
The Optic Nerve at Stake: Update on Environmental Factors Modulating Expression of Leber’s Hereditary Optic Neuropathy

 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
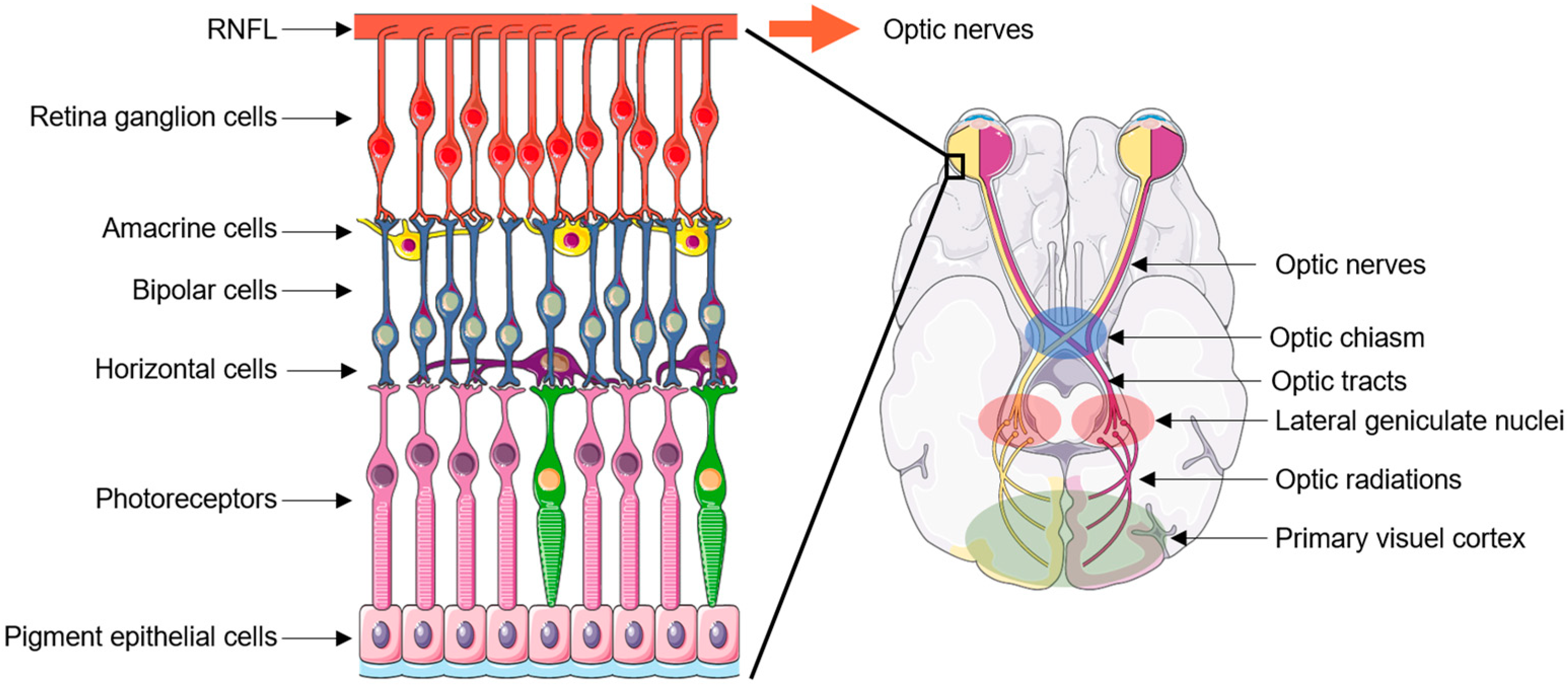
2. Neurodegeneration Pattern
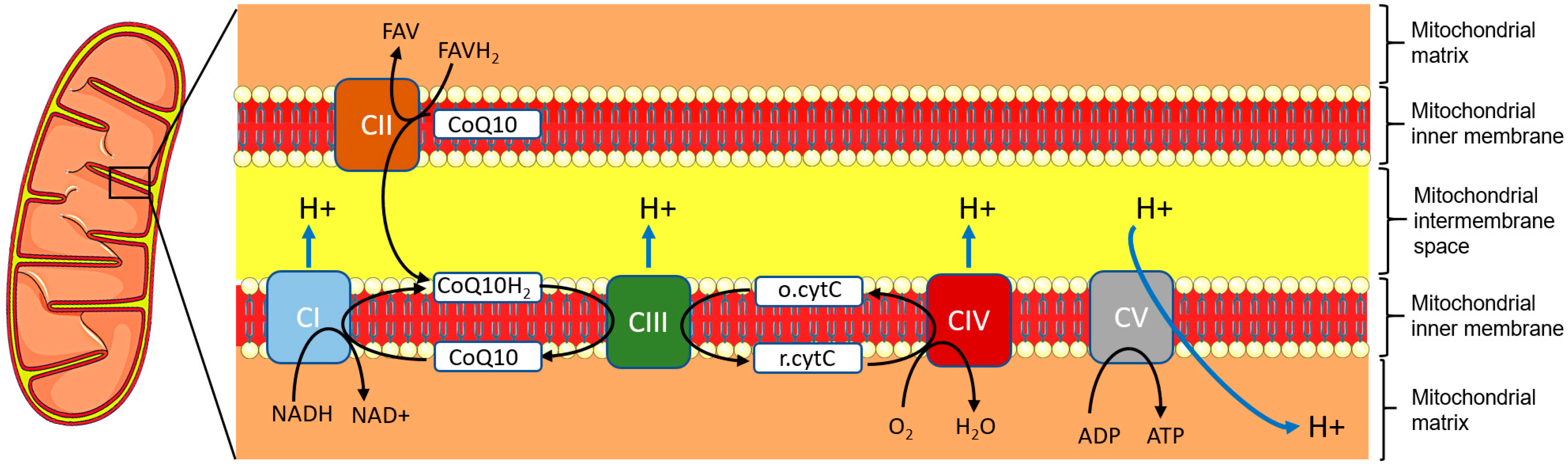
3. Genetics and Pathophysiology
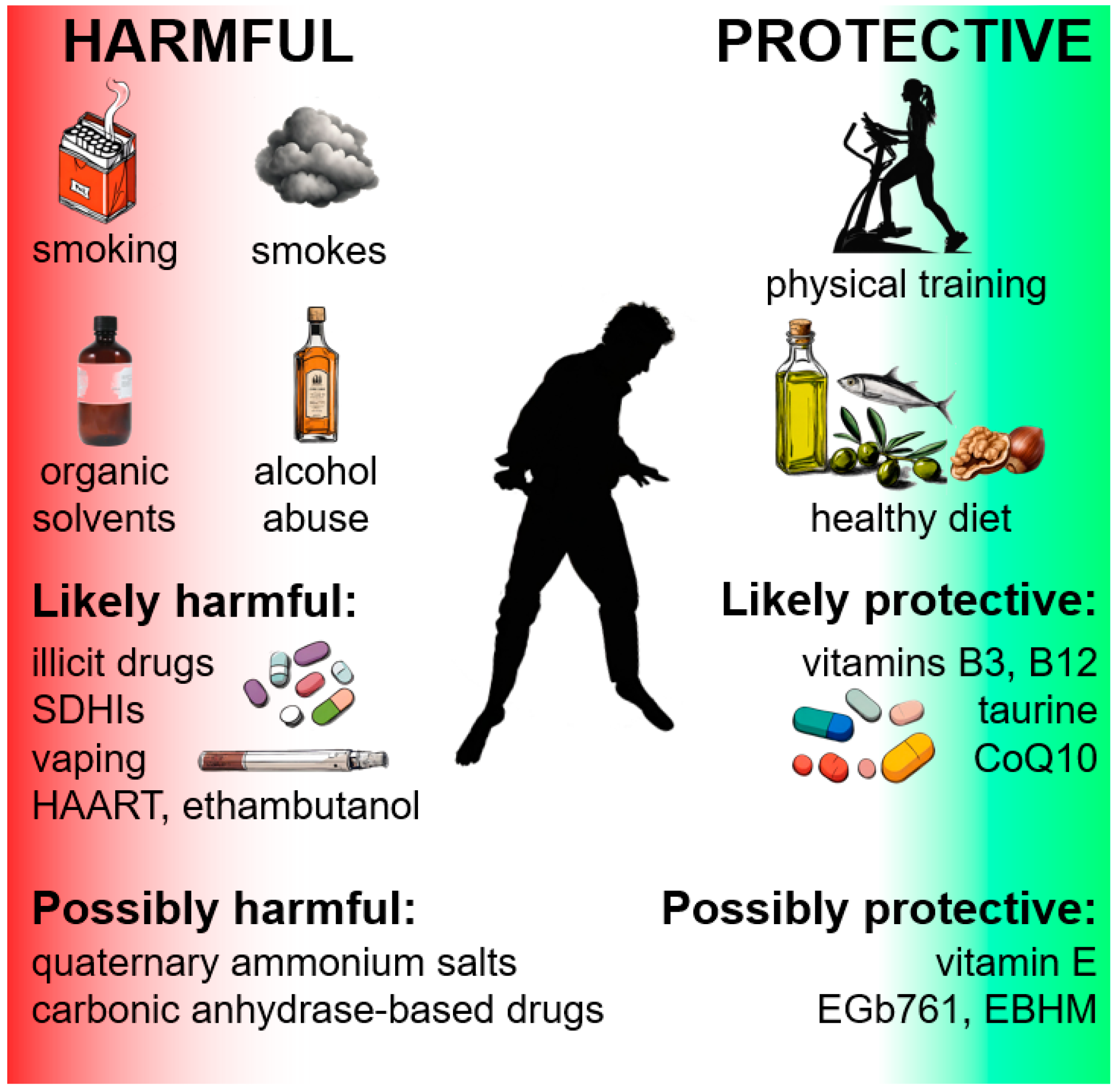
4. Environmental Protective Factors
4.1. Diet
4.2. Food Supplements or Inputs
4.3. Exercise
5. Environmental Risk Factors
5.1. Smoke, Smoking, and Vaping
5.2. Alcohol Abuse
5.3. Illicit Drug Abuse
5.4. Organic Solvents
5.5. ETC Inhibitors in Food
5.6. Quaternary Ammonium Salts
6. Drugs
6.1. Authorized or Candidate Therapeutic Drugs for LHON
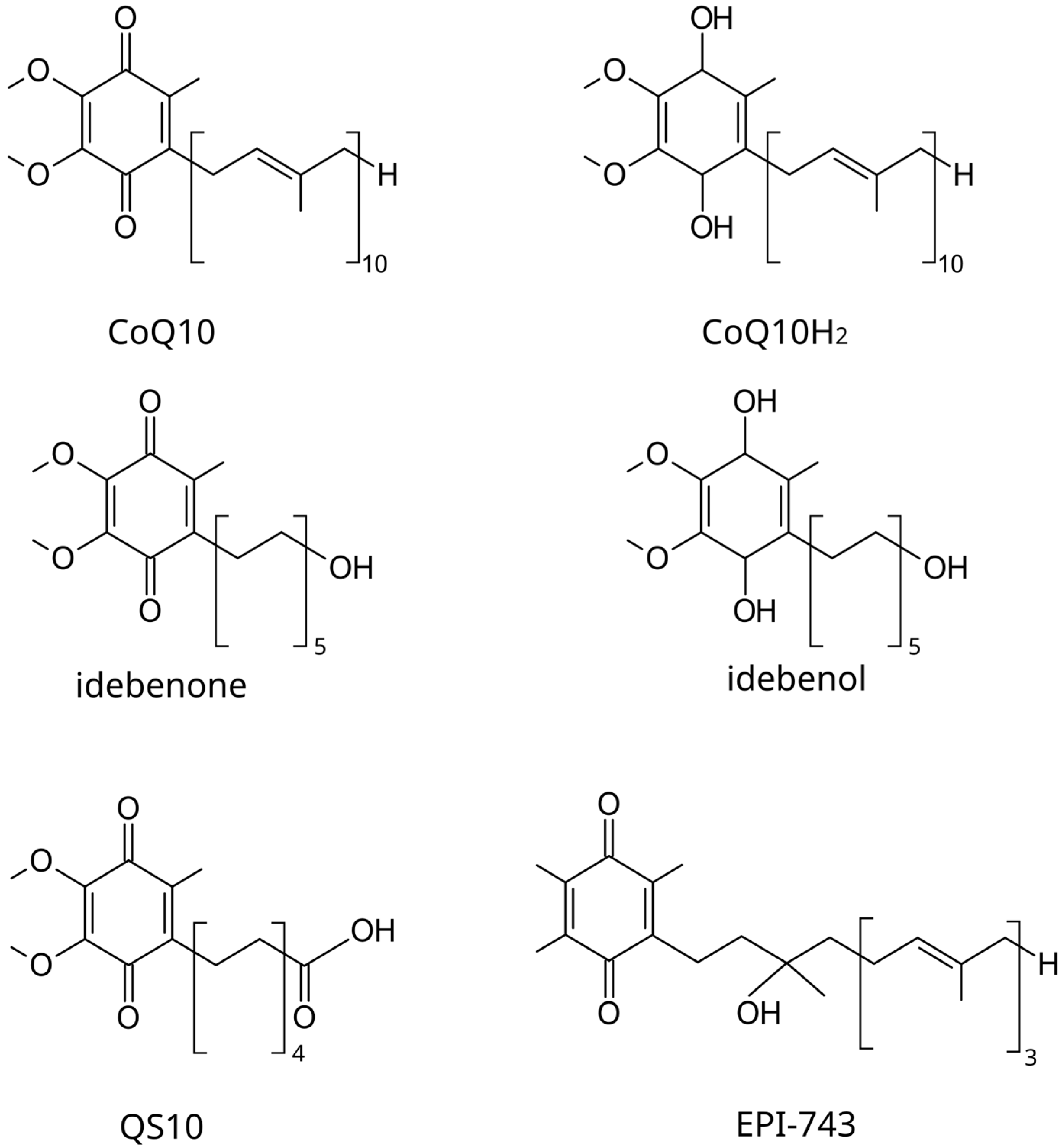
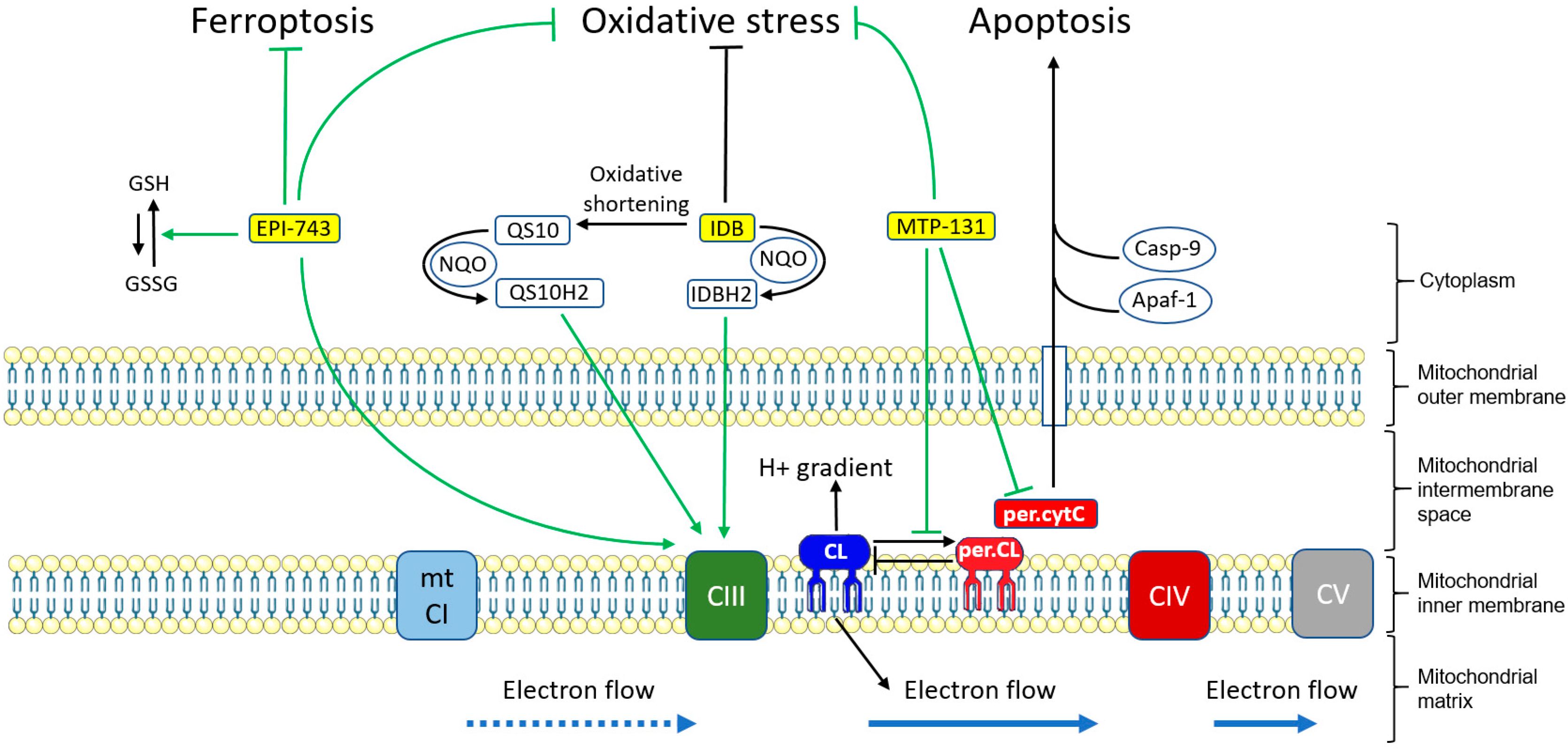
6.1.1. Idebenone
6.1.2. EPI-743
6.1.3. Elamipretide
6.2. Other Drugs with Possibly Adverse Outcomes on the Optic Nerve
7. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hayreh, S.S. Ischemic Optic Neuropathy. Prog. Retin. Eye Res. 2009, 28, 34–62. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhang, H.; Zhai, Q.; Li, H.; Wang, C.; Wang, Y. Traumatic Optic Neuropathy: A Review of Current Studies. Neurosurg. Rev. 2022, 45, 1895–1913. [Google Scholar] [CrossRef] [PubMed]
- Baj, J.; Forma, A.; Kobak, J.; Tyczyńska, M.; Dudek, I.; Maani, A.; Teresiński, G.; Buszewicz, G.; Januszewski, J.; Flieger, J. Toxic and Nutritional Optic Neuropathies—An Updated Mini-Review. Int. J. Environ. Res. Public Health 2022, 19, 3092. [Google Scholar] [CrossRef] [PubMed]
- Bourne, R.R.A.; Steinmetz, J.D.; Saylan, M.; Mersha, A.M.; Weldemariam, A.H.; Wondmeneh, T.G.; Sreeramareddy, C.T.; Pinheiro, M.; Yaseri, M.; Yu, C.; et al. Causes of Blindness and Vision Impairment in 2020 and Trends over 30 Years, and Prevalence of Avoidable Blindness in Relation to VISION 2020: The Right to Sight: An Analysis for the Global Burden of Disease Study. Lancet Glob. Health 2021, 9, e144. [Google Scholar] [CrossRef]
- Carelli, V.; La Morgia, C.; Ross-Cisneros, F.N.; Sadun, A.A. Optic Neuropathies: The Tip of the Neurodegeneration Iceberg. Hum. Mol. Genet. 2017, 26, R139–R150. [Google Scholar] [CrossRef] [PubMed]
- Hage, R.; Vignal-Clermont, C. Leber Hereditary Optic Neuropathy: Review of Treatment and Management. Front. Neurol. 2021, 12, 651639. [Google Scholar] [CrossRef]
- Lenaers, G.; Beaulieu, C.; Charif, M.; Gerber, S.; Kaplan, J.; Rozet, J.-M. Autosomal Recessive Leber Hereditary Optic Neuropathy, a New Neuro-Ophthalmo-Genetic Paradigm. Brain 2023, 146, 3156–3161. [Google Scholar] [CrossRef] [PubMed]
- Catarino, C.B.; von Livonius, B.; Priglinger, C.; Banik, R.; Matloob, S.; Tamhankar, M.A.; Castillo, L.; Friedburg, C.; Halfpenny, C.A.; Lincoln, J.A.; et al. Real-World Clinical Experience with Idebenone in the Treatment of Leber Hereditary Optic Neuropathy. J. Neuroophthalmol. 2020, 40, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Newman, N.J.; Yu-Wai-Man, P.; Carelli, V.; Moster, M.L.; Biousse, V.; Vignal-Clermont, C.; Sergott, R.C.; Klopstock, T.; Sadun, A.A.; Barboni, P.; et al. Efficacy and Safety of Intravitreal Gene Therapy for Leber Hereditary Optic Neuropathy Treated within 6 Months of Disease Onset. Ophthalmology 2021, 128, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Bhatti, M.T. Gene Therapy for Leber Hereditary Optic Neuropathy: Is Vision Truly RESCUED? Ophthalmology 2021, 128, 661–662. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Griffiths, P.G.; Chinnery, P.F. Mitochondrial Optic Neuropathies—Disease Mechanisms and Therapeutic Strategies. Prog. Retin. Eye Res. 2011, 30, 81–114. [Google Scholar] [CrossRef] [PubMed]
- Takano, F.; Ueda, K.; Godefrooij, D.A.; Yamagami, A.; Ishikawa, H.; Chuman, H.; Ishikawa, H.; Ikeda, Y.; Sakamoto, T.; Nakamura, M. Incidence of Leber Hereditary Optic Neuropathy in 2019 in Japan: A Second Nationwide Questionnaire Survey. Orphanet J. Rare Dis. 2022, 17, 319. [Google Scholar] [CrossRef] [PubMed]
- Watson, E.C.; Davis, R.L.; Ravishankar, S.; Copty, J.; Kummerfeld, S.; Sue, C.M. Low Disease Risk and Penetrance in Leber Hereditary Optic Neuropathy. Am. J. Hum. Genet. 2023, 110, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Lopez Sanchez, M.I.G.; Kearns, L.S.; Staffieri, S.E.; Clarke, L.; McGuinness, M.B.; Meteoukki, W.; Samuel, S.; Ruddle, J.B.; Chen, C.; Fraser, C.L.; et al. Establishing Risk of Vision Loss in Leber Hereditary Optic Neuropathy. Am. J. Hum. Genet. 2021, 108, 2159–2170. [Google Scholar] [CrossRef] [PubMed]
- Newman, N.J.; Yu-Wai-Man, P.; Biousse, V.; Carelli, V. Understanding the Molecular Basis and Pathogenesis of Hereditary Optic Neuropathies: Towards Improved Diagnosis and Management. Lancet Neurol. 2023, 22, 172–188. [Google Scholar] [CrossRef] [PubMed]
