevalence of Conformational Change
3.2. ADAMTS13 Interactions with Inflammatory States and Endothelial Damage
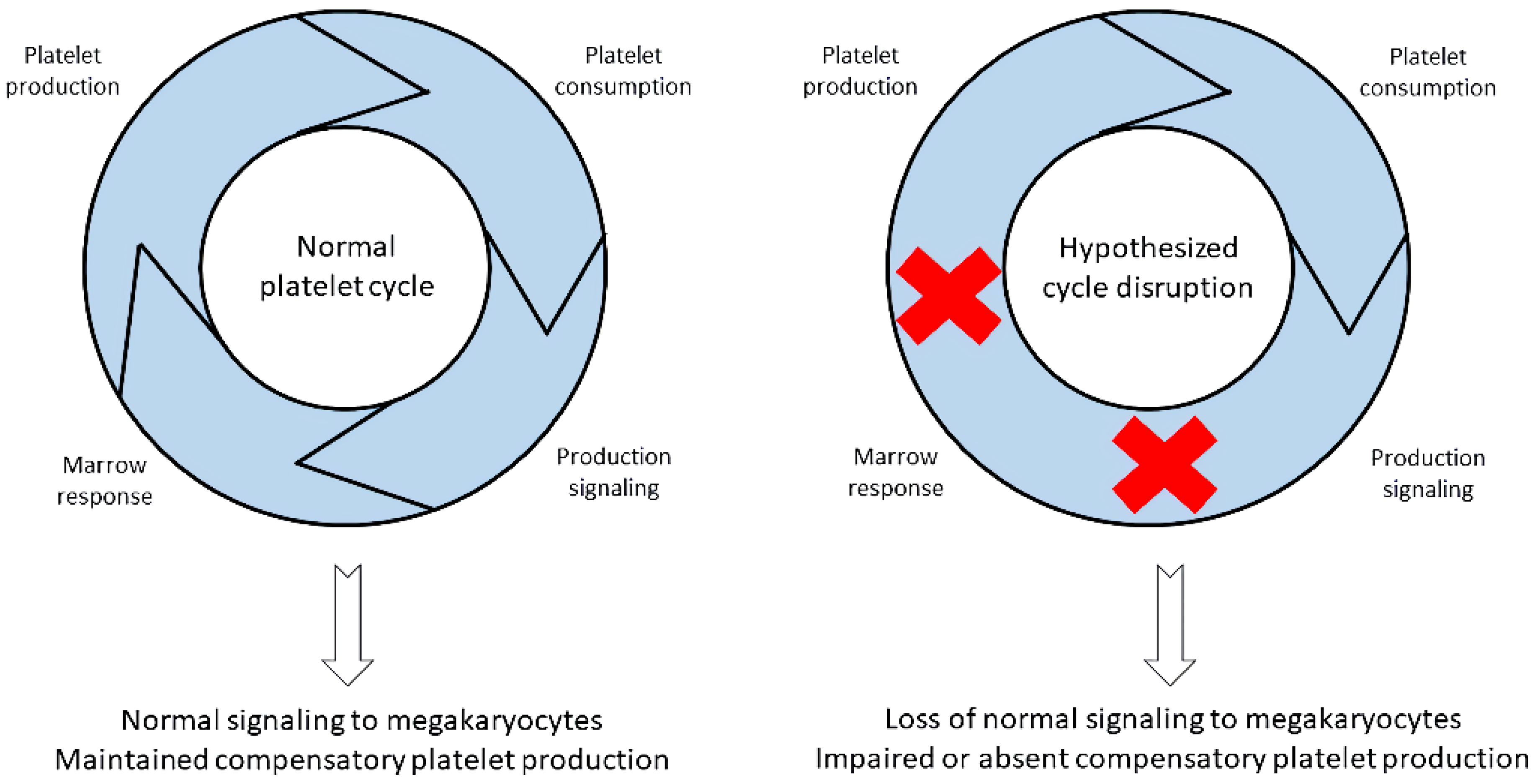
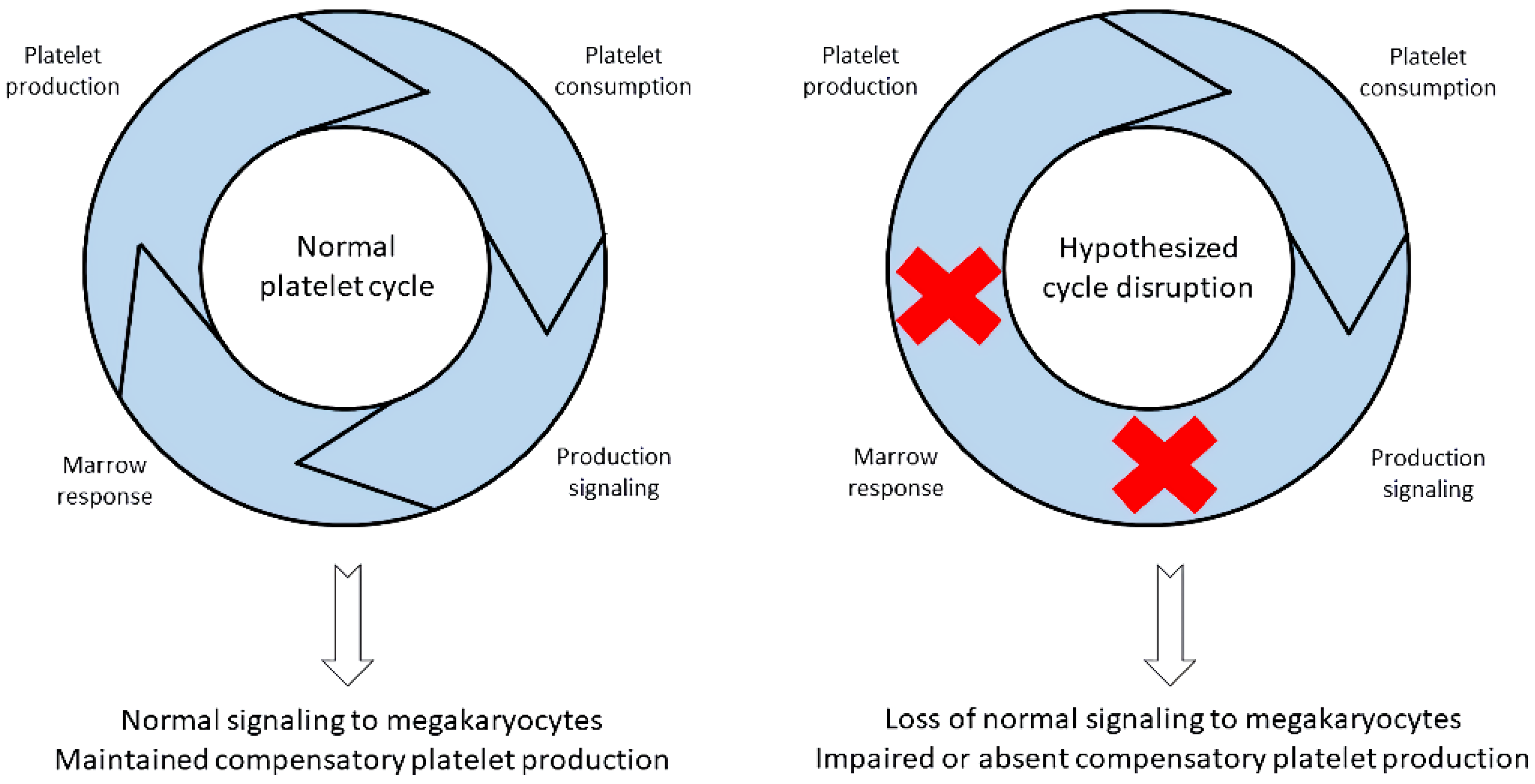
4. Absolute Immature Platelet Count and Potential Platelet Production Suppression
5. Importance of Low A-IPC and Possible Additional Pathologic Insult
6. Other Contributors to TTP Pathogenesis and Outcome
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gómez-Seguí, I.; Pascual Izquierdo, C.; de la Rubia Comos, J. Best practices and recommendations for drug regimens and plasma exchange for immune thrombotic thrombocytopenic purpura. Expert Rev. Hematol. 2021, 14, 707–719. [Google Scholar] [CrossRef]
- Lancellotti, S.; Sacco, M.; Tardugno, M.; Ferretti, A.; De Cristofaro, R. Immune and Hereditary Immune Thrombocytopenic Purpura: Can ADAMTS13 Deficiency Alone Explain the Different Clinical Phenotypes? J. Clin. Med. 2023, 12, 3111. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.A.; Waldrum, M.R.; Marques, M.B. Platelet Count and Prothrombin Type Help Distinguish Thrombotic Thrombocytopenic Purpura-Hemolytic Uremic Syndrome from Disseminated Intravascular Coagulation in Adults. Hematopathology 2010, 133, 460–465. [Google Scholar]
- Chiasakul, T.; Cuker, A. Clinical and laboratory diagnosis of TTP: An integrated approach. Am. Soc. Hematol. 2018, 1, 530–538. [Google Scholar] [CrossRef]
- Lin, H.; Huang, J.; Huang, J.; Zhang, L.; Yin, X.; Yang, J.; Huang, X. Concurrence of immune thrombocytopenic purpura and thrombotic thrombocytopenic purpura: A case report and review of the literature. J. Med. Case Rep. 2023, 17, 38. [Google Scholar] [CrossRef]
- Bittencourt, C.E.; Ha, J.P.; Maitta, R.W. Re-Examination of 30-Day Survival and Relapse Rates in Patients with Thrombotic Thrombocytopenic Purpura-Hemolytic Uremic Syndrome. PLoS ONE 2015, 10, e0127744. [Google Scholar]
- Dolin, H.H.; Dziuba, M.; Pappada, S.M.; Papadimos, T.J. Presumed antiphospholipid syndrome and thrombotic thrombocytopenic purpura: An infrequent association. Clin. Case Rep. 2019, 7, 1984–1988. [Google Scholar] [CrossRef]
