
Cell Death in Liver Disease and Liver Surgery
 , , and
, , and
Abstract
1. Introduction
2. Types of Cell Death
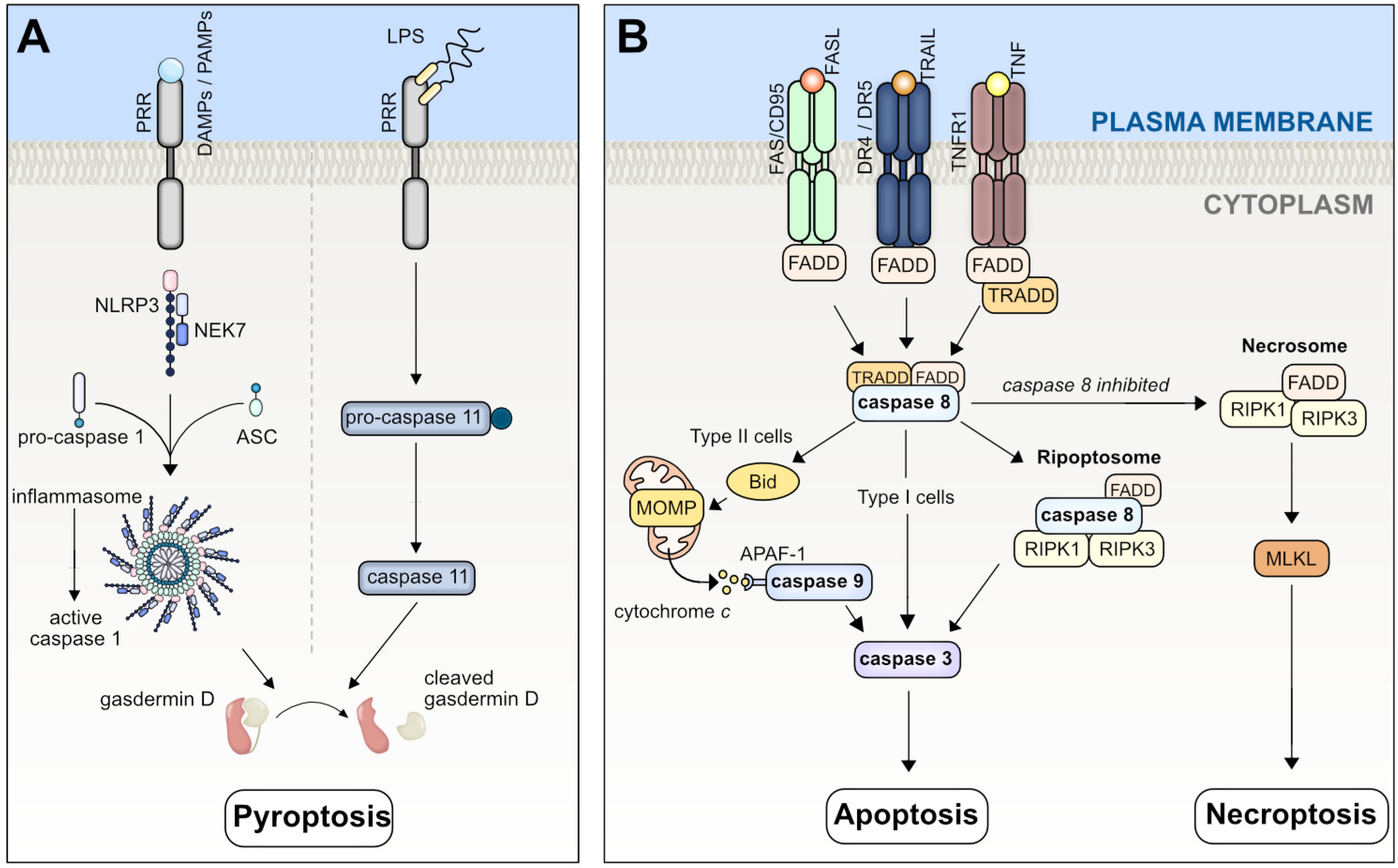
