
Overexpression of SMYD3 Promotes Autosomal Dominant Polycystic Kidney Disease by Mediating Cell Proliferation and Genome Instability
 and
and 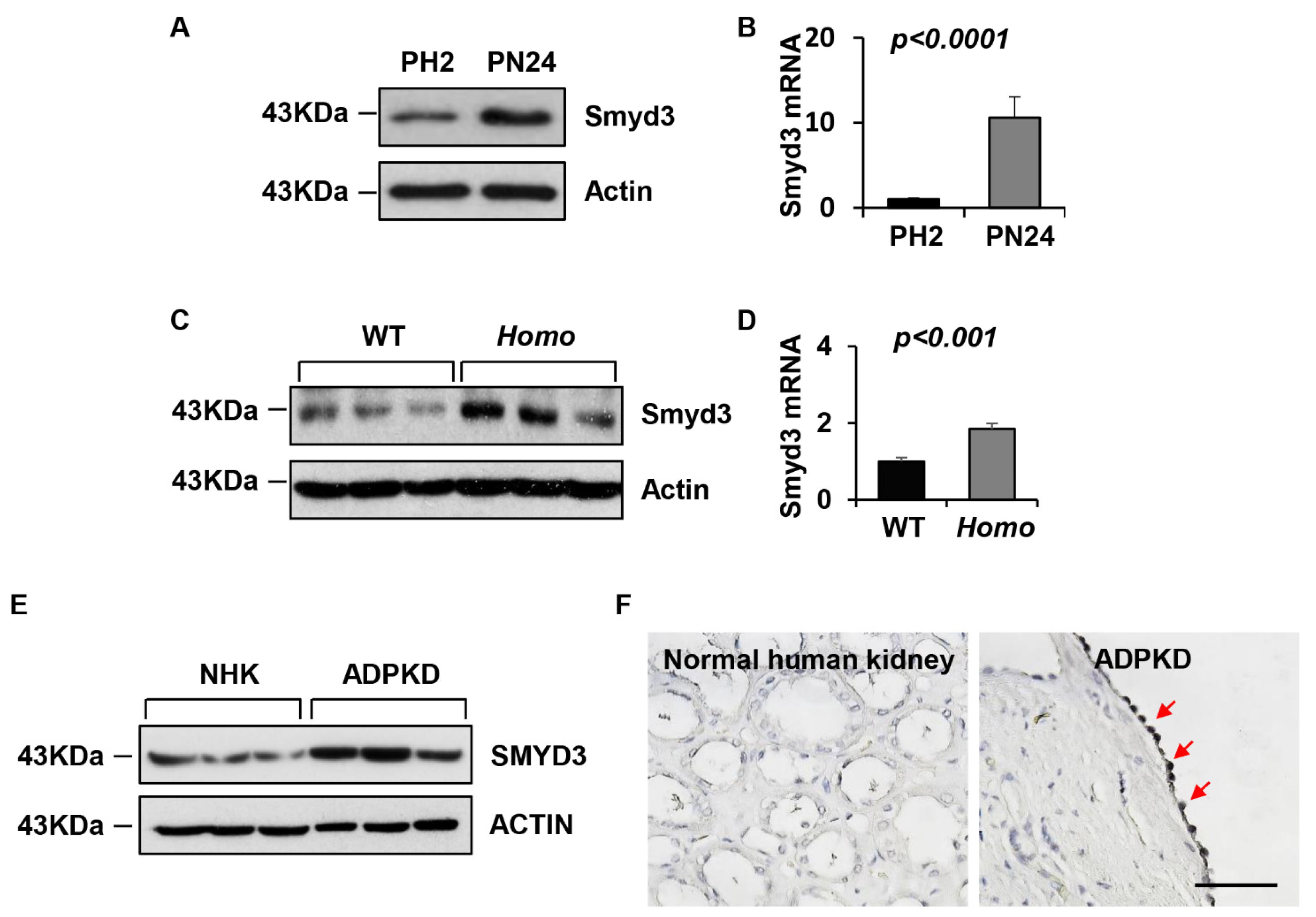{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmids
2.3. Generating a Stable SMYD3-Knockdown IMCD3 Cell Line
2.4. Antibodies and Reagents
2.5. Histology and Immunohistochemistry
2.6. Cell Immunofluorescence Staining
2.7. Western Blot Analysis and Immunoprecipitation
2.8. Glutathione S-Transferase (GST) Pull-Down Assay
2.9. Microscopy and Imaging
2.10. Real-Time Quantitative Reverse Transcription PCR (qRT-PCR)
2.11. RNA Interference
2.12. Mice
2.13. Study Approval
2.14. Measurement of Cyst Area
2.15. Quantitative BUN Determination
2.16. Statistics
3. Results
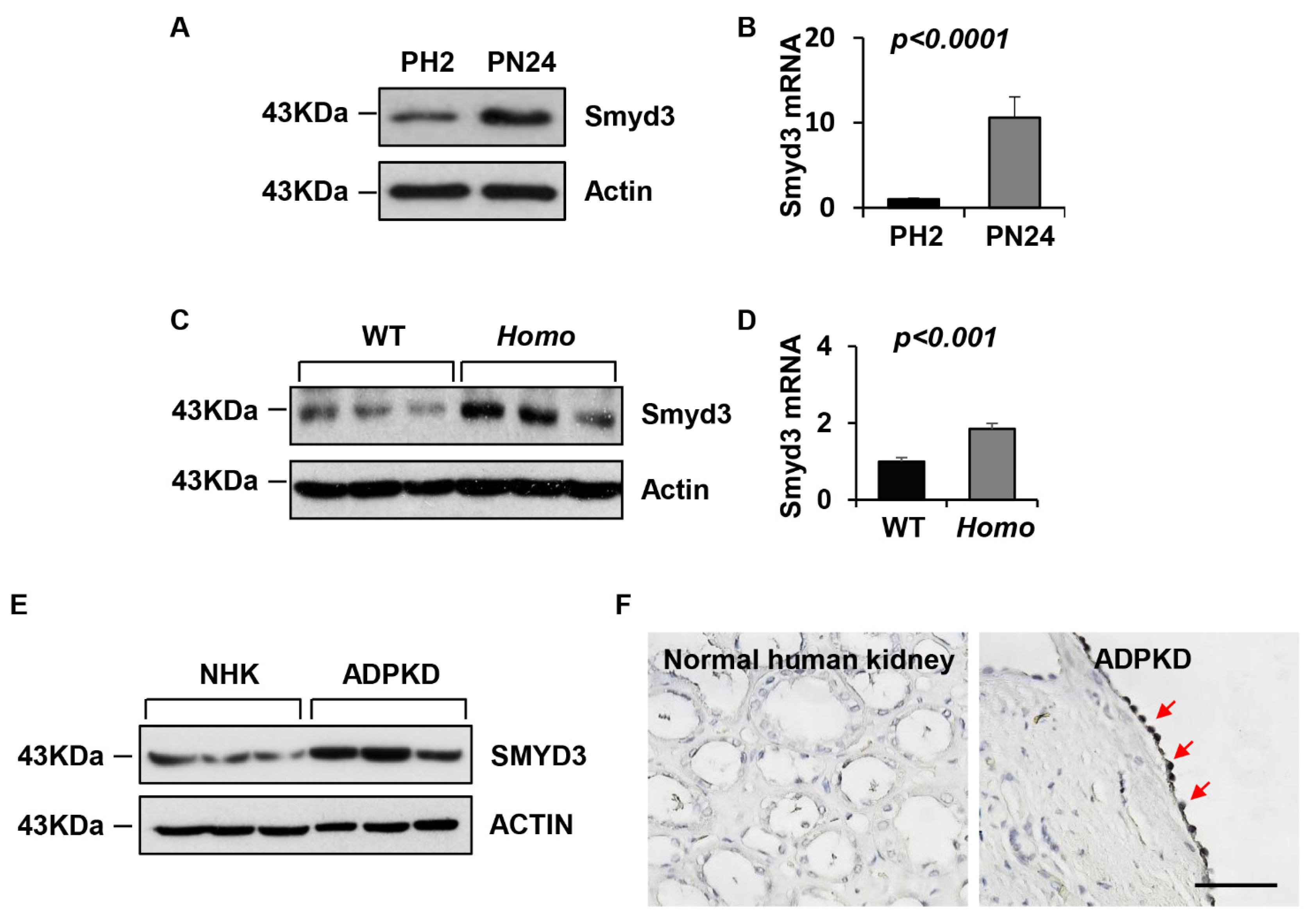
3.1. SMYD3 Is Upregulated in PKD1 Mutant Renal Epithelial Cells and ADPKD Tissues
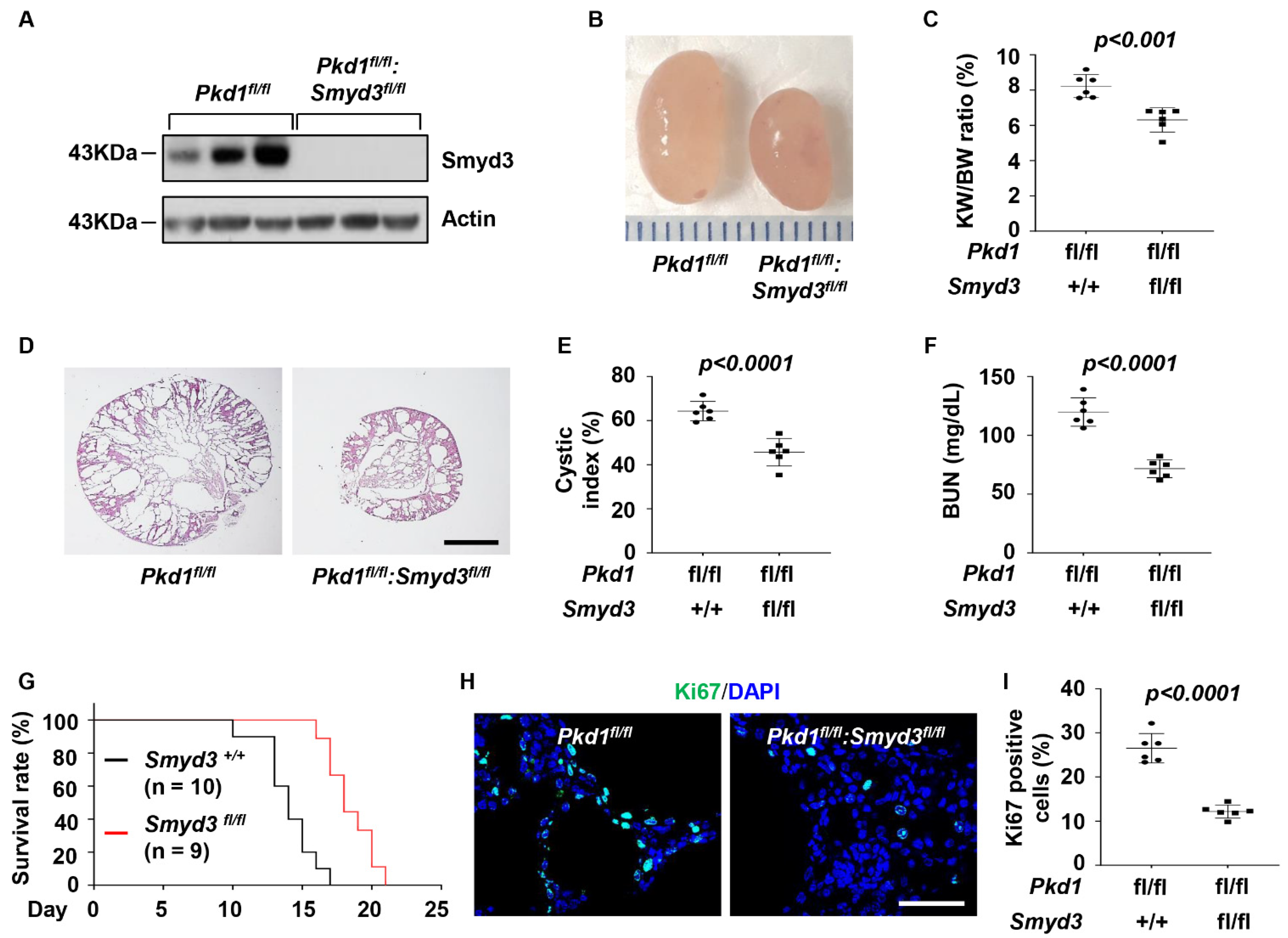
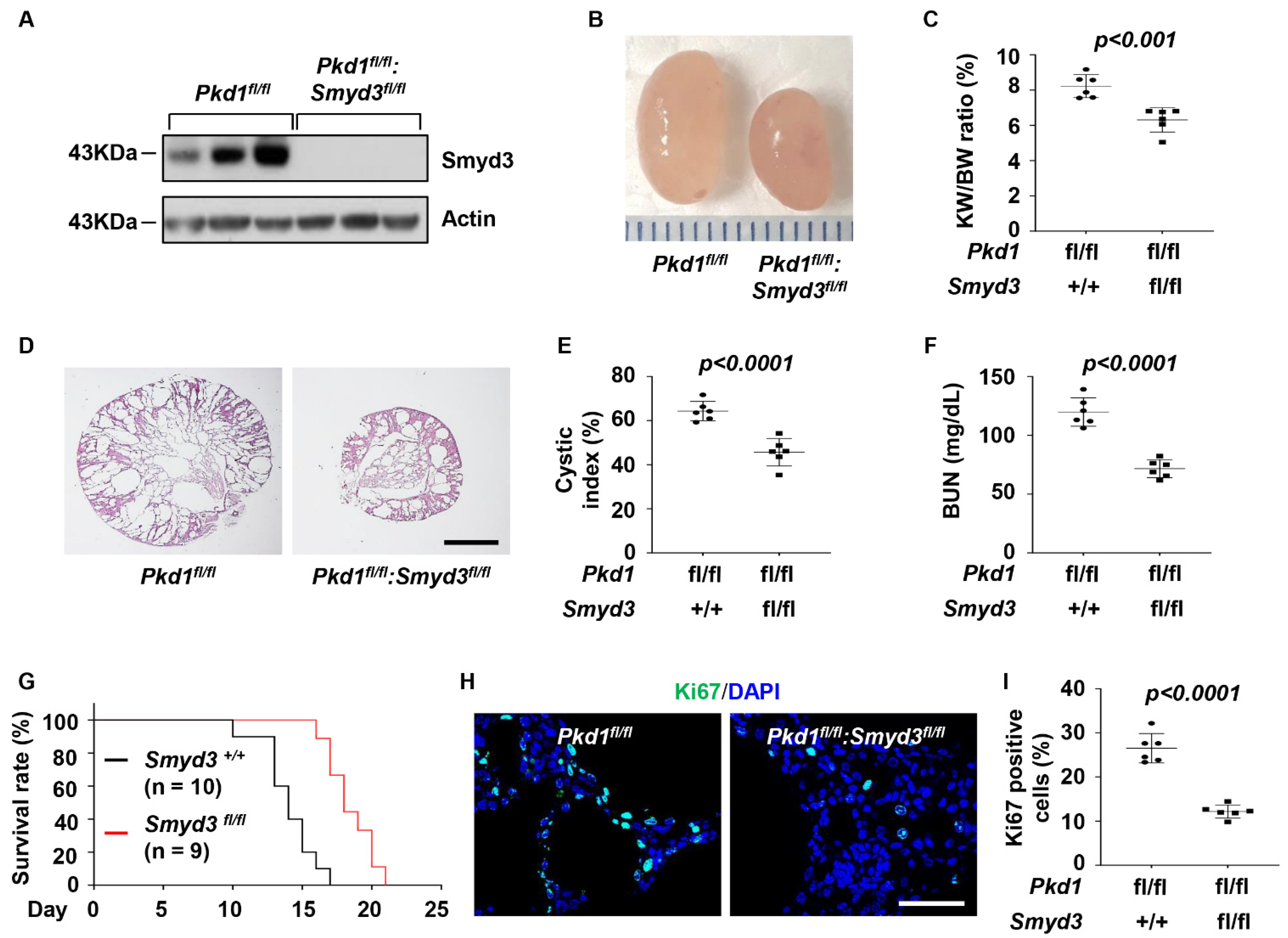
3.2. SMYD3 and PKD1 Double Conditional Knockout Delayed Renal Cyst Growth
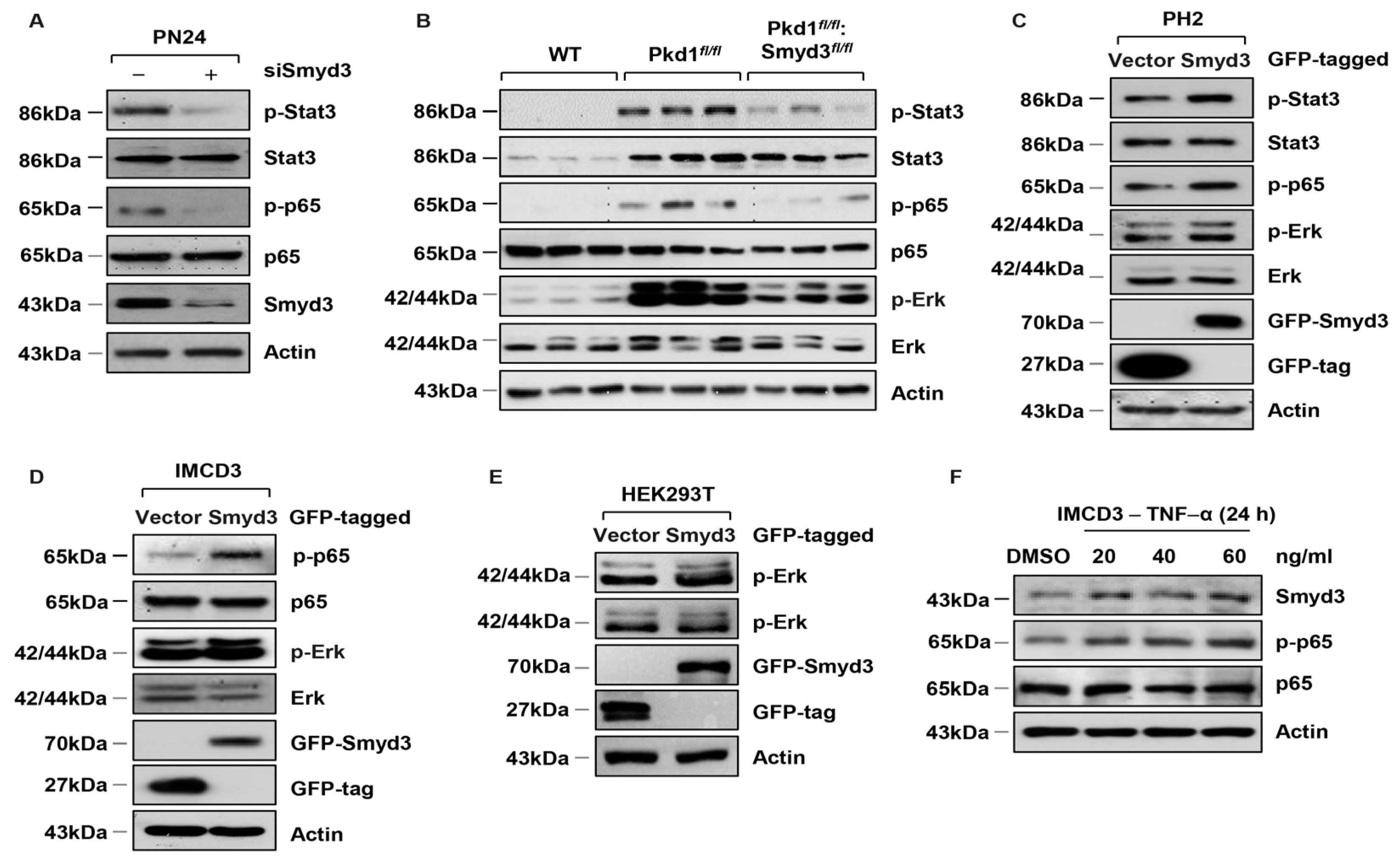
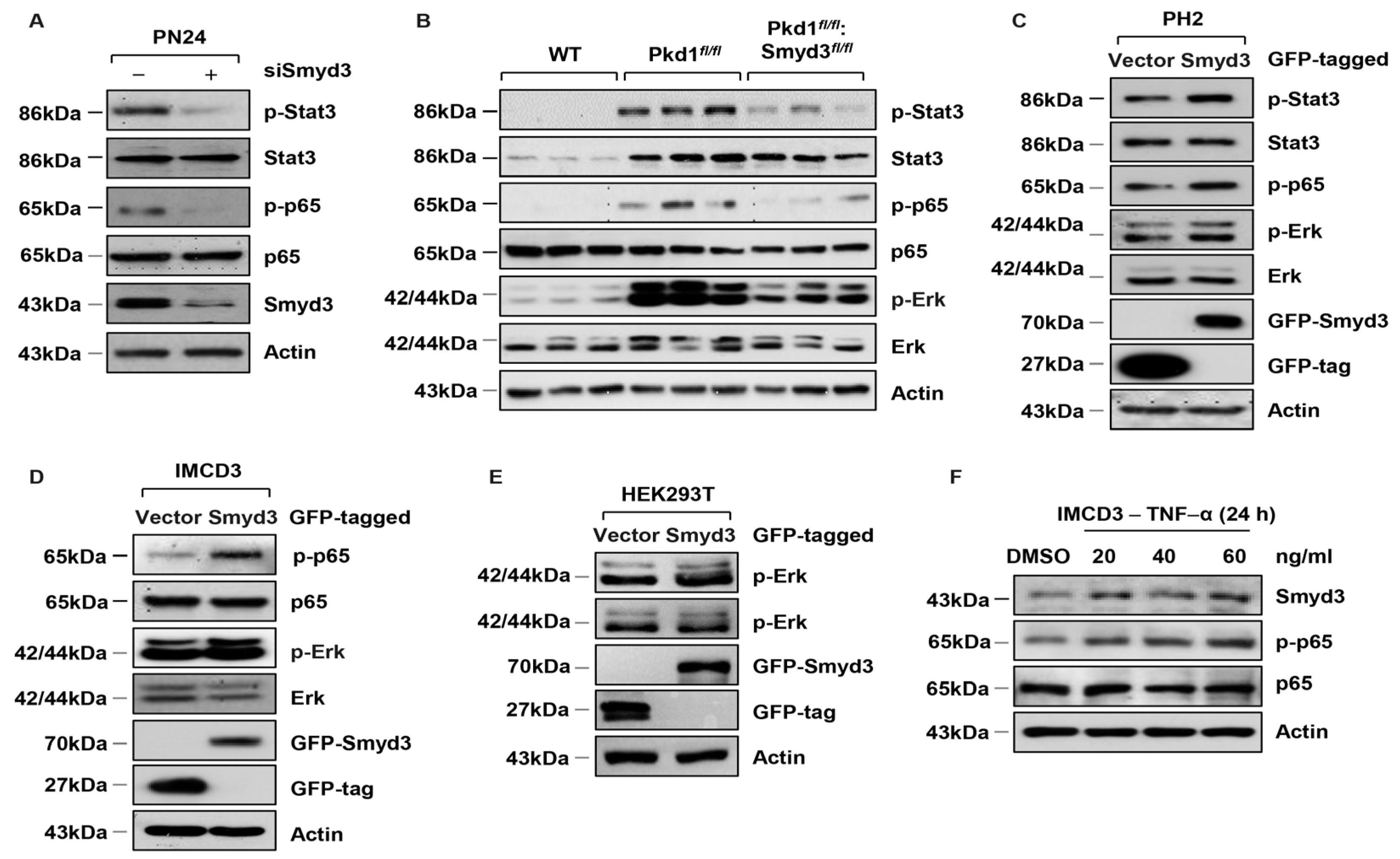
3.3. SMYD3 Regulates PKD-Associated Signaling Pathways in PKD1 Mutant Mouse Kidneys
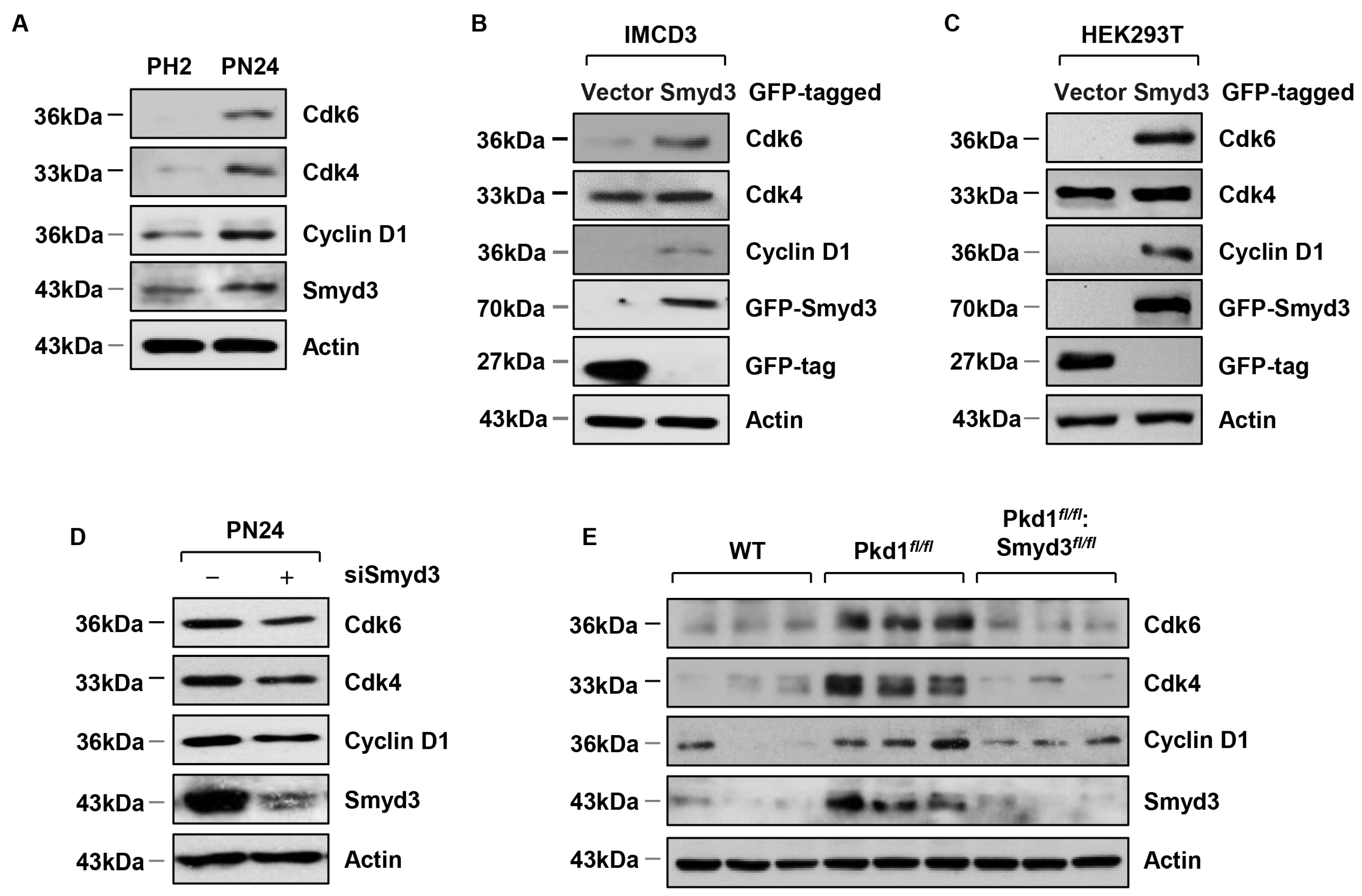
3.4. SMYD3 Regulates Cell Cycle Effectors
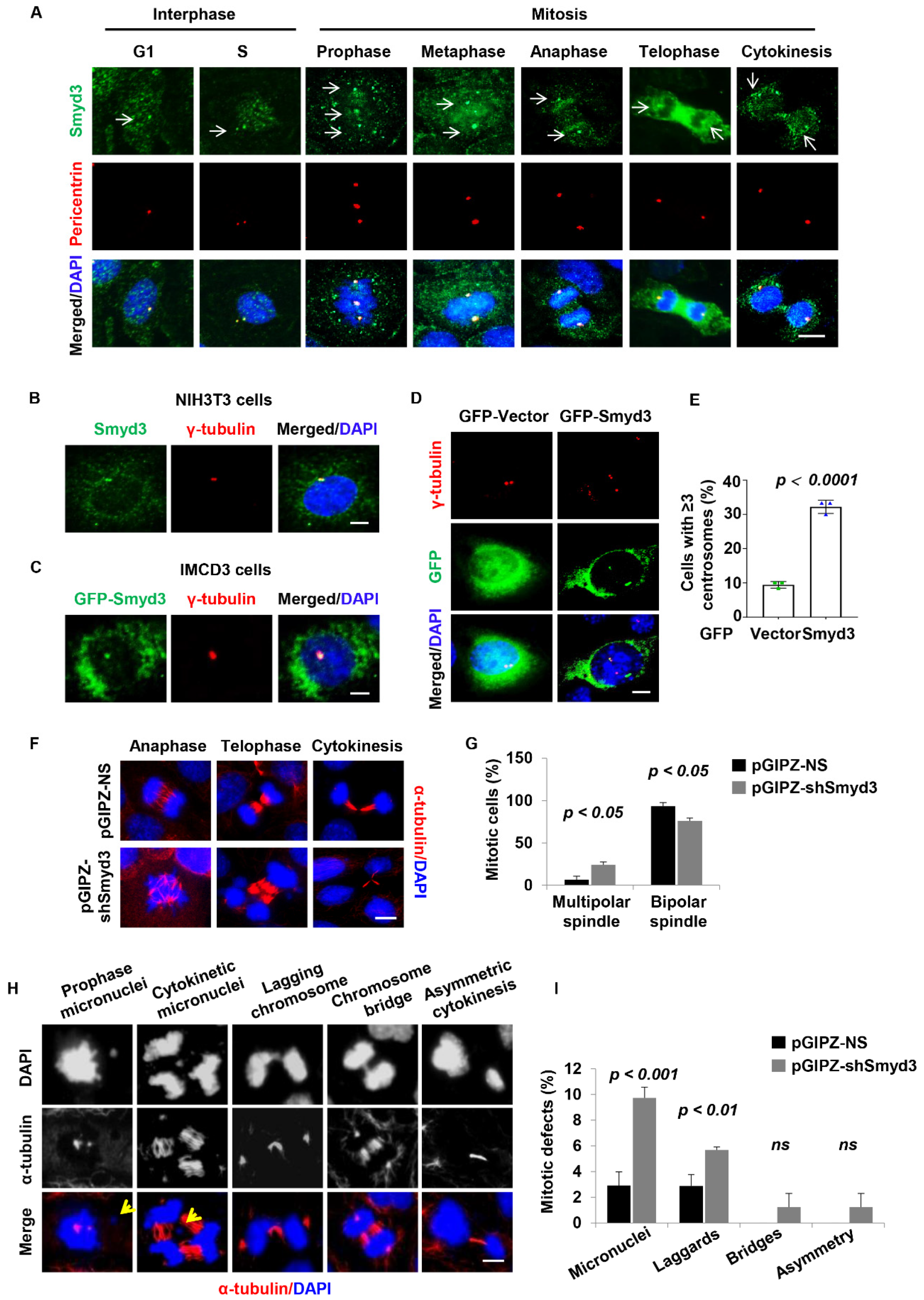
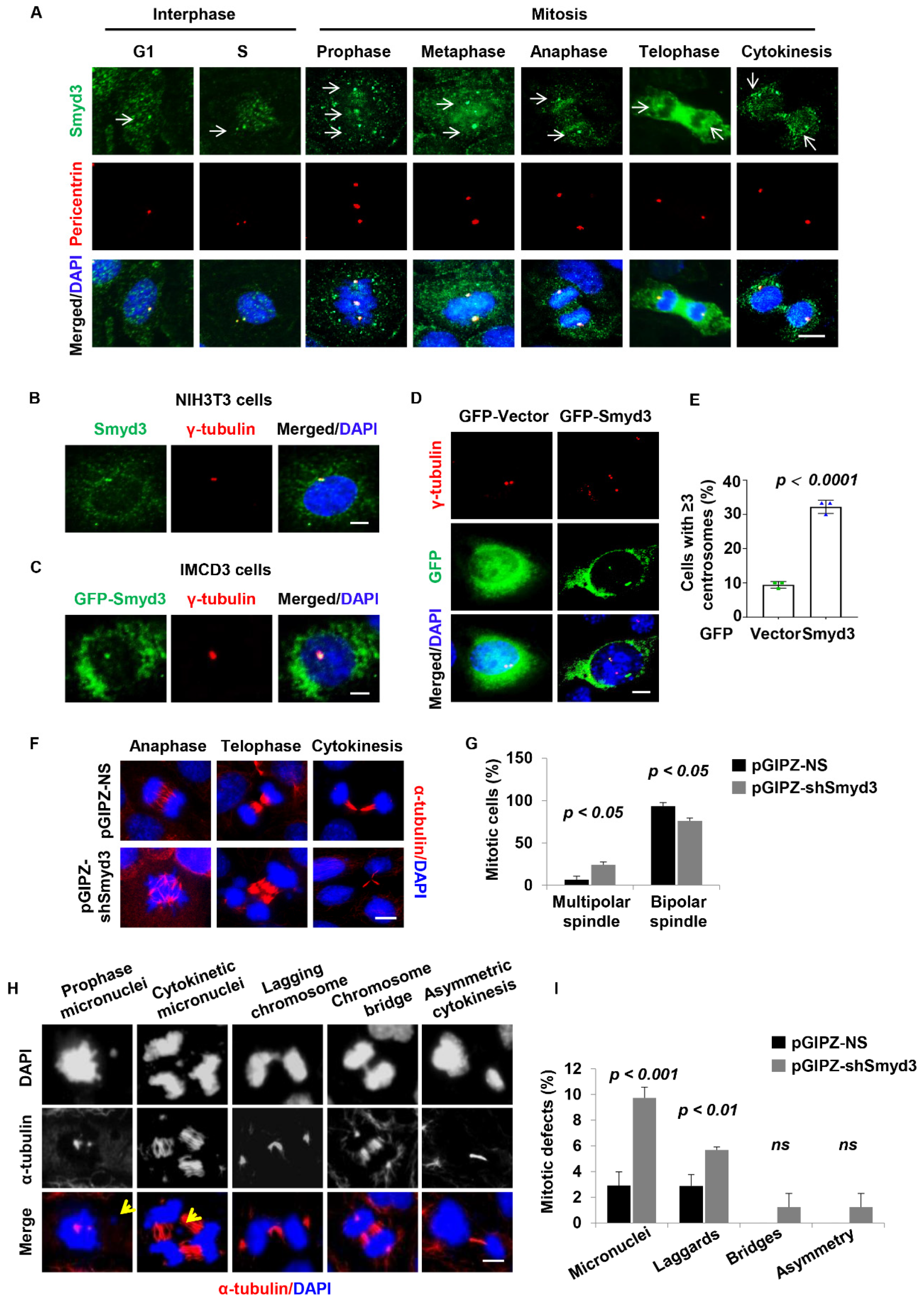
3.5. SMYD3 Is Located at the Centrosome and Regulates Centrosome Amplification
3.6. SMYD3 Is Required for Normal Mitosis and Cytokinesis
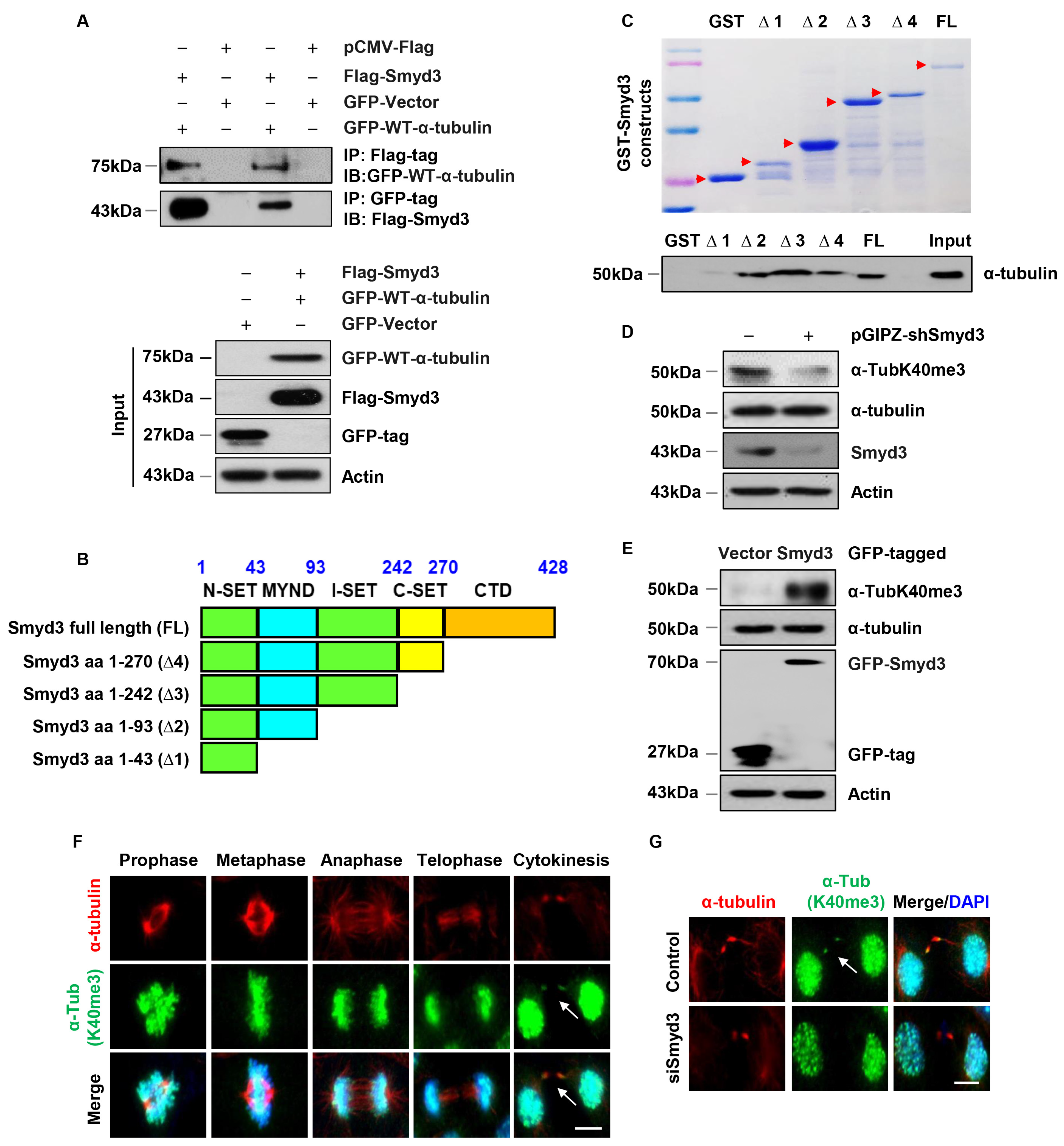
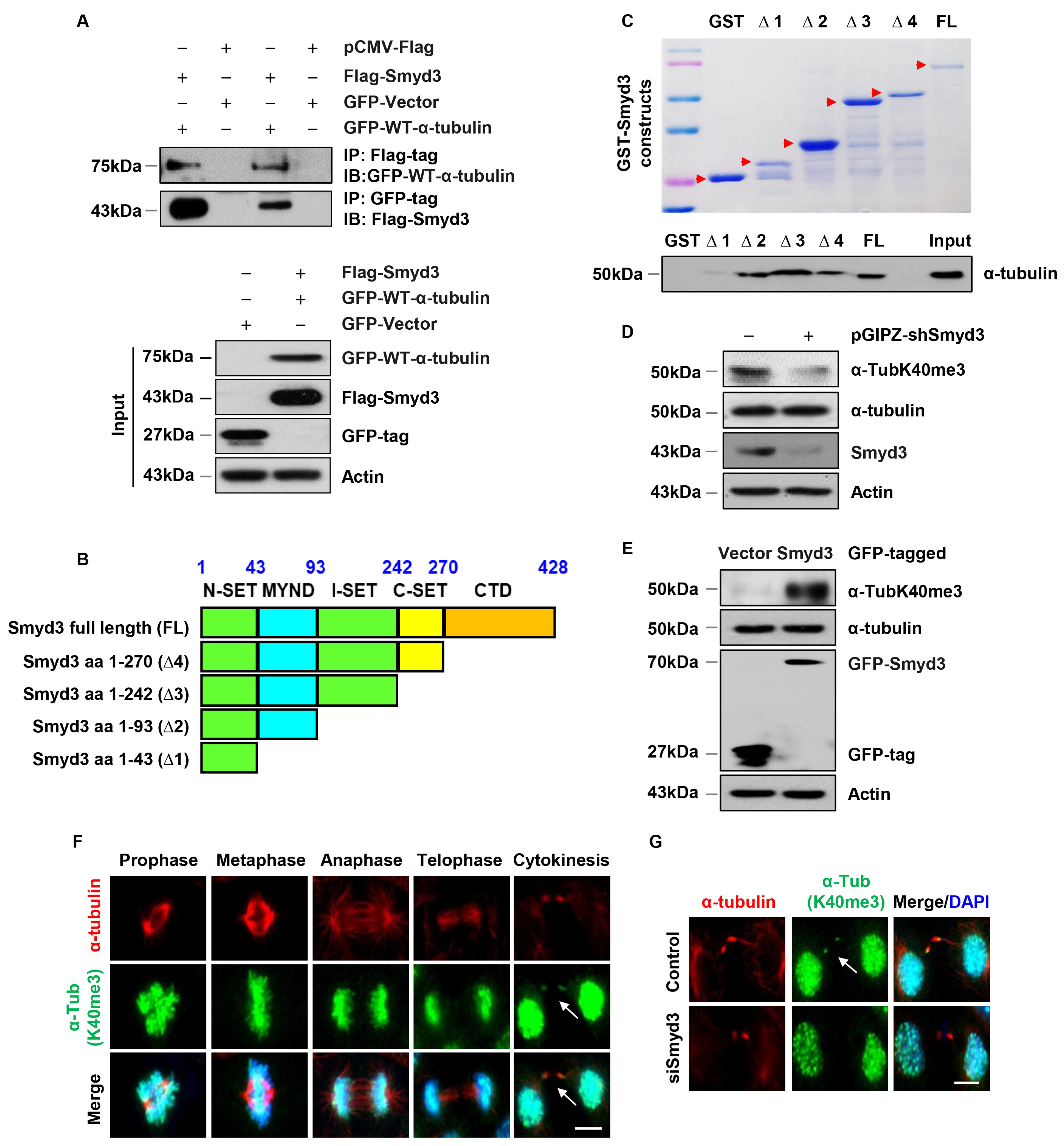
3.7. SMYD3 Interacts with Methylates α-Tubulin at Lysine 40
3.8. SMYD3 Is a Mitotic Microtubule Methyltransferase
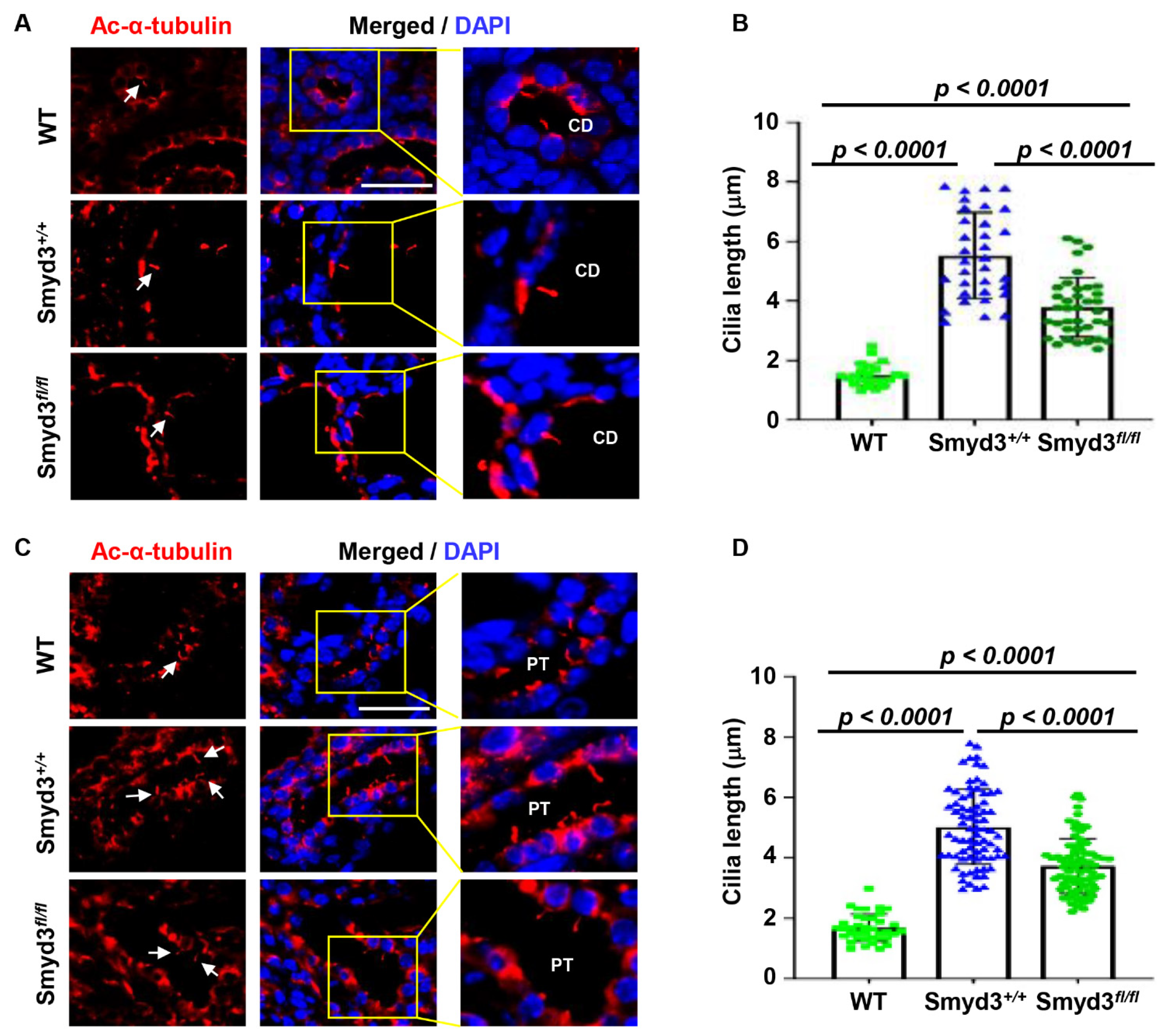