- Zhu, M.; Reeves, H.M.; Maitta, R.W. Immature platelet dynamics correlate with ADAMTS13 deficiency and predict therapy response in immune-mediated thrombotic thrombocytopenic purpura. Thromb. Res. 2021, 198, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Kier, Y.E.; Stempak, L.W.; Maitta, R.W. Immature platelet fraction can help adjust therapy in refractory thrombotic microangiopathic hemolytic anemia cases. Transfus. Apher. Sci. 2013, 49, 644–646. [Google Scholar] [CrossRef]
- Reeves, H.M.; Maitta, R.W. Immature Platelet Dynamics in Immune-Mediated Thrombocytopenic States. Front. Med. 2020, 7, 597734. [Google Scholar] [CrossRef]
- Zheng, Y.; Hong, H.; Reeves, H.M.; Maitta, R.W. Absolute immature platelet count helps differentiate thrombotic thrombocytopenic purpura from hypertension-induced thrombotic microangiopathy. Transf. Apher. Sci. 2014, 51, 54–57. [Google Scholar] [CrossRef]
- Hong, H.; Xiao, W.; Stempak, L.M.; Sandhaus, L.M.; Maitta, R.W. Absolute immature platelet count dynamics in diagnosing and monitoring the clinical course of thrombotic thrombocytopenic purpura. Transfusion 2014, 55, 756–765. [Google Scholar] [CrossRef]
- Lämmle, B. A third form of thrombotic thrombocytopenic purpura? Haematologica 2023, 108, 299–300. [Google Scholar] [CrossRef]
- Joly, B.S.; Roose, E.; Coppo, P.; Vanhoorelbeke, K.; Veyradier, A. ADAMTS13 conformation is closed in non-immune acquired thrombotic thrombocytopenic purpura of unidentified pathophysiology (uTTP). Haematologica 2023, 108, 638–644. [Google Scholar] [CrossRef]
- Béranger, N.; Coppo, P.; Tsatsaris, V.; Boisseau, P.; Provôt, F.; Delmas, Y.; Poullin, P.; Vanhoorelbeke, K.; Veyradier, A.; Joly, B.S. Management and follow-up of pregnancy-onset thrombotic thrombocytopenic purpura: The French experience. Blood Adv. 2024, 8, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Bonnez, Q.; Sakai, K.; Vanhoorelbeke, K. ADAMTS13 and Non-ADAMTS13 Biomarkers in Immune-Mediated Thrombotic Thrombocytopenic Purpura. J. Clin. Med. 2023, 12, 6169. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, S.; Lämmle, B.; Cataland, S.R. Thrombotic Thrombocytopenic Purpura: Pathophysiology, Diagnosis, and Management. J. Clin. Med. 2021, 10, 536. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Zheng, X.L. Animal models for thrombotic thrombocytopenic purpura: A narrative review. Ann. Blood 2023, 8, 23. [Google Scholar] [CrossRef]
- Cauchois, R.; Muller, R.; Lagarde, M.; Dignat-George, F.; Tellier, E.; Kaplanski, G. Is Endothelial Activation a Critical Event in Thrombotic Thrombocytopenic Purpura? J. Clin. Med. 2023, 12, 758. [Google Scholar] [CrossRef] [PubMed]