- Kirkman, M.A.; Yu-Wai-Man, P.; Korsten, A.; Leonhardt, M.; Dimitriadis, K.; De Coo, I.F.; Klopstock, T.; Chinnery, P.F. Gene–Environment Interactions in Leber Hereditary Optic Neuropathy. Brain 2009, 132, 2317–2326. [Google Scholar] [CrossRef] [PubMed]
- Mackey, D.A.; Ong, J.-S.; MacGregor, S.; Whiteman, D.C.; Craig, J.E.; Lopez Sanchez, M.I.G.; Kearns, L.S.; Staffieri, S.E.; Clarke, L.; McGuinness, M.B.; et al. Is the Disease Risk and Penetrance in Leber Hereditary Optic Neuropathy Actually Low? Am. J. Hum. Genet. 2023, 110, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Elliott, H.R.; Samuels, D.C.; Eden, J.A.; Relton, C.L.; Chinnery, P.F. Pathogenic Mitochondrial DNA Mutations Are Common in the General Population. Am. J. Hum. Genet. 2008, 83, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Hoon, M.; Okawa, H.; Della Santina, L.; Wong, R.O.L. Functional Architecture of the Retina: Development and Disease. Prog. Retin. Eye Res. 2014, 42, 44–84. [Google Scholar] [CrossRef]
- Smith, A.M.; Czyz, C.N. Neuroanatomy, Cranial Nerve 2 (Optic). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Maresca, A.; la Morgia, C.; Caporali, L.; Valentino, M.L.; Carelli, V. The Optic Nerve: A “Mito-Window” on Mitochondrial Neurodegeneration. Mol. Cell Neurosci. 2013, 55, 62–76. [Google Scholar] [CrossRef]
- Saadati, H.G.; Hsu, H.Y.; Heller, K.B.; Sadun, A.A. A Histopathologic and Morphometric Differentiation of Nerves in Optic Nerve Hypoplasia and Leber Hereditary Optic Neuropathy. Arch. Ophthalmol. 1998, 116, 911–916. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sadun, A.A.; Win, P.H.; Ross-Cisneros, F.N.; Walker, S.O.; Carelli, V. Leber’s Hereditary Optic Neuropathy Differentially Affects Smaller Axons in the Optic Nerve. Trans. Am. Ophthalmol. Soc. 2000, 98, 223–235. [Google Scholar] [PubMed]
- Carelli, V.; La Morgia, C.; Valentino, M.L.; Barboni, P.; Ross-Cisneros, F.N.; Sadun, A.A. Retinal Ganglion Cell Neurodegeneration in Mitochondrial Inherited Disorders. Biochim. Biophys. Acta (BBA) Bioenerg. 2009, 1787, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Chow-Wing-Bom, H.T.; Callaghan, M.F.; Wang, J.; Wei, S.; Dick, F.; Yu-Wai-Man, P.; Dekker, T.M. Neuroimaging in Leber Hereditary Optic Neuropathy: State-of-the-Art and Future Prospects. NeuroImage Clin. 2022, 36, 103240. [Google Scholar] [CrossRef] [PubMed]
- Matthews, L.; Enzinger, C.; Fazekas, F.; Rovira, A.; Ciccarelli, O.; Dotti, M.T.; Filippi, M.; Frederiksen, J.L.; Giorgio, A.; Küker, W.; et al. MRI in Leber’s Hereditary Optic Neuropathy: The Relationship to Multiple Sclerosis. J. Neurol. Neurosurg. Psychiatry 2015, 86, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial Electron Transport Chain: Oxidative Phosphorylation, Oxidant Production, and Methods of Measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.Y.-C.; Liu, P.-K.; Wen, Y.-T.; Quinn, P.M.J.; Levi, S.R.; Wang, N.-K.; Tsai, R.-K. Role of Oxidative Stress in Ocular Diseases Associated with Retinal Ganglion Cells Degeneration. Antioxidants 2021, 10, 1948. [Google Scholar] [CrossRef] [PubMed]
- Rovcanin, B.; Jancic, J.; Pajic, J.; Rovcanin, M.; Samardzic, J.; Djuric, V.; Nikolic, B.; Ivancevic, N.; Novakovic, I.; Kostic, V. Oxidative Stress Profile in Genetically Confirmed Cases of Leber’s Hereditary Optic Neuropathy. J. Mol. Neurosci. 2021, 71, 1070–1081. [Google Scholar] [CrossRef]
- Kyriazis, I.D.; Vassi, E.; Alvanou, M.; Angelakis, C.; Skaperda, Z.; Tekos, F.; Garikipati, V.N.S.; Spandidos, D.A.; Kouretas, D. The Impact of Diet upon Mitochondrial Physiology (Review). Int. J. Mol. Med. 2022, 50, 135. [Google Scholar] [CrossRef]
- Dyńka, D.; Kowalcze, K.; Paziewska, A. The Role of Ketogenic Diet in the Treatment of Neurological Diseases. Nutrients 2022, 14, 5003. [Google Scholar] [CrossRef] [PubMed]
- Jensen, N.J.; Wodschow, H.Z.; Nilsson, M.; Rungby, J. Effects of Ketone Bodies on Brain Metabolism and Function in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8767. [Google Scholar] [CrossRef]
- Storoni, M.; Robert, M.P.; Plant, G.T. The Therapeutic Potential of a Calorie-Restricted Ketogenic Diet for the Management of Leber Hereditary Optic Neuropathy. Nutr. Neurosci. 2019, 22, 156–164. [Google Scholar] [CrossRef]
- Venanzi, A.W.; Carmy-Bennun, T.; Marino, F.S.; Ribeiro, M.; Hackam, A.S. Context-Dependent Effects of the Ketogenic Diet on Retinal Ganglion Cell Survival and Axonal Regeneration After Optic Nerve Injury. J. Ocul. Pharmacol. Ther. 2023, 39, 509–518. [Google Scholar] [CrossRef]
- Emperador, S.; López-Gallardo, E.; Hernández-Ainsa, C.; Habbane, M.; Montoya, J.; Bayona-Bafaluy, M.P.; Ruiz-Pesini, E. Ketogenic Treatment Reduces the Percentage of a LHON Heteroplasmic Mutation and Increases mtDNA Amount of a LHON Homoplasmic Mutation. Orphanet J. Rare Dis. 2019, 14, 150. [Google Scholar] [CrossRef] [PubMed]
