
Rotundifuran Induces Ferroptotic Cell Death and Mitochondria Permeability Transition in Lung Cancer Cells
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Cell Culture
2.3. Cell Viability
2.4. Caspase Enzyme Activity
2.5. Cytosolic and Mitochondrial Ca2+ Levels
2.6. Transmission Electron Microscopy
2.7. Transfection of siRNA
2.8. Intracellular ROS Measurement
2.9. Cell Cycle Analysis
2.10. RHOD-2 Staining
2.11. Lipid Peroxidation and Intracellular Ferrous Levels
2.12. Mitochondrial Membrane Potential
2.13. Western Blotting
2.14. Immunocytochemistry
2.15. Statistical Analysis
3. Results
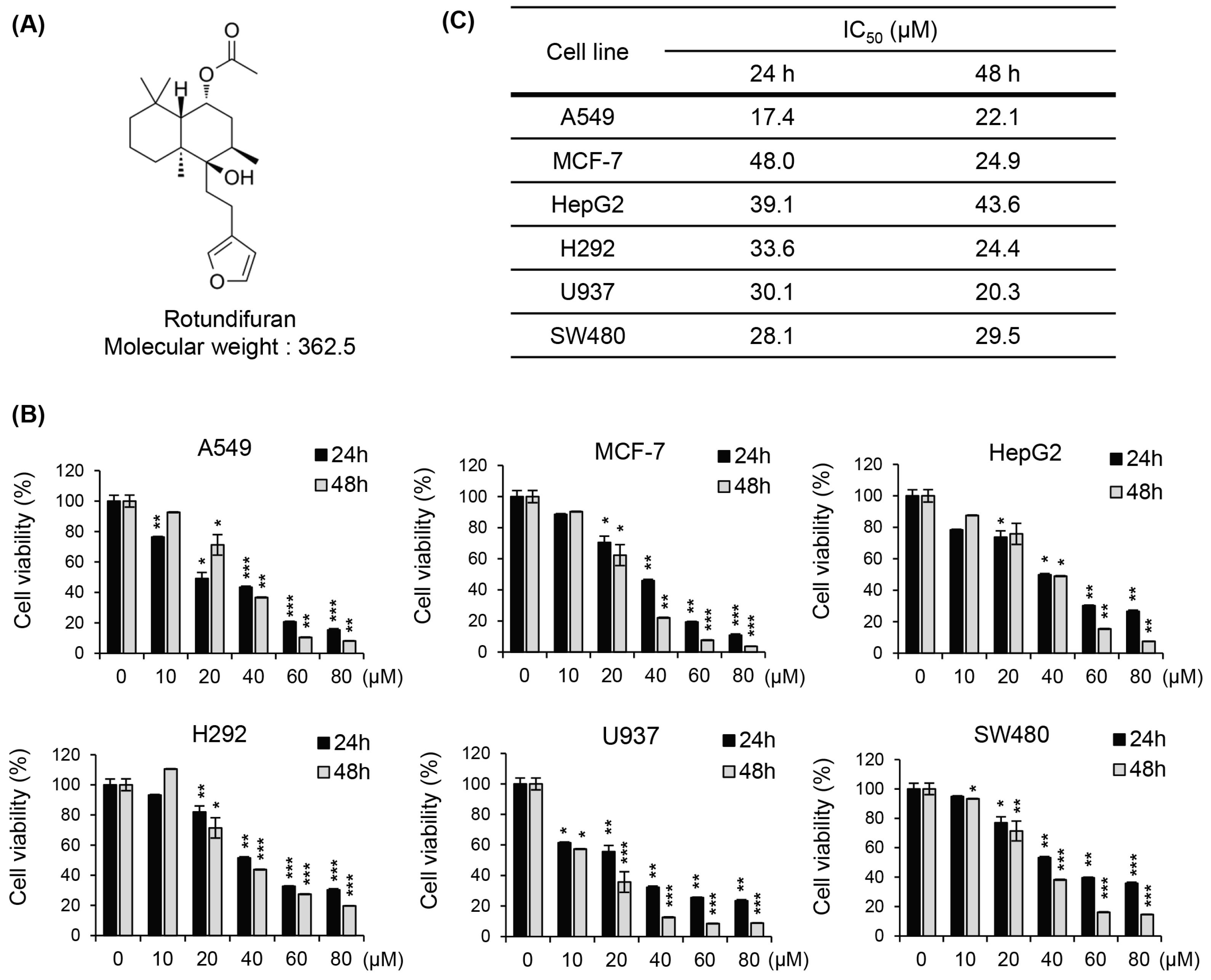
3.1. The Molecular Structures of RF and Its Cytotoxic Effects on Various Cancer Cell Lines
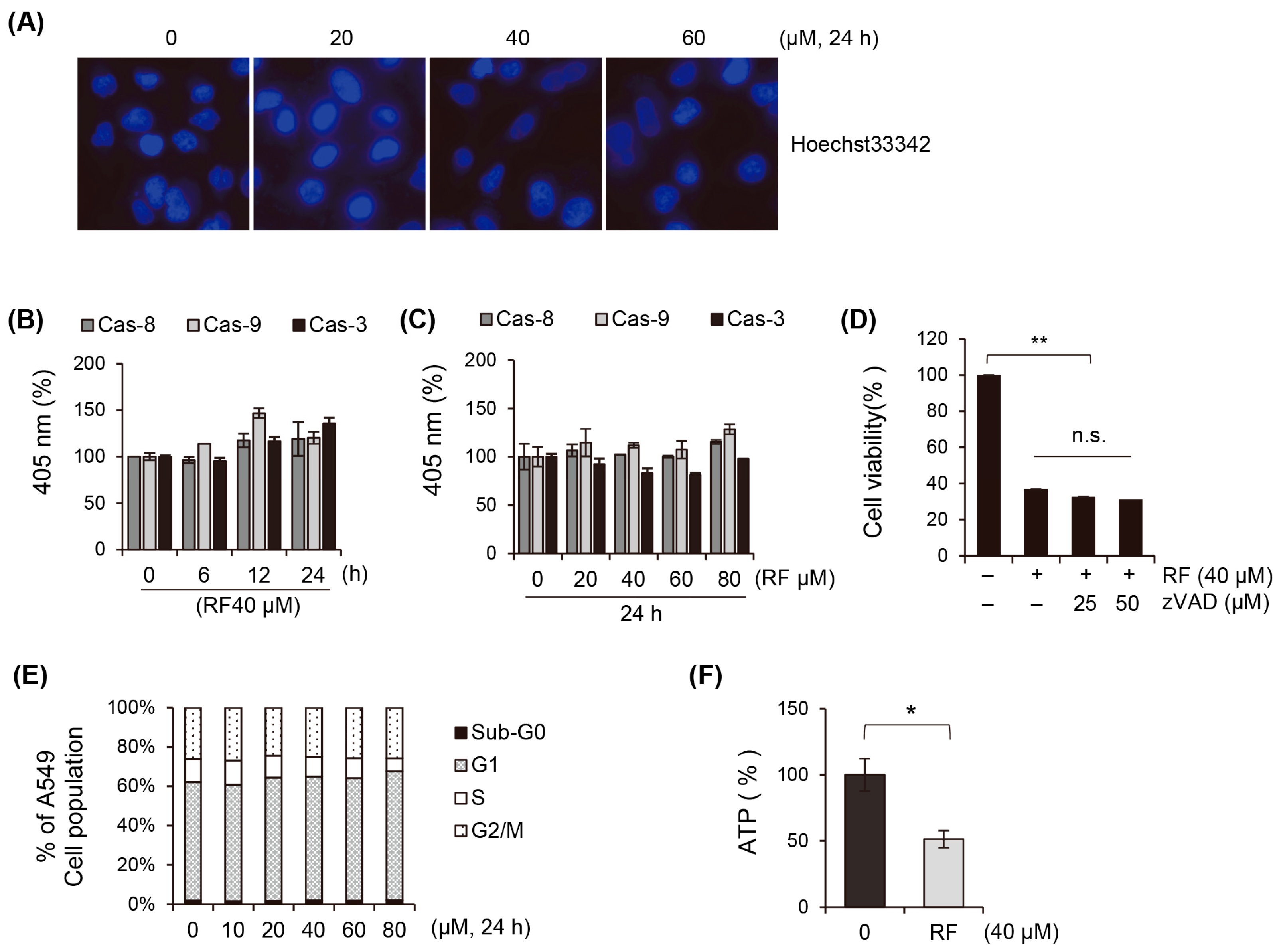
3.2. Apoptosis Does Not Play a Critical Role in the Cell Death of Cancer Cells Induced by RF
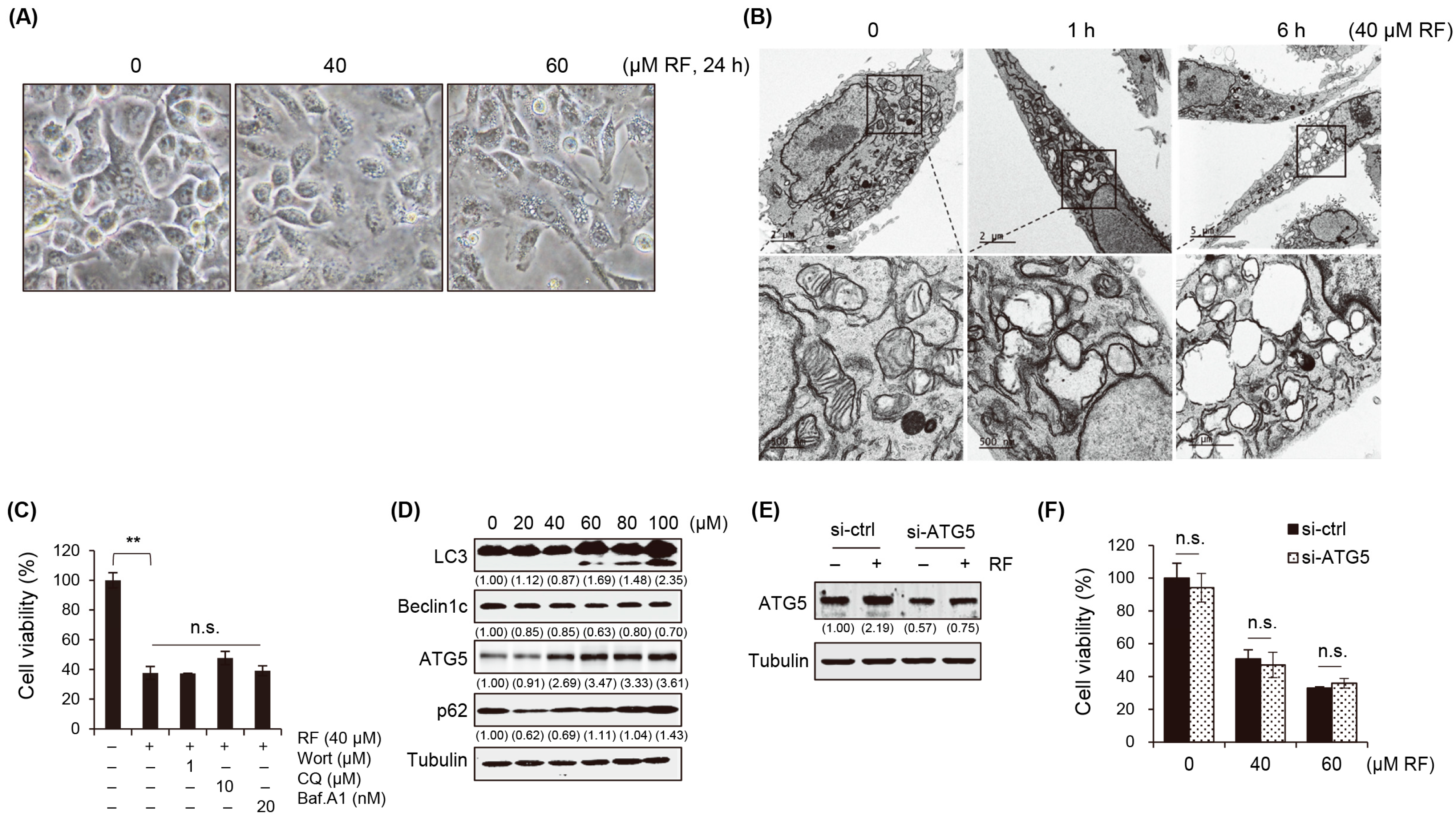
3.3. RF-Induced Autophagy Is Not Associated with Cell Death
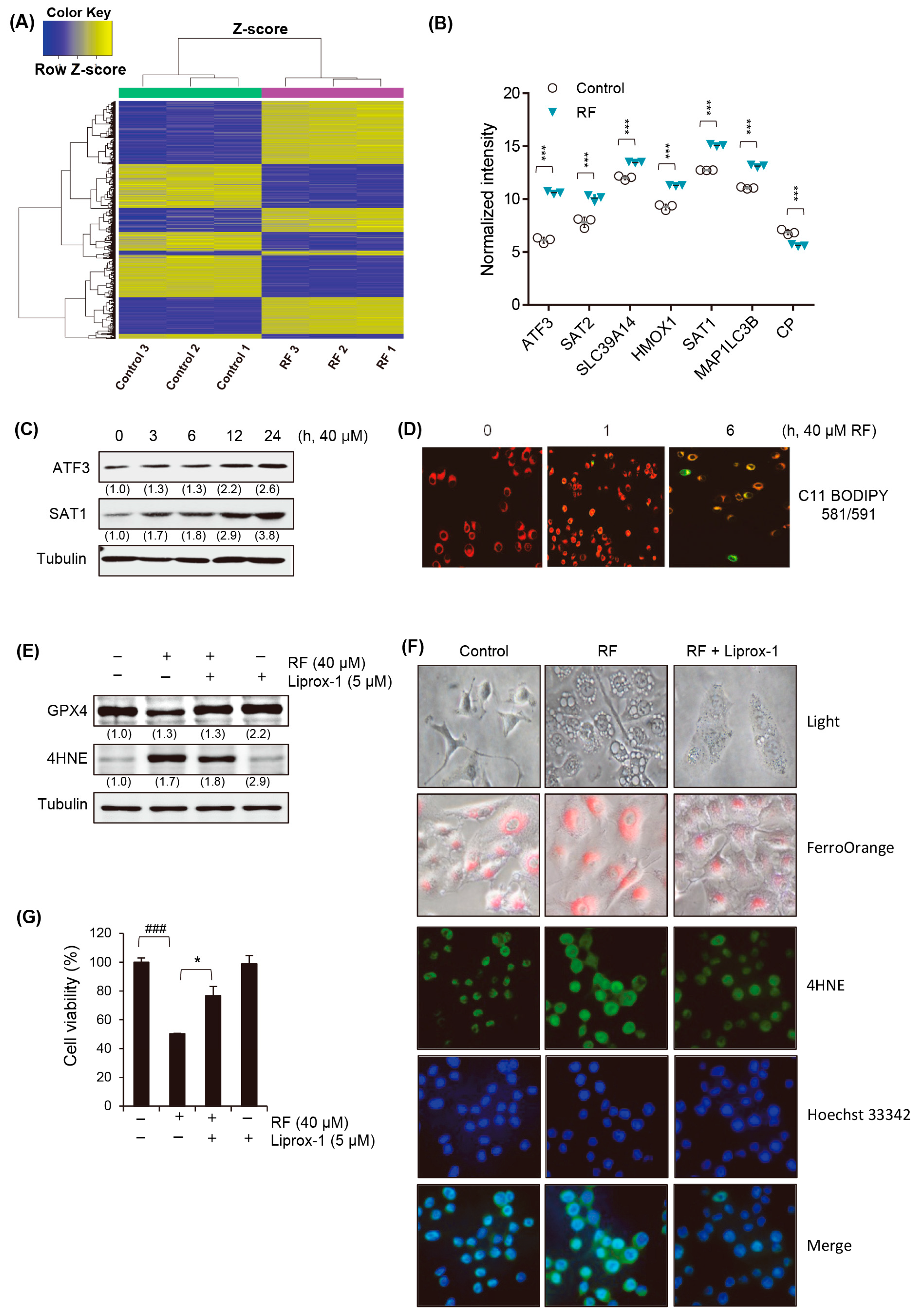
3.4. RF Induces Ferroptotic Cell Death