- Staley, E.M.; Cao, W.; Pham, H.P.; Kim, C.H.; Kocher, N.K.; Zheng, L.; Gangaraju, R.; Lorenz, R.G.; Williams, L.; Marques, M.B.; et al. Clinical factors and biomarkers predict outcome in patients with immune-mediated thrombotic thrombocytopenic purpura. Haematologica 2019, 104, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Hrdinová, J.; D’Angelo, S.; Graça, N.A.G.; Ercig, B.; Vanhoorelbeke, K.; Veyaradier, A.; Voorberg, J.; Coppo, P. Dissecting the pathophysiology of immune thrombotic thrombocytopenic purpura: Interplay between genes and environmental triggers. Haematologica 2018, 103, 1099–1109. [Google Scholar] [CrossRef]
- Li, A.; Khalighi, P.R.; Wu, Q.; Garcia, D.A. External validation of the PLASMIC score: A clinical prediction tool for thrombotic thrombocytopenic purpura diagnosis and treatment. J. Thromb. Haemost. 2018, 16, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Baysal, M.; Ümit, E.; Onur Kırkızlar, H.; Muzaffer Demir, A. Comparison of Clinical Scoring Systems in the Management of Patients with Microangiopathic Hemolytic Anemia and Thrombocytopenia. Turk. J. Hematol. 2021, 38, 64–68. [Google Scholar] [CrossRef]
- Liu, A.; Dhaliwal, N.; Upreti, H.; Kasmani, J.; Dane, K.; Moliterno, A.; Braunstein, E.; Brodsky, R.; Chaturvedi, S. Reduced sensitivity of PLASMIC and French scores for the diagnosis of Thrombotic Thrombocytopenic Purpura (TTP) in Older Individuals. Transfusion 2021, 61, 266–273. [Google Scholar] [CrossRef]
- Fage, N.; Orvain, C.; Henry, N.; Mellaza, C.; Beloncle, F.; Tuffigo, M.; Geneviève, F.; Coppo, P.; Augusto, J.F.; Brilland, B. Proteinuria Increases the PLASMIC and French Scores Performance to Predict Thrombotic Thrombocytopenic Purpura in Patients With Thrombotic Microangiopathy Syndrome. Kidney Int. Rep. 2022, 7, 221–231. [Google Scholar] [CrossRef]
- Laghmouchi, A.; Graça, N.A.G.; Voorberg, J. Emerging Concepts in Immune Thrombotic Thrombocytopenic Purpura. Front. Immunol. 2021, 12, 757192. [Google Scholar] [CrossRef] [PubMed]
- Abrams, C.S.; Barnes, G.D. SARS-CoV-2 Vaccination-Induced Thrombotic Thrombocytopenia: A Rare but Serious Immunologic Complication. Ann. Rev. Med. 2023, 74, 65–74. [Google Scholar] [CrossRef]