- Paoli, A.; Mancin, L.; Bianco, A.; Thomas, E.; Mota, J.F.; Piccini, F. Ketogenic Diet and Microbiota: Friends or Enemies? Genes 2019, 10, 534. [Google Scholar] [CrossRef] [PubMed]
- Shandilya, S.; Kumar, S.; Kumar Jha, N.; Kumar Kesari, K.; Ruokolainen, J. Interplay of Gut Microbiota and Oxidative Stress: Perspective on Neurodegeneration and Neuroprotection. J. Adv. Res. 2021, 38, 223–244. [Google Scholar] [CrossRef] [PubMed]
- Gardener, H.; Caunca, M.R. Mediterranean Diet in Preventing Neurodegenerative Diseases. Curr. Nutr. Rep. 2018, 7, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Benítez, M.; Calderón-Fernández, A.; Canales-Cortés, S.; Alegre-Cortés, E.; Uribe-Carretero, E.; Paredes-Barquero, M.; Gimenez-Bejarano, A.; Duque González, G.; Gómez-Suaga, P.; Ortega-Vidal, J.; et al. Biological Effects of Olive Oil Phenolic Compounds on Mitochondria. Mol. Cell. Oncol. 2022, 9, 2044263. [Google Scholar] [CrossRef] [PubMed]
- Solch, R.J.; Aigbogun, J.O.; Voyiadjis, A.G.; Talkington, G.M.; Darensbourg, R.M.; O’Connell, S.; Pickett, K.M.; Perez, S.R.; Maraganore, D.M. Mediterranean Diet Adherence, Gut Microbiota, and Alzheimer’s or Parkinson’s Disease Risk: A Systematic Review. J. Neurol. Sci. 2022, 434, 120166. [Google Scholar] [CrossRef] [PubMed]
- Pollicino, F.; Veronese, N.; Dominguez, L.J.; Barbagallo, M. Mediterranean Diet and Mitochondria: New Findings. Exp. Gerontol. 2023, 176, 112165. [Google Scholar] [CrossRef]
- Butt, M.S.; Tariq, U.; Iahtisham-Ul-Haq; Naz, A.; Rizwan, M. Neuroprotective Effects of Oleuropein: Recent Developments and Contemporary Research. J. Food Biochem. 2021, 45, e13967. [Google Scholar] [CrossRef] [PubMed]
- Grewal, R.; Reutzel, M.; Dilberger, B.; Hein, H.; Zotzel, J.; Marx, S.; Tretzel, J.; Sarafeddinov, A.; Fuchs, C.; Eckert, G.P. Purified Oleocanthal and Ligstroside Protect against Mitochondrial Dysfunction in Models of Early Alzheimer’s Disease and Brain Ageing. Exp. Neurol. 2020, 328, 113248. [Google Scholar] [CrossRef] [PubMed]
- Amick, K.A.; Mahapatra, G.; Bergstrom, J.; Gao, Z.; Craft, S.; Register, T.C.; Shively, C.A.; Molina, A.J.A. Brain Region-Specific Disruption of Mitochondrial Bioenergetics in Cynomolgus Macaques Fed a Western versus a Mediterranean Diet. Am. J. Physiol. Endocrinol. Metab. 2021, 321, E652–E664. [Google Scholar] [CrossRef] [PubMed]
- Fahmideh, F.; Marchesi, N.; Barbieri, A.; Govoni, S.; Pascale, A. Non-Drug Interventions in Glaucoma: Putative Roles for Lifestyle, Diet and Nutritional Supplements. Surv. Ophthalmol. 2022, 67, 675–696. [Google Scholar] [CrossRef] [PubMed]
- Karaa, A.; Kriger, J.; Grier, J.; Holbert, A.; Thompson, J.L.P.; Parikh, S.; Hirano, M. Mitochondrial Disease Patients’ Perception of Dietary Supplements’ Use. Mol. Genet. Metab. 2016, 119, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Swarnakar, N.K.; Jain, A.K.; Singh, R.P.; Godugu, C.; Das, M.; Jain, S. Oral Bioavailability, Therapeutic Efficacy and Reactive Oxygen Species Scavenging Properties of Coenzyme Q10-Loaded Polymeric Nanoparticles. Biomaterials 2011, 32, 6860–6874. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, S.; Haddadi, R.; Saki, S.; Kourosh-Arami, M.; Rashno, M.; Mojaver, A.; Komaki, A. Neuroprotective Effects of Coenzyme Q10 on Neurological Diseases: A Review Article. Front. Neurosci. 2023, 17, 1188839. [Google Scholar] [CrossRef] [PubMed]
- Fuller, J.T.; Barnes, S.; Sadun, L.A.; Ajmera, P.; Alexandrova, A.N.; Sadun, A.A. Coenzyme Q10 Trapping in Mitochondrial Complex I Underlies Leber’s Hereditary Optic Neuropathy. Proc. Natl. Acad. Sci. USA 2023, 120, e2304884120. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-C.; Kuo, H.-C.; Chu, C.-C.; Kao, L.-Y. Rapid Visual Recovery After Coenzyme Q10 Treatment of Leber Hereditary Optic Neuropathy. J. Neuro-Ophthalmol. 2002, 22, 66. [Google Scholar] [CrossRef]
- Boreková, M.; Hojerová, J.; Koprda, V.; Bauerová, K. Nourishing and Health Benefits of Coenzyme Q10. Czech J. Food Sci. 2008, 26, 229–241. [Google Scholar] [CrossRef]
- Zibold, J.; von Livonius, B.; Kolarova, H.; Rudolph, G.; Priglinger, C.S.; Klopstock, T.; Catarino, C.B. Vitamin B12 in Leber Hereditary Optic Neuropathy Mutation Carriers: A Prospective Cohort Study. Orphanet J. Rare Dis. 2022, 17, 310. [Google Scholar] [CrossRef]