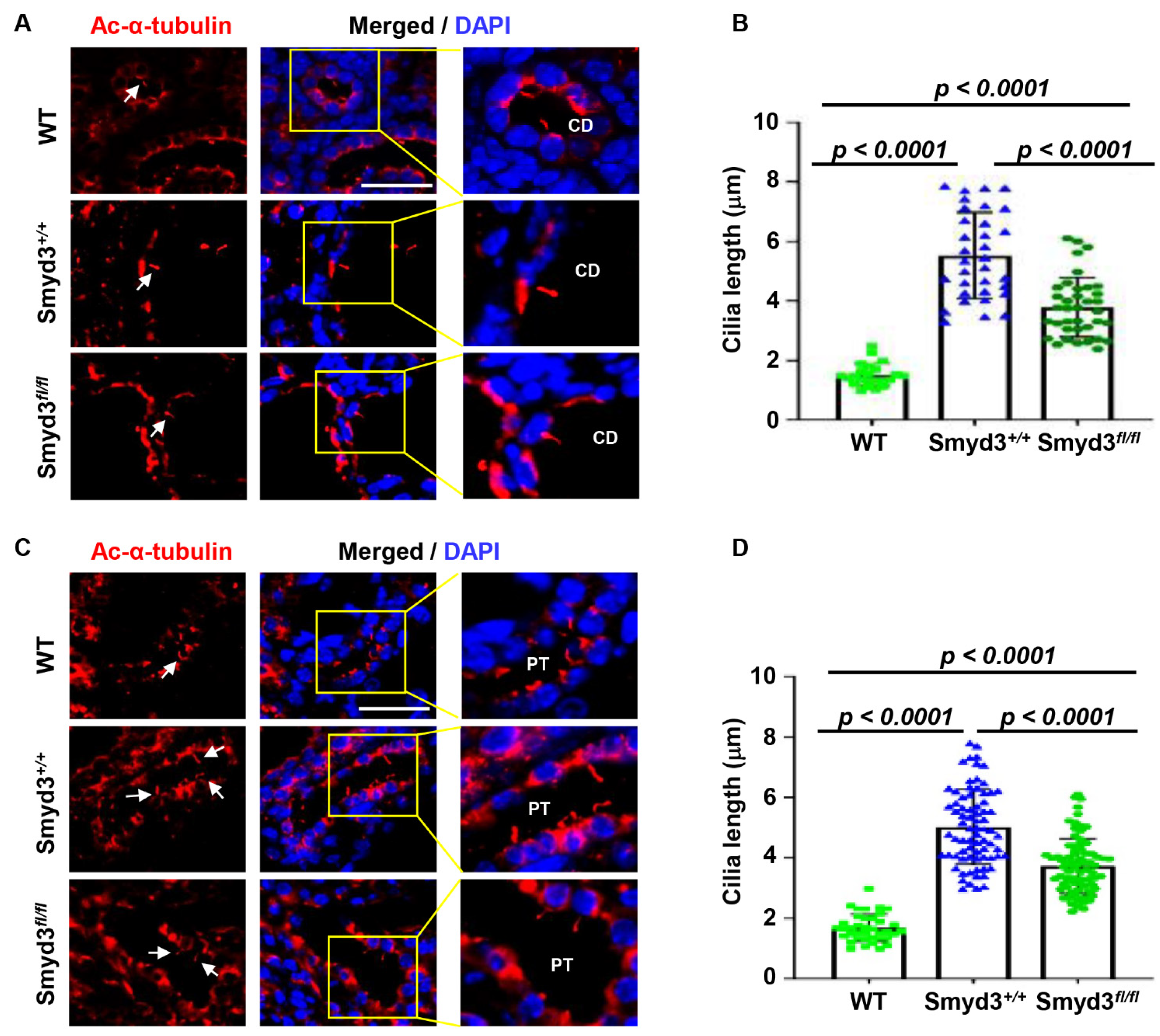
3.9. SMYD3 Regulates Primary Cilium Assembly in PKD1 Mutant Mouse Kidneys
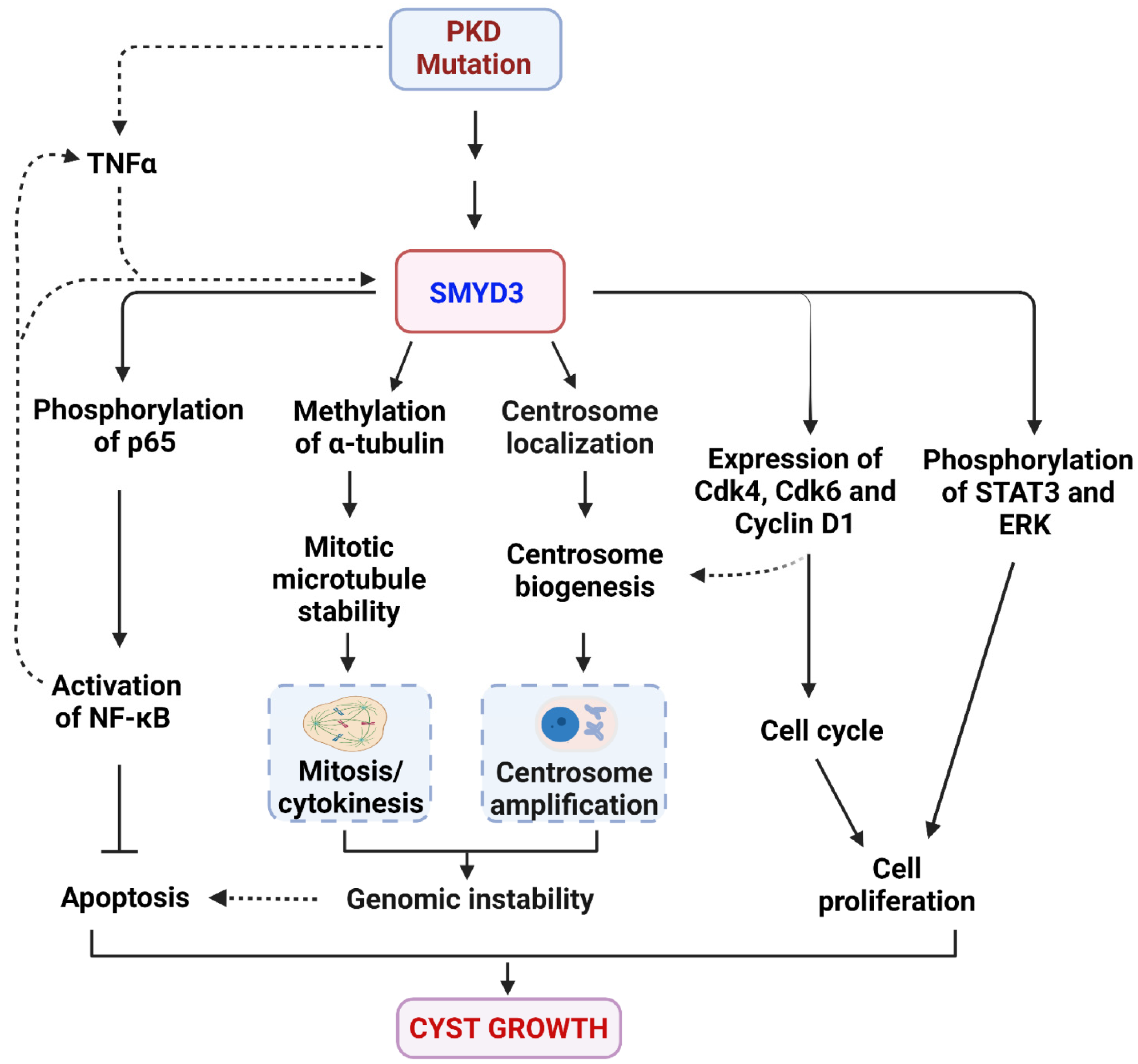
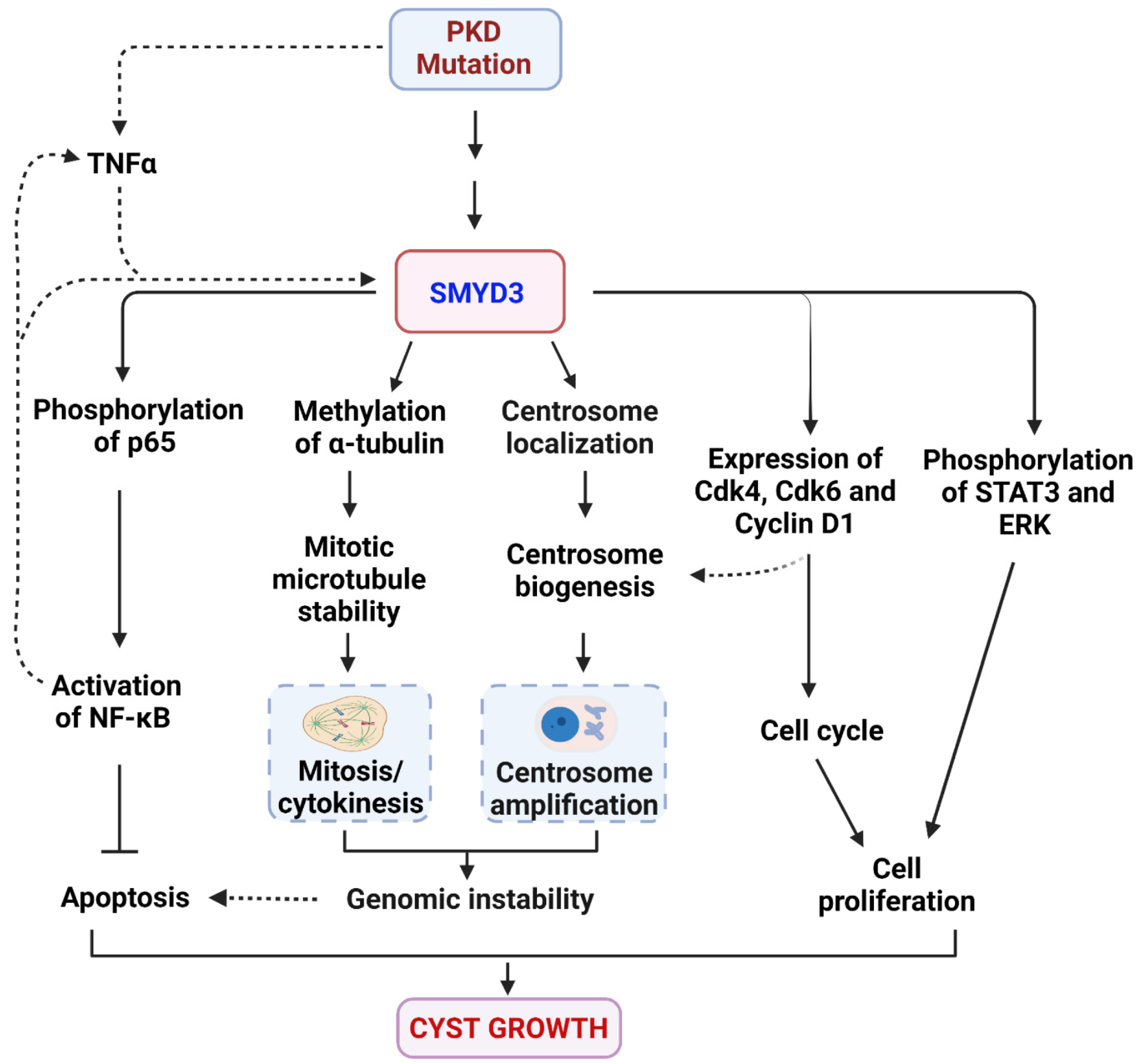
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rangan, G.K.; Tchan, M.C.; Tong, A.; Wong, A.T.; Nankivell, B.J. Recent advances in autosomal-dominant polycystic kidney disease. Intern. Med. J. 2016, 46, 883–892. [Google Scholar] [CrossRef]
- Ferreira, F.M.; Watanabe, E.H.; Onuchic, L.F. Polycystins and Molecular Basis of Autosomal Dominant Polycystic Kidney Disease. In Polycystic Kidney Disease; Li, X., Ed.; Codon Publications: Brisbane, Australia, 2015. [Google Scholar]
- Qian, F.; Watnick, T.J.; Onuchic, L.F.; Germino, G.G. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell 1996, 87, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Germino, G.G. Autosomal dominant polycystic kidney disease: A two-hit model. Hosp. Pract. 1997, 32, 81–102. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, S.; Burton, S.; Strmecki, L.; Pond, G.R.; San Millan, J.L.; Zerres, K.; Barratt, T.M.; Ozen, S.; Torres, V.E.; Bergstralh, E.J.; et al. The position of the polycystic kidney disease 1 (PKD1) gene mutation correlates with the severity of renal disease. J. Am. Soc. Nephrol. 2002, 13, 1230–1237. [Google Scholar] [CrossRef]
- Battini, L.; Macip, S.; Fedorova, E.; Dikman, S.; Somlo, S.; Montagna, C.; Gusella, G.L. Loss of polycystin-1 causes centrosome amplification and genomic instability. Hum. Mol. Genet. 2008, 17, 2819–2833. [Google Scholar] [CrossRef]
- Nigg, E.A. Centrosome aberrations: Cause or consequence of cancer progression? Nat. Rev. Cancer 2002, 2, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A.; Stearns, T. The centrosome cycle: Centriole biogenesis, duplication and inherent asymmetries. Nat. Cell Biol. 2011, 13, 1154–1160. [Google Scholar] [CrossRef]