- Vorster, L.; Kirk, S.E.; Muscal, E.; Despotovic, J.M.; Cohen, C.T.; Sartain, S.E. COVID-19 vaccine (mRNA BNT162b2) and COVID-19 infection-induced thrombotic thrombocytopenic purpura in adolescents. Pediatr. Blood Cancer 2022, 69, e29681. [Google Scholar] [CrossRef]
- Mannucci, P.M.; Cugno, M. The complex differential diagnosis between thrombotic thrombocytopenic purpura and the atypical hemolytic uremic syndrome: Laboratory weapons and their impact on treatment choice and monitoring. Thromb. Res. 2015, 136, 851–854. [Google Scholar] [CrossRef]
- Shankar, K.; Huffman, D.L.; Peterson, C.; Yasir, M.; Kaplan, R. A Case of COVID-19 Induced Thrombotic Thrombocytopenic Purpura. Cureus 2021, 13, e16311. [Google Scholar] [CrossRef] [PubMed]
- Joly, B.S.; Coppo, P.; Veyradier, A. Thrombotic thrombocytopenic purpura. Blood 2017, 129, 2836–2846. [Google Scholar] [CrossRef]
- Colonne, C.K.; Favaloro, E.J.; Pasalic, L. The Intriguing Connections Between von Willebrand Factor, ADAMTS13, and Cancer. Healthcare 2022, 10, 557. [Google Scholar] [CrossRef]
- Meikle, C.K.S.; Kelly, C.A.; Garg, P.; Wuescher, L.M.; Ali, R.A.; Worth, R.G. Cancer and Thrombosis: The Platelet Perspective. Front. Cell Dev. Biol. 2017, 4, 147. [Google Scholar] [CrossRef]
- Reeves, H.M.; Maitta, R.W. Comparison of absolute immature platelet count to the PLASMIC score at presentation in predicting ADAMTS13 deficiency in suspected thrombotic thrombocytopenic purpura. Thromb. Res. 2022, 215, 30–36. [Google Scholar] [CrossRef]
- Gokozan, H.M.; Reeves, H.M.; Maitta, R.W. Absolute immature platelet count dynamics of thrombotic thrombocytopenic purpura patients with high ADAMTS13 inhibitor. Thromb. Res. 2019, 179, 128–131. [Google Scholar] [CrossRef]
- Chen, W.; Ha, J.P.; Hong, H.; Maitta, R.W. Absolute immature platelet counts in the setting of suspected heparin-induced thrombocytopenia may predict anti-PF4-heparin immunoassay testing results. Transf. Apheresis Sci. 2018, 57, 507–511. [Google Scholar] [CrossRef]
- Briggs, C.; Kunka, S.; Hart, D.; Oguni, S.; Machin, S.J. Assessment of an immature platelet fraction (IPF) in peripheral thrombocytopenia. Br. J. Haematol. 2004, 126, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Seery, C.; Imahiyerobo, A.M.; Bussel, J.B. Beyond the platelet count: Immature platelet fraction and thromboelastometry correlate with bleeding in patients with immune thrombocytopenia. Br. J. Haematol. 2014, 166, 592–600. [Google Scholar]