- Bocca, C.; Le Paih, V.; Chao de la Barca, J.M.; Kouassy Nzoughet, J.; Amati-Bonneau, P.; Blanchet, O.; Védie, B.; Géromin, D.; Simard, G.; Procaccio, V.; et al. A Plasma Metabolomic Signature of Leber Hereditary Optic Neuropathy Showing Taurine and Nicotinamide Deficiencies. Hum. Mol. Genet. 2021, 30, 21–29. [Google Scholar] [CrossRef]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cochran, K.E.; Philip, V.M.; Porciatti, V.; Smithies, O.; John, S.W.M. Vitamin B3 Modulates Mitochondrial Vulnerability and Prevents Glaucoma in Aged Mice. Science 2017, 355, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Lawson, E.C.; Han, M.K.; Sellers, J.T.; Chrenek, M.A.; Hanif, A.; Gogniat, M.A.; Boatright, J.H.; Pardue, M.T. Aerobic Exercise Protects Retinal Function and Structure from Light-Induced Retinal Degeneration. J. Neurosci. 2014, 34, 2406–2412. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.; Goldstein, A.; Koenig, M.K.; Scaglia, F.; Enns, G.M.; Saneto, R.; Anselm, I.; Cohen, B.H.; Falk, M.J.; Greene, C.; et al. Diagnosis and Management of Mitochondrial Disease: A Consensus Statement from the Mitochondrial Medicine Society. Genet. Med. 2015, 17, 689–701. [Google Scholar] [CrossRef] [PubMed]
- He, Y.-Y.; Wang, L.; Zhang, T.; Weng, S.-J.; Lu, J.; Zhong, Y.-M. Aerobic Exercise Delays Retinal Ganglion Cell Death after Optic Nerve Injury. Exp. Eye Res. 2020, 200, 108240. [Google Scholar] [CrossRef] [PubMed]
- Kerrison, J.B.; Miller, N.R.; Hsu, F.-C.; Beaty, T.H.; Maumenee, I.H.; Smith, K.H.; Savino, P.J.; Stone, E.M.; Newman, N.J. A Case-Control Study of Tobacco and Alcohol Consumption in Leber Hereditary Optic Neuropathy. Am. J. Ophthalmol. 2000, 130, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Sadun, A.A.; Carelli, V.; Salomao, S.R.; Berezovsky, A.; Quiros, P.A.; Sadun, F.; DeNegri, A.-M.; Andrade, R.; Moraes, M.; Passos, A.; et al. Extensive Investigation of a Large Brazilian Pedigree of 11778/Haplogroup J Leber Hereditary Optic Neuropathy. Am. J. Ophthalmol. 2003, 136, 231–238. [Google Scholar] [CrossRef]
- Malińska, D.; Więckowski, M.R.; Michalska, B.; Drabik, K.; Prill, M.; Patalas-Krawczyk, P.; Walczak, J.; Szymański, J.; Mathis, C.; Van der Toorn, M.; et al. Mitochondria as a Possible Target for Nicotine Action. J. Bioenerg. Biomembr. 2019, 51, 259–276. [Google Scholar] [CrossRef]
- Arruebarrena, M.A.; Hawe, C.T.; Lee, Y.M.; Branco, R.C. Mechanisms of Cadmium Neurotoxicity. Int. J. Mol. Sci. 2023, 24, 16558. [Google Scholar] [CrossRef]
- Stucki, D.; Stahl, W. Carbon Monoxide—Beyond Toxicity? Toxicol. Lett. 2020, 333, 251–260. [Google Scholar] [CrossRef]
- Tulen, C.B.M.; Opperhuizen, A.; van Schooten, F.-J.; Remels, A.H.V. Disruption of the Molecular Regulation of Mitochondrial Metabolism in Airway and Lung Epithelial Cells by Cigarette Smoke: Are Aldehydes the Culprit? Cells 2023, 12, 299. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, R.N.; Smith, A.J.; Carelli, V.; Sadun, A.A.; Keltner, J.L. Leber Hereditary Optic Neuropathy Possibly Triggered by Exposure to Tire Fire. J. Neuro-Ophthalmol. 2006, 26, 268. [Google Scholar] [CrossRef] [PubMed]
- Johns, D.R.; Smith, K.H.; Miller, N.R.; Sulewski, M.E.; Bias, W.B. Identical Twins Who Are Discordant for Leber’s Hereditary Optic Neuropathy. Arch. Ophthalmol. 1993, 111, 1491–1494. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Ventura, J.; Loza, A.; Wang, Y.; Talbot, P. Chemical Elements in Electronic Cigarette Solvents and Aerosols Inhibit Mitochondrial Reductases and Induce Oxidative Stress. Nicotine Tob. Res. 2020, 22, S14–S24. [Google Scholar] [CrossRef] [PubMed]
- Basma, H.; Tatineni, S.; Dhar, K.; Qiu, F.; Rennard, S.; Lowes, B.D. Electronic Cigarette Extract Induced Toxic Effect in iPS-Derived Cardiomyocytes. BMC Cardiovasc. Disord. 2020, 20, 357. [Google Scholar] [CrossRef]
- Jabba, S.V.; Diaz, A.N.; Erythropel, H.C.; Zimmerman, J.B.; Jordt, S.-E. Chemical Adducts of Reactive Flavor Aldehydes Formed in E-Cigarette Liquids Are Cytotoxic and Inhibit Mitochondrial Function in Respiratory Epithelial Cells. Nicotine Tob. Res. 2020, 22, S25–S34. [Google Scholar] [CrossRef] [PubMed]
- Rabenstein, A.; Catarino, C.B.; Rampeltshammer, V.; Schindler, D.; Gallenmüller, C.; Priglinger, C.; Pogarell, O.; Rüther, T.; Klopstock, T. Smoking and Alcohol, Health-Related Quality of Life and Psychiatric Comorbidities in Leber’s Hereditary Optic Neuropathy Mutation Carriers: A Prospective Cohort Study. Orphanet J. Rare Dis. 2021, 16, 127. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.K.; Ahmad, M.H.; Sahu, M.R.; Subba, R.; Mondal, A.C. Detrimental Effects of Alcohol-Induced Inflammation on Brain Health: From Neurogenesis to Neurodegeneration. Cell Mol. Neurobiol. 2023, 43, 1885–1904. [Google Scholar] [CrossRef] [PubMed]
- Sarić, N.; Hashimoto-Torii, K.; Jevtović-Todorović, V.; Ishibashi, N. Non-Apoptotic Caspases in Neural Development and in Anesthesia-Induced Neurotoxicity. Trends Neurosci. 2022, 45, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.D.; Padmavathi, P.; Kavitha, G.; Saradamma, B.; Varadacharyulu, N. Alcohol-Induced Oxidative/Nitrosative Stress Alters Brain Mitochondrial Membrane Properties. Mol. Cell Biochem. 2013, 375, 39–47. [Google Scholar] [CrossRef]