- Nigg, E.A.; Holland, A.J. Once and only once: Mechanisms of centriole duplication and their deregulation in disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 297–312. [Google Scholar] [CrossRef]
- Fukasawa, K. Centrosome amplification, chromosome instability and cancer development. Cancer Lett. 2005, 230, 6–19. [Google Scholar] [CrossRef]
- Mauro, M.; Catanzaro, I.; Naselli, F.; Sciandrello, G.; Caradonna, F. Abnormal mitotic spindle assembly and cytokinesis induced by D-Limonene in cultured mammalian cells. Mutagenesis 2013, 28, 631–635. [Google Scholar] [CrossRef]
- Ganem, N.J.; Godinho, S.A.; Pellman, D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009, 460, 278–282. [Google Scholar] [CrossRef]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Valent, A.; Raslova, H.; Yakushijin, K.; Horne, D.; Feunteun, J.; Lenoir, G.; Medema, R.; et al. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene 2004, 23, 4362–4370. [Google Scholar] [CrossRef]
- Burtey, S.; Riera, M.; Ribe, E.; Pennenkamp, P.; Rance, R.; Luciani, J.; Dworniczak, B.; Mattei, M.G.; Fontes, M. Centrosome overduplication and mitotic instability in PKD2 transgenic lines. Cell Biol. Int. 2008, 32, 1193–1198. [Google Scholar] [CrossRef]
- Zhang, Q.H.; Taulman, P.D.; Yoder, B.K. Cystic kidney diseases: All roads lead to the cilium. Physiology 2004, 19, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Badano, J.L.; Teslovich, T.M.; Katsanis, N. The centrosome in human genetic disease. Nat. Rev. Genet. 