- Lee, G.M.; Arepally, G.M. Heparin-Induced Thrombocytopenia. Hematol. Am. Soc. Hematol. Educ. Program 2013, 2013, 668–674. [Google Scholar] [CrossRef]
- Stefaniuk, C.M.; Reeves, H.M.; Maitta, R.W. Dynamic changes in absolute immature platelet count suggest the presence of a coexisting immune process in the setting of thrombotic thrombocytopenic purpura. Transfusion 2017, 57, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Maitta, R.W.; Reeves, H.M.; Downes, K.A.; He, X.; Hackney, L.R.; Ahuja, S.P. Immature platelet dynamics in management of protracted response to therapy of a young pediatric patient with immune-mediated thrombotic thrombocytopenic purpura. Thromb. Res. 2023, 228, 145–147. [Google Scholar] [CrossRef]
- Siniard, R.C.; Gangaraju, R.; May, J.E.; Marques, M.B. Challenges in the diagnosis of thrombotic thrombocytopenic purpura. Exp. Rev. Hematol. 2023, 16, 861–869. [Google Scholar] [CrossRef]
- Gómez Seguí, I.; Mingot Castellano, M.E.; Pascal Izquierdo, C. Should we consider caplacizumab as routine treatment for acute thrombotic thrombocytopenic purpura? An expert perspective on the pros and cons. Exp. Rev. Hematol. 2024, 17, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Chait, Y.; Germain, M.J.; Hollot, C.V.; Horowitz, J. The Role of Feedback Control Design in Developing Anemia Management Protocols. Ann. Biomed. Eng. 2021, 49, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Koulnis, M.; Porpiglia, E.; Hidalgo, D.; Socolovsky, M. Erythropoiesis: From molecular pathways to system properties. Adv. Exp. Med. Biol. 2014, 844, 37–58. [Google Scholar] [PubMed]
- Van de Wyngaert, Z.; Fournier, E.; Bera, E.; Carrette, M.; Soenen, V.; Gauthier, J.; Preudhomme, C.; Boyer, T. Immature platelet fraction (IPF): A reliable tool to predict peripheral thrombocytopenia. Curr. Res. Transl. Med. 2020, 68, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Pavenski, K.; Huang, S.S.; Patriquin, C.J. Predictors of relapse and preventative strategies in immune thrombotic thrombocytopenic purpura. Exp. Rev. Hematol. 2021, 14, 1027–1040. [Google Scholar] [CrossRef] [PubMed]