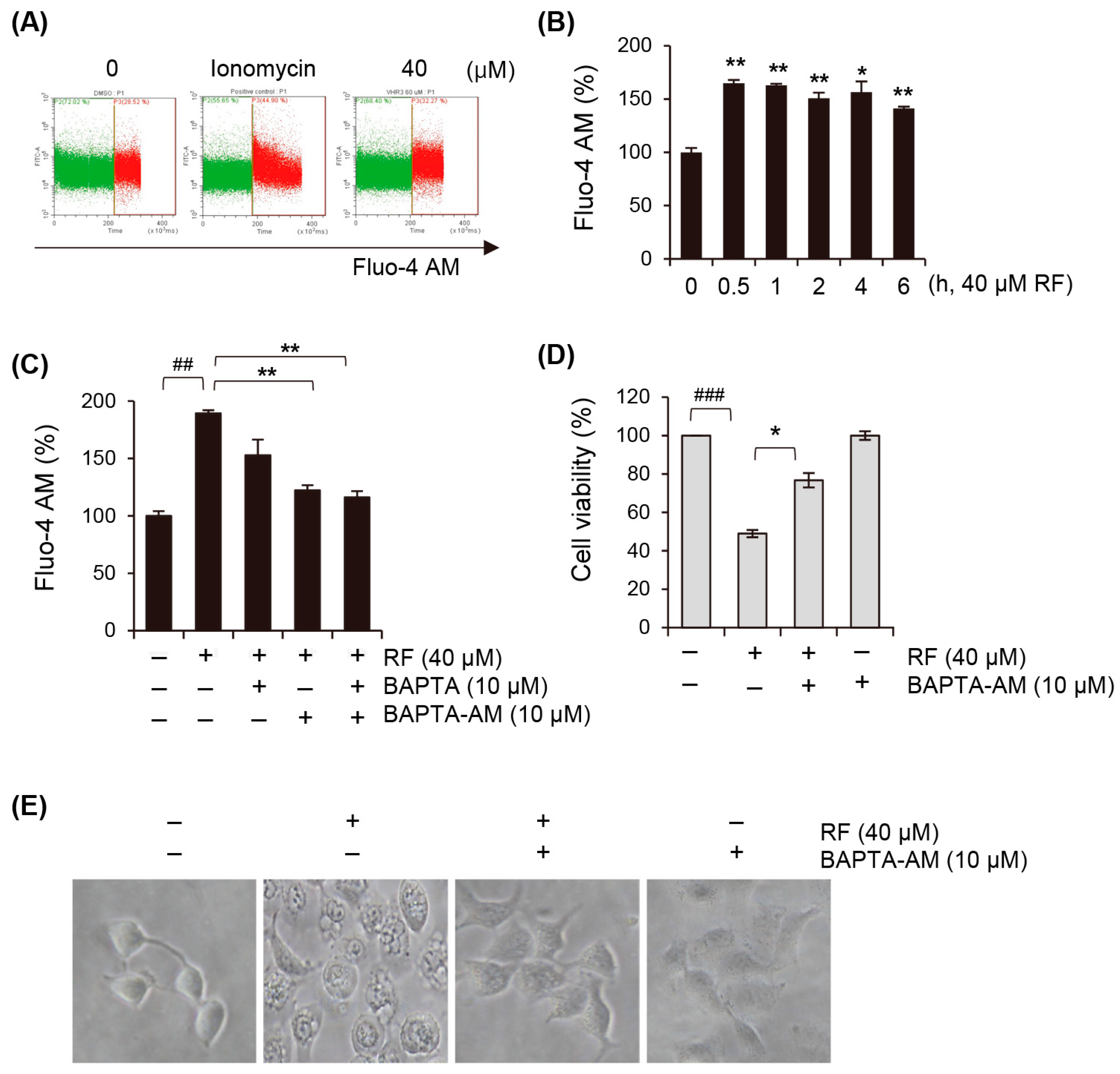
3.5. Cytosolic Calcium Concentration Is Elevated upon RF-Induced Ferroptosis
3.6. RF Induces Mitochondrial Dysfunction by Excess Calcium
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef] [PubMed]
- Gridelli, C.; Rossi, A.; Carbone, D.P.; Guarize, J.; Karachaliou, N.; Mok, T.; Petrella, F.; Spaggiari, L.; Rosell, R. Non-small-cell lung cancer. Nat. Rev. Dis. Primers 2015, 1, 15009. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Jiang, X. A physiological function for ferroptosis in tumor suppression by the immune system. Cell Metab. 2019, 29, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting ferroptosis to iron out cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Benqin, H.; Sun, X.; Li, Y.; Li, X.; Wang, Q.; Liu, K.; Li, X.; Song, L. Ferroptosis: An emerging target for human diseases. Front. Pharmacol. 2019, 10, 1228. [Google Scholar]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Badgley, M.A.; Kremer, D.M.; Maurer, H.C.; DelGiorno, K.E.; Lee, H.J.; Purohit, V.; Sagalovskiy, I.R.; Ma, A.; Kapilian, J.; Firl, C.E.; et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020, 368, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Wang, X.; Liu, Y.; Wang, M.; Yan, B.; Jiang, Y.; Shi, Y.; Shen, Y.; Liu, X.; Lai, W.; et al. A G3BP1-Interacting lncRNA Promotes Ferroptosis and Apoptosis in Cancer via Nuclear Sequestration of p53. Cancer Res. 2018, 78, 3484–3496. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Mao, L.; Zhao, T.; Song, Y.; Lin, L.; Fan, X.; Cui, B.; Feng, H.; Wang, X.; Yu, Q.; Zhang, J.; et al. The emerging role of ferroptosis in non-cancer liver diseases: Hype or increasing hope? Cell Death Dis. 2020, 11, 518. [Google Scholar] [CrossRef]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Lee, Y.M.; Jeong, Y.T.; Kim, Y.H.; Kim, J.H.; Baek, N.I.; Lee, J.W.; Cho, J.Y. Rotundifuran, isolated from the roots of Angelica gigas, inhibits the production of inflammatory mediators by suppressing NF-κB and MAPK pathways in LPS-stimulated RAW 264.7 cells. Int. Immunopharmacol. 2014, 21, 229–235. [Google Scholar]
- Kim, J.H.; Jeong, Y.T.; Lee, Y.M.; Kim, Y.H.; Hong, S.H.; Cho, J.Y. Anti-inflammatory effects of rotundifuran, an active component isolated from Angelica gigas, in RAW 264.7 cells. Int. Immunopharmacol. 2013, 16, 352–358. [Google Scholar]
- Kim, Y.H.; Kim, J.H.; Lee, Y.M.; Jeong, Y.T.; Hong, S.H.; Cho, J.Y. Rotundifuran, isolated from Angelica gigas, inhibits the production of inflammatory mediators in HaCaT cells and macrophages. Biol. Pharm. Bull. 2012, 35, 1262–1268. [Google Scholar]
- Kim, D.H.; Choi, S.S.; Kim, S.H.; Shin, J.W.; Park, H.J.; Cho, Y.W.; Cho, J.Y. Rotundifuran, isolated from Angelica gigas, ameliorates acute inflammation through inhibition of NF-κB signaling. J. Ethnopharmacol. 2014, 153, 133–140. [Google Scholar]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations With Regulated Cell Death: A Review. Front Physiol. 2019, 10, 139. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Feng, D.; Wang, Z.; Zhao, Y.; Sun, R.; Tian, D.; Liu, D.; Zhang, F.; Ning, S.; Yao, J.; et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019, 26, 2284–2299. [Google Scholar] [CrossRef] [PubMed]