2005, 6, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Jonassen, J.A.; Agustin, J.S.; Follit, J.A.; Pazour, G.J. Deletion of IFT20 in the mouse kidney causes misorientation of the mitotic spindle and cystic kidney disease. J. Cell Biol. 2008, 183, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Hiesberger, T.; Cordes, K.; Sinclair, A.M.; Goldstein, L.S.; Somlo, S.; Igarashi, P. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2003, 100, 5286–5291. [Google Scholar] [CrossRef]
- Yoder, B.K.; Hou, X.; Guay-Woodford, L.M. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J. Am. Soc. Nephrol. 2002, 13, 2508–2516. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Somlo, S. Cyst growth, polycystins, and primary cilia in autosomal dominant polycystic kidney disease. Kidney Res. Clin. Pract. 2014, 33, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Duldulao, N.A.; Lee, S.; Sun, Z. Cilia localization is essential for in vivo functions of the Joubert syndrome protein Arl13b/Scorpion. Development 2009, 136, 4033–4042. [Google Scholar] [CrossRef]
- Cantagrel, V.; Silhavy, J.L.; Bielas, S.L.; Swistun, D.; Marsh, S.E.; Bertrand, J.Y.; Audollent, S.; Attie-Bitach, T.; Holden, K.R.; Dobyns, W.B.; et al. Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am. J. Hum. Genet. 2008, 83, 170–179. [Google Scholar] [CrossRef]
- Ma, M.; Tian, X.; Igarashi, P.; Pazour, G.J.; Somlo, S. Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat. Genet. 2013, 45, 1004–1012. [Google Scholar] [CrossRef]
- Shao, L.; El-Jouni, W.; Kong, F.; Ramesh, J.; Kumar, R.S.; Shen, X.; Ren, J.; Devendra, S.; Dorschel, A.; Wu, M.; et al. Genetic reduction of cilium length by targeting intraflagellar transport 88 protein impedes kidney and liver cyst formation in mouse models of autosomal polycystic kidney disease. Kidney Int. 2020, 98, 1225–1241. [Google Scholar] [CrossRef]