- Behtaj, M.; Zhu, M.; Bittencourt, C.E.; Ha, J.P.; Maitta, R.W. Non-O blood group thrombotic thrombocytopenic purpura patients take longer to recover as measured by number of therapeutic plasma exchanges needed for platelet recovery. Thromb. Res. 2020, 185, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Hussein, E.; Teruya, J. Evaluating the impact of the ABO blood group on the clinical outcome of thrombotic thrombocytopenic purpura associated with severe ADAMTS13 deficiency. Vox Sang. 2017, 112, 434–442. [Google Scholar] [CrossRef]
- Yıldırım, M.; Sayın, S.; Güneş, A.K.; Aras, M.R.; Yılmaz, E.S.; Albayrak, M.; Özet, G.; Aylı, M. Effect of Blood Groups on Clinical Presentations and Treatment Outcomes in Immune Thrombotic Thrombocytopenic Purpura Patients with Severe ADAMTS13 Deficiency: A Multi-Center Experience. Transfus. Med. Hemotherapy 2023, 50, 18–24. [Google Scholar] [CrossRef]
- Kremer Hovinga, J.A.; Coppo, P.; Lämmle, B.; Moake, J.L.; Miyata, T.; Vanhoorelbeke, K. Thrombotic thrombocytopenic purpura. Nat. Rev. Dis. Primers 2017, 3, 17020. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Owais Subhan, M.; Cambridge, G.; Guo, Y.; de Groot, R.; Scully, M.; Thomas, M. Alterations in B- and circulating T-follicular helper cell subsets in immune thrombotic thrombocytopenic purpura. Blood Adv. 2022, 6, 3792–3802. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Cambridge, G.; Guo, Y.; Scully, M.; Thomas, M. Increased Activated Circulating T Follicular Helper Cells and Changes in B Cell Subsets in Immune TTP (iTTP) and in Response to Rituximab Treatment. Blood 2020, 136 (Suppl. S1), 23–24. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dolin, H.H.; Maitta, R.W. Pathological Mechanisms and Novel Testing Methods in Thrombotic Thrombocytopenic Purpura. Biomedicines 2024, 12, 621. https://doi.org/10.3390/biomedicines12030621
Dolin HH, Maitta RW. Pathological Mechanisms and Novel Testing Methods in Thrombotic Thrombocytopenic Purpura. Biomedicines. 2024; 12(3):621. https://doi.org/10.3390/biomedicines12030621
Chicago/Turabian StyleDolin, Hallie H., and Robert W. Maitta. 2024. "Pathological Mechanisms and Novel Testing Methods in Thrombotic Thrombocytopenic Purpura" Biomedicines 12, no. 3: 621. https://doi.org/10.3390/biomedicines12030621
APA StyleDolin, H. H., & Maitta, R. W. (2024). Pathological Mechanisms and Novel Testing Methods in Thrombotic Thrombocytopenic Purpura. Biomedicines, 12(3), 621. https://doi.org/10.3390/biomedicines12030621