- Mena, C.; Cabrera, C.; Lorenzo, M.L.; López, M.C. Cadmium Levels in Wine, Beer and Other Alcoholic Beverages: Possible Sources of Contamination. Sci. Total Environ. 1996, 181, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Morris, N.L.; Harris, F.L.; Brown, L.A.S.; Yeligar, S.M. Alcohol Induces Mitochondrial Derangements in Alveolar Macrophages by Upregulating NADPH Oxidase 4. Alcohol 2021, 90, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Park, M.W.; Cha, H.W.; Kim, J.; Kim, J.H.; Yang, H.; Yoon, S.; Boonpraman, N.; Yi, S.S.; Yoo, I.D.; Moon, J.-S. NOX4 Promotes Ferroptosis of Astrocytes by Oxidative Stress-Induced Lipid Peroxidation via the Impairment of Mitochondrial Metabolism in Alzheimer’s Diseases. Redox Biol. 2021, 41, 101947. [Google Scholar] [CrossRef] [PubMed]
- Kabanovski, A.; Donaldson, L.; Margolin, E. Neuro-Ophthalmological Manifestations of Wolfram Syndrome: Case Series and Review of the Literature. J. Neurol. Sci. 2022, 437, 120267. [Google Scholar] [CrossRef] [PubMed]
- Thoudam, T.; Chanda, D.; Lee, J.Y.; Jung, M.-K.; Sinam, I.S.; Kim, B.-G.; Park, B.-Y.; Kwon, W.H.; Kim, H.-J.; Kim, M.; et al. Enhanced Ca2+-Channeling Complex Formation at the ER-Mitochondria Interface Underlies the Pathogenesis of Alcohol-Associated Liver Disease. Nat. Commun. 2023, 14, 1703. [Google Scholar] [CrossRef] [PubMed]
- Chao de la Barca, J.M.; Simard, G.; Amati-Bonneau, P.; Safiedeen, Z.; Prunier-Mirebeau, D.; Chupin, S.; Gadras, C.; Tessier, L.; Gueguen, N.; Chevrollier, A.; et al. The Metabolomic Signature of Leber’s Hereditary Optic Neuropathy Reveals Endoplasmic Reticulum Stress. Brain 2016, 139, 2864–2876. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, J.; Kaun, K.R. Alcohol, Neuronal Plasticity, and Mitochondrial Trafficking. Proc. Natl. Acad. Sci. USA 2022, 119, e2208744119. [Google Scholar] [CrossRef]
- Dhingra, D.; Kaur, S.; Ram, J. Illicit Drugs: Effects on Eye. Indian J. Med. Res. 2019, 150, 228–238. [Google Scholar] [CrossRef]
- Carelli, V.; Franceschini, F.; Venturi, S.; Barboni, P.; Savini, G.; Barbieri, G.; Pirro, E.; La Morgia, C.; Valentino, M.L.; Zanardi, F.; et al. Grand Rounds: Could Occupational Exposure to n-Hexane and Other Solvents Precipitate Visual Failure in Leber Hereditary Optic Neuropathy? Environ. Health Perspect. 2007, 115, 113–115. [Google Scholar] [CrossRef]
- Ghelli, A.; Porcelli, A.M.; Zanna, C.; Vidoni, S.; Mattioli, S.; Barbieri, A.; Iommarini, L.; Pala, M.; Achilli, A.; Torroni, A.; et al. The Background of Mitochondrial DNA Haplogroup J Increases the Sensitivity of Leber’s Hereditary Optic Neuropathy Cells to 2,5-Hexanedione Toxicity. PLoS ONE 2009, 4, e7922. [Google Scholar] [CrossRef] [PubMed]
- Liberski, S.; Kaluzny, B.J.; Kocięcki, J. Methanol-Induced Optic Neuropathy: A Still-Present Problem. Arch. Toxicol. 2022, 96, 431–451. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.R.; Palmer, C.A.; Curé, J.K.; Balos, L.L.; Lincoff, N.S.; Kline, L.B. Toluene Optic Neurotoxicity: Magnetic Resonance Imaging and Pathologic Features. Hum. Pathol. 2011, 42, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Kiyokawa, M.; Mizota, A.; Takasoh, M.; Adachi-Usami, E. Pattern Visual Evoked Cortical Potentials in Patients with Toxic Optic Neuropathy Caused by Toluene Abuse. Jpn. J. Ophthalmol. 1999, 43, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Ehyai, A.; Freemon, F.R. Progressive Optic Neuropathy and Sensorineural Hearing Loss Due to Chronic Glue Sniffing. J. Neurol. Neurosurg. Psychiatry 1983, 46, 349–351. [Google Scholar] [CrossRef] [PubMed]
- López-Gallardo, E.; Emperador, S.; Hernández-Ainsa, C.; Montoya, J.; Bayona-Bafaluy, M.P.; Ruiz-Pesini, E. Food Derived Respiratory Complex I Inhibitors Modify the Effect of Leber Hereditary Optic Neuropathy Mutations. Food Chem. Toxicol. 2018, 120, 89–97. [Google Scholar] [CrossRef]
- Schmidt, F.; Douaron, G.L.; Champy, P.; Amar, M.; Séon-Méniel, B.; Raisman-Vozari, R.; Figadère, B. Tryptamine-Derived Alkaloids from Annonaceae Exerting Neurotrophin-like Properties on Primary Dopaminergic Neurons. Bioorganic Med. Chem. 2010, 18, 5103–5113. [Google Scholar] [CrossRef] [PubMed]
- Bénit, P.; Kahn, A.; Chretien, D.; Bortoli, S.; Huc, L.; Schiff, M.; Gimenez-Roqueplo, A.-P.; Favier, J.; Gressens, P.; Rak, M.; et al. Evolutionarily Conserved Susceptibility of the Mitochondrial Respiratory Chain to SDHI Pesticides and Its Consequence on the Impact of SDHIs on Human Cultured Cells. PLoS ONE 2019, 14, e0224132. [Google Scholar] [CrossRef]
- Kogachi, K.; Ter-Zakarian, A.; Asanad, S.; Sadun, A.; Karanjia, R. Toxic Medications in Leber’s Hereditary Optic Neuropathy. Mitochondrion 2019, 46, 270–277. [Google Scholar] [CrossRef]
- Park, E.-J.; Jin, S.-W.; Kang, M.-S.; Yang, M.-J.; Kim, S.-H.; Han, H.-Y.; Kang, J.W. Pulmonary Inflammation and Cellular Responses Following Exposure to Benzalkonium Chloride: Potential Impact of Disrupted Pulmonary Surfactant Homeostasis. Toxicol. Appl. Pharmacol. 2022, 440, 115930. [Google Scholar] [CrossRef]