- Zhou, X.; Fan, L.X.; Sweeney, W.E., Jr.; Denu, J.M.; Avner, E.D.; Li, X. Sirtuin 1 inhibition delays cyst formation in autosomal-dominant polycystic kidney disease. J. Clin. Investig. 2013, 123, 3084–3098. [Google Scholar] [CrossRef] [PubMed]
- Li, L.X.; Fan, L.X.; Zhou, J.X.; Grantham, J.J.; Calvet, J.P.; Sage, J.; Li, X.G. Lysine methyltransferase SMYD2 promotes cyst growth in autosomal dominant polycystic kidney disease. J. Clin. Investig. 2017, 127, 2751–2764. [Google Scholar] [CrossRef] [PubMed]
- Tracy, C.; Warren, J.S.; Szulik, M.; Wang, L.; Garcia, J.; Makaju, A.; Russell, K.; Miller, M.; Franklin, S. The Smyd Family of Methyltransferases: Role in Cardiac and Skeletal Muscle Physiology and Pathology. Curr. Opin. Physiol. 2018, 1, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Liu, Y.; Chen, Q.; Yang, J.; Jiang, Z.; Zhang, H.; Liu, Z.; Jin, B. Expression patterns and the prognostic value of the SMYD family members in human breast carcinoma using integrative bioinformatics analysis. Oncol. Lett. 2019, 17, 3851–3861. [Google Scholar] [CrossRef] [PubMed]
- Asuthkar, S.; Venkataraman, S.; Avilala, J.; Shishido, K.; Vibhakar, R.; Veo, B.; Purvis, I.J.; Guda, M.R.; Velpula, K.K. SMYD3 Promotes Cell Cycle Progression by Inducing Cyclin D3 Transcription and Stabilizing the Cyclin D1 Protein in Medulloblastoma. Cancers 2022, 14, 1673. [Google Scholar] [CrossRef] [PubMed]
- Bernard, B.J.; Nigam, N.; Burkitt, K.; Saloura, V. SMYD3: A regulator of epigenetic and signaling pathways in cancer. Clin. Epigenetics 2021, 13, 45. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Tsai, C.H.; Wang, P.Y.; Teng, S.C. SMYD3 Promotes Homologous Recombination via Regulation of H3K4-mediated Gene Expression. Sci. Rep. 2017, 7, 3842. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, R.; Furukawa, Y.; Morita, M.; Iimura, Y.; Silva, F.P.; Li, M.; Yagyu, R.; Nakamura, Y. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat. Cell Biol. 2004, 6, 731–740. [Google Scholar] [CrossRef]