- Bano, I.; Horky, P.; Abbas, S.Q.; Majid, M.; Bilal, A.H.; Ali, F.; Behl, T.; Hassan, S.S.U.; Bungau, S. Ferroptosis: A New Road towards Cancer Management. Molecules 2022, 27, 2129. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhang, S.; Jiao, J.; Zhao, S. Ferroptosis and Its Potential Role in Lung Cancer: Updated Evidence from Pathogenesis to Therapy. J. Inflamm. Res. 2021, 14, 7079–7090. [Google Scholar] [CrossRef] [PubMed]
- Patron, M.; Raffaello, A.; Granatiero, V.; Tosatto, A.; Merli, G.; De Stefani, D.; Rizzuto, R. The mitochondrial calcium uniporter (MCU): Molecular identity and physiological roles. J. Endocrinol. Investig. 2018, 41, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Kostic, M.; Ludtmann, M.H.; Bading, H. Mitochondrial Ca2+ uptake and not mitochondrial motility is required for STIM1-Orai1-dependent store-operated Ca2+ entry. J. Cell Sci. 2019, 132, jcs221812. [Google Scholar]
- Celsi, F.; Pizzo, P.; Brini, M.; Leo, S. Mitochondria, calcium, and ROS in cell death and aging. Cells 2020, 9, 1511. [Google Scholar]
- Anibal, E.V.; Roger, F.C.; Alicia, J.K.; Helena, C.F.; Nadja, C.; Tiago, R.F.; Estela, N.B. Mitochondrial calcium transport and the redox nature of the calcium-induced membrane permeability transition. Free Radic. Biol. Med. 2018, 129, 1–24. [Google Scholar]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef]
- Osada-Oka, M.; Takahashi, M.; Akiba, S.; Sato, T. Involvement of Ca2+-independent phospholipase A2 in the translocation of hypoxia-inducible factor-1α to the nucleus under hypoxic conditions. Eur. J. Pharmacol. 2006, 549, 58–62. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, H.; Daniels, J.D.; Zandkarimi, F.; Liu, H.; Brown, L.M.; Uchida, K.; O’Connor, O.A.; Stockwell, B.R. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem. Biol. 2019, 26, 623–633. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, M.-J.; Moon, D.-O.; Park, J.-Y.; Kim, N.; Lee, S.H.; Ryu, H.W.; Huh, Y.H.; Lee, H.-S.; Kim, M.-O. Rotundifuran Induces Ferroptotic Cell Death and Mitochondria Permeability Transition in Lung Cancer Cells. Biomedicines 2024, 12, 576. https://doi.org/10.3390/biomedicines12030576