- Rogov, A.G.; Goleva, T.N.; Sukhanova, E.I.; Epremyan, K.K.; Trendeleva, T.A.; Ovchenkova, A.P.; Aliverdieva, D.A.; Zvyagilskaya, R.A. Mitochondrial Dysfunctions May Be One of the Major Causative Factors Underlying Detrimental Effects of Benzalkonium Chloride. Oxid. Med. Cell Longev. 2020, 2020, 8956504. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Baudouin, C.; Brignole-Baudouin, F.; Denoyer, A.; Cortopassi, G.A. The Eye Drop Preservative Benzalkonium Chloride Potently Induces Mitochondrial Dysfunction and Preferentially Affects LHON Mutant Cells. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2406–2412. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; Carbonelli, M.; de Coo, I.F.; Kawasaki, A.; Klopstock, T.; Lagrèze, W.A.; La Morgia, C.; Newman, N.J.; Orssaud, C.; Pott, J.W.R.; et al. International Consensus Statement on the Clinical and Therapeutic Management of Leber Hereditary Optic Neuropathy. J. Neuro-Ophthalmol. 2017, 37, 371. [Google Scholar] [CrossRef] [PubMed]
- Esposti, M.D.; Ngo, A.; Ghelli, A.; Benelli, B.; Carelli, V.; McLennan, H.; Linnane, A.W. The Interaction of Q Analogs, Particularly Hydroxydecyl Benzoquinone (Idebenone), with the Respiratory Complexes of Heart Mitochondria. Arch. Biochem. Biophys. 1996, 330, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Haefeli, R.H.; Erb, M.; Gemperli, A.C.; Robay, D.; Fruh, I.; Anklin, C.; Dallmann, R.; Gueven, N. NQo1-Dependent Redox Cycling of Idebenone: Effects on Cellular Redox Potential and Energy Levels. PLoS ONE 2011, 6, e17963. [Google Scholar] [CrossRef] [PubMed]
- Tiefenbach, J.; Magomedova, L.; Liu, J.; Reunov, A.A.; Tsai, R.; Eappen, N.S.; Jockusch, R.A.; Nislow, C.; Cummins, C.L.; Krause, H.M. Idebenone and Coenzyme Q10 Are Novel PPARα/γ Ligands, with Potential for Treatment of Fatty Liver Diseases. DMM Dis. Models Mech. 2018, 11, dmm034801. [Google Scholar] [CrossRef]
- Giorgio, V.; Schiavone, M.; Galber, C.; Carini, M.; Da Ros, T.; Petronilli, V.; Argenton, F.; Carelli, V.; Acosta Lopez, M.J.; Salviati, L.; et al. The Idebenone Metabolite QS10 Restores Electron Transfer in Complex I and Coenzyme Q Defects. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 901–908. [Google Scholar] [CrossRef]
- Mashima, Y.; Hiida, Y.; Oguchi, Y. Remission of Leber’s Hereditary Optic Neuropathy with Idebenone. Lancet 1992, 340, 368–369. [Google Scholar] [CrossRef]
- Klopstock, T.; Yu-Wai-Man, P.; Dimitriadis, K.; Rouleau, J.; Heck, S.; Bailie, M.; Atawan, A.; Chattopadhyay, S.; Schubert, M.; Garip, A.; et al. A Randomized Placebo-Controlled Trial of Idebenone in Leber’s Hereditary Optic Neuropathy. Brain 2011, 134, 2677–2686. [Google Scholar] [CrossRef]
- Carelli, V.; Morgia, C.L.; Valentino, M.L.; Rizzo, G.; Carbonelli, M.; De Negri, A.M.; Sadun, F.; Carta, A.; Guerriero, S.; Simonelli, F.; et al. Idebenone Treatment in Leber’s Hereditary Optic Neuropathy. Brain 2011, 134, e188. [Google Scholar] [CrossRef]
- van Everdingen, J.A.M.; Pott, J.W.R.; Bauer, N.J.C.; Krijnen, A.M.; Lushchyk, T.; Wubbels, R.J. Clinical Outcomes of Treatment with Idebenone in Leber’s Hereditary Optic Neuropathy in the Netherlands: A National Cohort Study. Acta Ophthalmol. 2022, 100, 700–706. [Google Scholar] [CrossRef]
- Pemp, B.; Mitsch, C.; Kircher, K.; Reitner, A. Changes in Visual Function and Correlations with Inner Retinal Structure in Acute and Chronic Leber’s Hereditary Optic Neuropathy Patients after Treatment with Idebenone. J. Clin. Med. 2021, 10, 151. [Google Scholar] [CrossRef]
- Enns, G.M.; Kinsman, S.L.; Perlman, S.L.; Spicer, K.M.; Abdenur, J.E.; Cohen, B.H.; Amagata, A.; Barnes, A.; Kheifets, V.; Shrader, W.D.; et al. Initial Experience in the Treatment of Inherited Mitochondrial Disease with EPI-743. Mol. Genet. Metab. 2012, 105, 91–102. [Google Scholar] [CrossRef]
- Zesiewicz, T.; Salemi, J.L.; Perlman, S.; Sullivan, K.L.; Shaw, J.D.; Huang, Y.; Isaacs, C.; Gooch, C.; Lynch, D.R.; Klein, M.B. Double-Blind, Randomized and Controlled Trial of EPI-743 in Friedreich’s Ataxia. Neurodegener. Dis. Manag. 2018, 8, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Shrader, W.D.; Amagata, A.; Barnes, A.; Enns, G.M.; Hinman, A.; Jankowski, O.; Kheifets, V.; Komatsuzaki, R.; Lee, E.; Mollard, P.; et al. α-Tocotrienol Quinone Modulates Oxidative Stress Response and the Biochemistry of Aging. Bioorganic Med. Chem. Lett. 2011, 21, 3693–3698. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, D.; Catteruccia, M.; Piemonte, F.; Pastore, A.; Tozzi, G.; Dionisi-Vici, C.; Pontrelli, G.; Corsetti, T.; Livadiotti, S.; Kheifets, V.; et al. EPI-743 Reverses the Progression of the Pediatric Mitochondrial Disease—Genetically Defined Leigh Syndrome. Mol. Genet. Metab. 