- Van Aller, G.S.; Reynoird, N.; Barbash, O.; Huddleston, M.; Liu, S.; Zmoos, A.F.; McDevitt, P.; Sinnamon, R.; Le, B.; Mas, G.; et al. Smyd3 regulates cancer cell phenotypes and catalyzes histone H4 lysine 5 methylation. Epigenetics 2012, 7, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Foreman, K.W.; Brown, M.; Park, F.; Emtage, S.; Harriss, J.; Das, C.; Zhu, L.; Crew, A.; Arnold, L.; Shaaban, S.; et al. Structural and functional profiling of the human histone methyltransferase SMYD3. PLoS ONE 2011, 6, e22290. [Google Scholar] [CrossRef] [PubMed]
- Mazur, P.K.; Reynoird, N.; Khatri, P.; Jansen, P.W.T.C.; Wilkinson, A.W.; Liu, S.C.; Barbash, O.; Van Aller, G.S.; Huddleston, M.; Dhanak, D.; et al. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature 2014, 510, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Kunizaki, M.; Hamamoto, R.; Silva, F.P.; Yamaguchi, K.; Nagayasu, T.; Shibuya, M.; Nakamura, Y.; Furukawa, Y. The lysine 831 of vascular endothelial growth factor receptor 1 is a novel target of methylation by SMYD3. Cancer Res. 2007, 67, 10759–10765. [Google Scholar] [CrossRef]
- Shibazaki, S.; Yu, Z.; Nishio, S.; Tian, X.; Thomson, R.B.; Mitobe, M.; Louvi, A.; Velazquez, H.; Ishibe, S.; Cantley, L.G.; et al. Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum. Mol. Genet. 2008, 17, 1505–1516. [Google Scholar] [CrossRef]
- Li, L.X.Y.; Zhou, J.X.; Wang, X.D.; Zhang, H.B.; Harris, P.C.; Calvet, J.P.; Li, X.G. Cross-talk between CDK4/6 and SMYD2 regulates gene transcription, tubulin methylation, and ciliogenesis. Sci. Adv. 2020, 6, eabb3154. [Google Scholar] [CrossRef]