Kang M-J, Moon D-O, Park J-Y, Kim N, Lee SH, Ryu HW, Huh YH, Lee H-S, Kim M-O. Rotundifuran Induces Ferroptotic Cell Death and Mitochondria Permeability Transition in Lung Cancer Cells. Biomedicines. 2024; 12(3):576. https://doi.org/10.3390/biomedicines12030576
Chicago/Turabian StyleKang, Myung-Ji, Dong-Oh Moon, Ji-Yoon Park, Namho Kim, Su Hyeon Lee, Hyung Won Ryu, Yang Hoon Huh, Hyun-Sun Lee, and Mun-Ock Kim. 2024. "Rotundifuran Induces Ferroptotic Cell Death and Mitochondria Permeability Transition in Lung Cancer Cells" Biomedicines 12, no. 3: 576. https://doi.org/10.3390/biomedicines12030576
APA StyleKang, M.-J., Moon, D.-O., Park, J.-Y., Kim, N., Lee, S. H., Ryu, H. W., Huh, Y. H., Lee, H.-S., & Kim, M.-O. (2024). Rotundifuran Induces Ferroptotic Cell Death and Mitochondria Permeability Transition in Lung Cancer Cells. Biomedicines, 12(3), 576. https://doi.org/10.3390/biomedicines12030576