2012, 107, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Zesiewicz, T.A.; Hancock, J.; Ghanekar, S.D.; Kuo, S.-H.; Dohse, C.A.; Vega, J. Emerging Therapies in Friedreich’s Ataxia. Expert. Rev. Neurother. 2020, 20, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Sadun, A.A.; Chicani, C.F.; Ross-Cisneros, F.N.; Barboni, P.; Thoolen, M.; Shrader, W.D.; Kubis, K.; Carelli, V.; Miller, G. Effect of EPI-743 on the Clinical Course of the Mitochondrial Disease Leber Hereditary Optic Neuropathy. Arch. Neurol. 2012, 69, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Chicani, C.F.; Chu, E.R.; Miller, G.; Kelman, S.E.; Sadun, A.A. Comparing EPI-743 Treatment in Siblings with Leber’s Hereditary Optic Neuropathy Mt14484 Mutation. Can. J. Ophthalmol. 2013, 48, e130–e133. [Google Scholar] [CrossRef] [PubMed]
- Kahn-Kirby, A.H.; Amagata, A.; Maeder, C.I.; Mei, J.J.; Sideris, S.; Kosaka, Y.; Hinman, A.; Malone, S.A.; Bruegger, J.J.; Wang, L.; et al. Targeting Ferroptosis: A Novel Therapeutic Strategy for the Treatment of Mitochondrial Disease-Related Epilepsy. PLoS ONE 2019, 14, e0214250. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Zhu, Y.; Shi, Y.; Meng, X.; Dong, X.; Zhang, H.; Wang, X.; Du, M.; Yan, H. Inhibition of Ferroptosis Promotes Retina Ganglion Cell Survival in Experimental Optic Neuropathies. Redox Biol. 2022, 58, 102541. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Gao, W.; Zhu, C.; Lou, Q.; Ye, C.; Ren, Y.; Mehmood, R.; Huang, B.; Nan, K. Efficiently Suppress of Ferroptosis Using Deferoxamine Nanoparticles as a New Method for Retinal Ganglion Cell Protection after Traumatic Optic Neuropathy. Biomater. Adv. 2022, 138, 212936. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Liu, K.; Su, Y.; Wang, F.; Feng, T. Research Progress of Ferroptosis in Glaucoma and Optic Nerve Damage. Mol. Cell Biochem. 2023, 478, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Suo, L.; Dai, W.; Chen, X.; Qin, X.; Li, G.; Song, S.; Zhang, D.; Zhang, C. Proteomics Analysis of N-Methyl-d-Aspartate-Induced Cell Death in Retinal and Optic Nerves. J. Proteom. 2022, 252, 104427. [Google Scholar] [CrossRef] [PubMed]
- Nhu, N.T.; Xiao, S.-Y.; Liu, Y.; Kumar, V.B.; Cui, Z.-Y.; Lee, S.-D. Neuroprotective Effects of a Small Mitochondrially-Targeted Tetrapeptide Elamipretide in Neurodegeneration. Front. Integr. Neurosci. 2022, 15, 747901. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Zeng, Q.; Luo, Y.; He, L.; Zhao, Y.; Li, N.; Han, C.; Zhang, G.; Liu, W. Application Research of Novel Peptide Mitochondrial-Targeted Antioxidant SS-31 in Mitigating Mitochondrial Dysfunction. Mitochondrion 2024, 75, 101846. [Google Scholar] [CrossRef] [PubMed]
- Karaa, A.; Bertini, E.; Carelli, V.; Cohen, B.H.; Enns, G.M.; Falk, M.J.; Goldstein, A.; Gorman, G.S.; Haas, R.; Hirano, M.; et al. Efficacy and Safety of Elamipretide in Individuals with Primary Mitochondrial Myopathy. Neurology 2023, 101, e238–e252. [Google Scholar] [CrossRef] [PubMed]
- Tse, B.C.; Dvoriantchikova, G.; Tao, W.; Gallo, R.A.; Lee, J.Y.; Ivanov, D.; Tse, D.T.; Pelaez, D. Mitochondrial Targeted Therapy with Elamipretide (MTP-131) as an Adjunct to Tumor Necrosis Factor Inhibition for Traumatic Optic Neuropathy in the Acute Setting. Exp. Eye Res. 2020, 199, 108178. [Google Scholar] [CrossRef] [PubMed]
- Karanjia, R.; Sadun, A.A. Elamipretide Topical Ophthalmic Solution for the Treatment of Subjects with Leber’s Hereditary Optic Neuropathy: A Randomized Trial. Ophthalmology, 2023; in press. [Google Scholar] [CrossRef]
- De Vries, M.C.; Brown, D.A.; Allen, M.E.; Bindoff, L.; Gorman, G.S.; Karaa, A.; Keshavan, N.; Lamperti, C.; McFarland, R.; Ng, Y.S.; et al. Safety of Drug Use in Patients with a Primary Mitochondrial Disease: An International Delphi-based Consensus. J. Inherit. Metab. Dis. 2020, 43, 800–818. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, K.; ter Hofstede, H.J.M.; Burger, D.M.; Smeitink, J.A.M.; Koopmans, P.P. Adverse Effects of Reverse Transcriptase Inhibitors: Mitochondrial Toxicity as Common Pathway. Aids 1998, 12, 1735. [Google Scholar] [CrossRef]
- Mackey, D.A.; Fingert, J.H.; Luzhansky, J.Z.; McCluskey, P.J.; Howell, N.; Hall, A.J.H.; Pierce, A.B.; Hoy, J.F. Leber’s Hereditary Optic Neuropathy Triggered by Antiretroviral Therapy for Human Immunodeficiency Virus. Eye 2003, 17, 312–317. [Google Scholar] [CrossRef]