- Agborbesong, E.; Zhou, J.X.; Li, L.X.; Calvet, J.P.; Li, X.G. Antioxidant enzyme peroxiredoxin 5 regulates cyst growth and ciliogenesis via modulating Plk1 stability. FASEB J. 2022, 36, e22089. [Google Scholar] [CrossRef]
- Shao, X.; Somlo, S.; Igarashi, P. Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J. Am. Soc. Nephrol. 2002, 13, 1837–1846. [Google Scholar] [CrossRef]
- Adon, A.M.; Zeng, X.; Harrison, M.K.; Sannem, S.; Kiyokawa, H.; Kaldis, P.; Saavedra, H.I. Cdk2 and Cdk4 regulate the centrosome cycle and are critical mediators of centrosome amplification in p53-null cells. Mol. Cell. Biol. 2010, 30, 694–710. [Google Scholar] [CrossRef]
- Park, I.Y.; Powell, R.T.; Tripathi, D.N.; Dere, R.; Ho, T.H.; Blasius, T.L.; Chiang, Y.C.; Davis, I.J.; Fahey, C.C.; Hacker, K.E.; et al. Dual Chromatin and Cytoskeletal Remodeling by SETD2. Cell 2016, 166, 950–962. [Google Scholar] [CrossRef] [PubMed]
- Li, X.G.; Magenheimer, B.S.; Xia, S.; Johnson, T.; Wallace, D.P.; Calvet, J.P.; Li, R. A tumor necrosis factor-alpha-mediated pathway promoting autosomal dominant polycystic kidney disease. Nat. Med. 2008, 14, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Jusino, S.; Fernandez-Padin, F.M.; Saavedra, H.I. Centrosome aberrations and chromosome instability contribute to tumorigenesis and intra-tumor heterogeneity. J. Cancer Metastasis Treat. 2018, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- Leontovich, A.A.; Salisbury, J.L.; Veroux, M.; Tallarita, T.; Billadeau, D.; McCubrey, J.; Ingle, J.; Galanis, E.; D’Assoro, A.B. Inhibition of Cdk2 activity decreases Aurora-A kinase centrosomal localization and prevents centrosome amplification in breast cancer cells. Oncol. Rep. 2013, 29, 1785–1788. [Google Scholar] [CrossRef]