- Luke, C.; Cornely, O.; Lehrer, E.; Bartz-Schmidt, U.; Wissinger, B.; Brunner, R. Late Onset of Leber’s Hereditary Optic Neuropathy in HIV Infection—PMC. Br. J. Ophtalmol. 1999, 83, 1194. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Luzhansky, J.Z.; Pierce, A.B.; Hoy, J.F.; Hall, A.J.H. Leber’s Hereditary Optic Neuropathy in the Setting of Nucleoside Analogue Toxicity. Aids 2001, 15, 1588. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.; Ta, C.; Basham, A.A.; Mansour, S. Leber Hereditary Optic Neuropathy Associated with Antiretroviral Therapy for Human Immunodeficiency Virus Infection. Am. J. Ophthalmol. 2001, 131, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Warner, J.E.A.; Ries, K.M. Optic Neuropathy in a Patient with AIDS. J. Neuro-Ophthalmol. 2001, 21, 92. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Oliveira, C. Toxic-Metabolic and Hereditary Optic Neuropathies. CONTINUUM Lifelong Learn. Neurol. 2019, 25, 1265. [Google Scholar] [CrossRef] [PubMed]
- Reinert, M.-C.; Pacheu-Grau, D.; Catarino, C.B.; Klopstock, T.; Ohlenbusch, A.; Schittkowski, M.; Wilichowski, E.; Rehling, P.; Brockmann, K. Sulthiame Impairs Mitochondrial Function in Vitro and May Trigger Onset of Visual Loss in Leber Hereditary Optic Neuropathy. Orphanet J. Rare Dis. 2021, 16, 64. [Google Scholar] [CrossRef] [PubMed]
- Rinalduzzi, S.; Cipriani, A.M.; Accornero, N. Topiramate and Visual Loss in a Patient Carrying a Leber Hereditary Optic Neuropathy Mutation. Neurol. Sci. 2012, 33, 419–421. [Google Scholar] [CrossRef] [PubMed]
- Bergamini, C.; Moruzzi, N.; Volta, F.; Faccioli, L.; Gerdes, J.; Mondardini, M.C.; Fato, R. Role of Mitochondrial Complex I and Protective Effect of CoQ10 Supplementation in Propofol Induced Cytotoxicity. J. Bioenerg. Biomembr. 2016, 48, 413–423. [Google Scholar] [CrossRef]
- Grzybowski, A.; Zülsdorff, M.; Wilhelm, H.; Tonagel, F. Toxic Optic Neuropathies: An Updated Review. Acta Ophthalmol. 2015, 93, 402–410. [Google Scholar] [CrossRef]
- Cornish, K.S.; Barras, C. Leber’s Hereditary Optic Neuropathy Precipitated by Tadalafil Use for Erectile Dysfunction. Semin. Ophthalmol. 2011, 26, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Roff Hilton, E.J.; Hosking, S.L.; Betts, T. The Effect of Antiepileptic Drugs on Visual Performance. Seizure 2004, 13, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Rasool, N.; Boudreault, K.; Lessell, S.; Prasad, S.; Cestari, D.M. Tacrolimus Optic Neuropathy. J. Neuro-Ophthalmol. 2018, 38, 160. [Google Scholar] [CrossRef] [PubMed]
- Varricchio, C.; Beirne, K.; Aeschlimann, P.; Heard, C.; Rozanowska, M.; Votruba, M.; Brancale, A. Discovery of Novel 2-Aniline-1,4-Naphthoquinones as Potential New Drug Treatment for Leber’s Hereditary Optic Neuropathy (LHON). J. Med. Chem. 2020, 63, 13638–13655. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Jin, X.; Peng, Y.; Wang, M.; Liu, H.; Liu, X.; Zhang, Z.; Ji, Y.; Zhang, J.; Liang, M.; et al. The Exome Sequencing Identified the Mutation in YARS2 Encoding the Mitochondrial Tyrosyl-tRNA Synthetase as a Nuclear Modifier for the Phenotypic Manifestation of Leber’s Hereditary Optic Neuropathy-Associated Mitochondrial DNA Mutation. Hum. Mol. Genet. 2016, 25, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Liang, X.; Ji, Y.; Ai, C.; Liu, J.; Zhu, L.; Nie, Z.; Jin, X.; Wang, C.; Zhang, J.; et al. PRICKLE3 Linked to ATPase Biogenesis Manifested Leber’s Hereditary Optic Neuropathy. J. Clin. Investig. 2020, 130, 4935–4946. [Google Scholar] [CrossRef] [PubMed]
- Gale, J.; Khoshnevis, M.; Frousiakis, S.E.; Karanjia, R.; Poincenot, L.; Sadun, A.A.; Baron, D.A. An International Study of Emotional Response to Bilateral Vision Loss Using a Novel Graphical Online Assessment Tool. Psychosomatics 2017, 58, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.S.; Holzinger, E.; Taiel, M.; Yu-Wai-Man, P. The Impact of Leber Hereditary Optic Neuropathy on the Quality of Life of Patients and Their Relatives: A Qualitative Study. J. Neuro-Ophthalmol. 2022, 42, 316. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Layrolle, P.; Orssaud, C.; Leleu, M.; Payoux, P.; Chavanas, S. The Optic Nerve at Stake: Update on Environmental Factors Modulating Expression of Leber’s Hereditary Optic Neuropathy. Biomedicines 2024, 12, 584. https://doi.org/10.3390/biomedicines12030584
Layrolle P, Orssaud C, Leleu M, Payoux P, Chavanas S. The Optic Nerve at Stake: Update on Environmental Factors Modulating Expression of Leber’s Hereditary Optic Neuropathy. Biomedicines. 2024; 12(3):584. https://doi.org/10.3390/biomedicines12030584
Chicago/Turabian StyleLayrolle, Pierre, Christophe Orssaud, Maryse Leleu, Pierre Payoux, and Stéphane Chavanas. 2024. "The Optic Nerve at Stake: Update on Environmental Factors Modulating Expression of Leber’s Hereditary Optic Neuropathy" Biomedicines 12, no. 3: 584. https://doi.org/10.3390/biomedicines12030584
APA StyleLayrolle, P., Orssaud, C., Leleu, M., Payoux, P., & Chavanas, S. (2024). The Optic Nerve at Stake: Update on Environmental Factors Modulating Expression of Leber’s Hereditary Optic Neuropathy. Biomedicines, 12(3), 584. https://doi.org/10.3390/biomedicines12030584