- Ma, M.; Gallagher, A.R.; Somlo, S. Ciliary Mechanisms of Cyst Formation in Polycystic Kidney Disease. Cold Spring Harb. Perspect. Biol. 2017, 9, a028209. [Google Scholar] [CrossRef]
- Walker, R.V.; Maranto, A.; Palicharla, V.R.; Hwang, S.H.; Mukhopadhyay, S.; Qian, F. Cilia-Localized Counterregulatory Signals as Drivers of Renal Cystogenesis. Front. Mol. Biosci. 2022, 9, 936070. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agborbesong, E.; Zhou, J.X.; Zhang, H.; Li, L.X.; Harris, P.C.; Calvet, J.P.; Li, X. Overexpression of SMYD3 Promotes Autosomal Dominant Polycystic Kidney Disease by Mediating Cell Proliferation and Genome Instability. Biomedicines 2024, 12, 603. https://doi.org/10.3390/biomedicines12030603
Agborbesong E, Zhou JX, Zhang H, Li LX, Harris PC, Calvet JP, Li X. Overexpression of SMYD3 Promotes Autosomal Dominant Polycystic Kidney Disease by Mediating Cell Proliferation and Genome Instability. Biomedicines. 2024; 12(3):603. https://doi.org/10.3390/biomedicines12030603
Chicago/Turabian StyleAgborbesong, Ewud, Julie Xia Zhou, Hongbing Zhang, Linda Xiaoyan Li, Peter C. Harris, James P. Calvet, and Xiaogang Li. 2024. "Overexpression of SMYD3 Promotes Autosomal Dominant Polycystic Kidney Disease by Mediating Cell Proliferation and Genome Instability" Biomedicines 12, no. 3: 603. https://doi.org/10.3390/biomedicines12030603
APA StyleAgborbesong, E., Zhou, J. X., Zhang, H., Li, L. X., Harris, P. C., Calvet, J. P., & Li, X. (2024). Overexpression of SMYD3 Promotes Autosomal Dominant Polycystic Kidney Disease by Mediating Cell Proliferation and Genome Instability. Biomedicines, 12(3), 603. https://doi.org/10.3390/biomedicines12030603