2.1. Pyroptosis
2.2. Apoptosis
2.3. Necroptosis
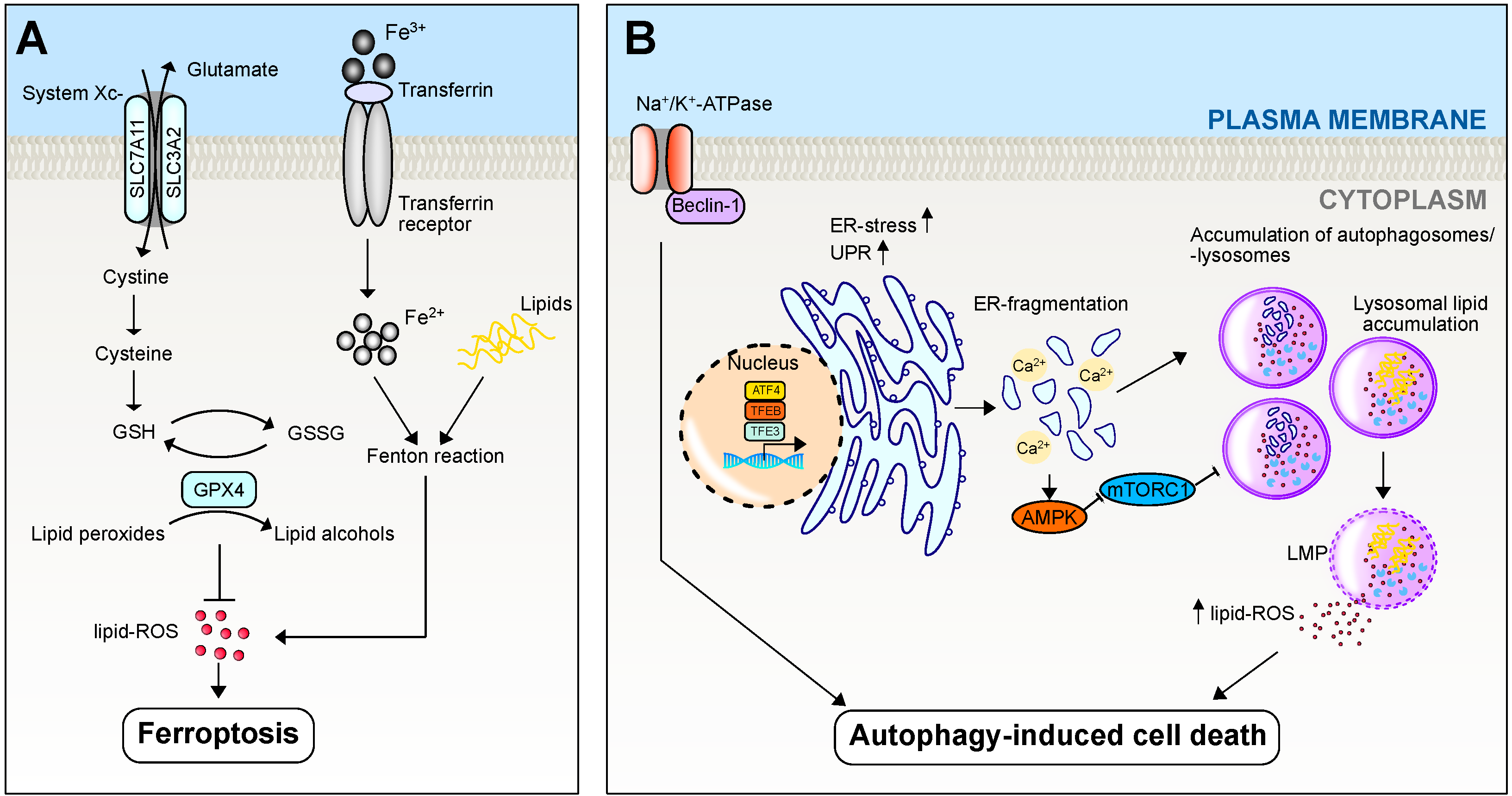
2.4. Ferroptosis
2.5. Autophagy/Autophagy-Induced Cell Death
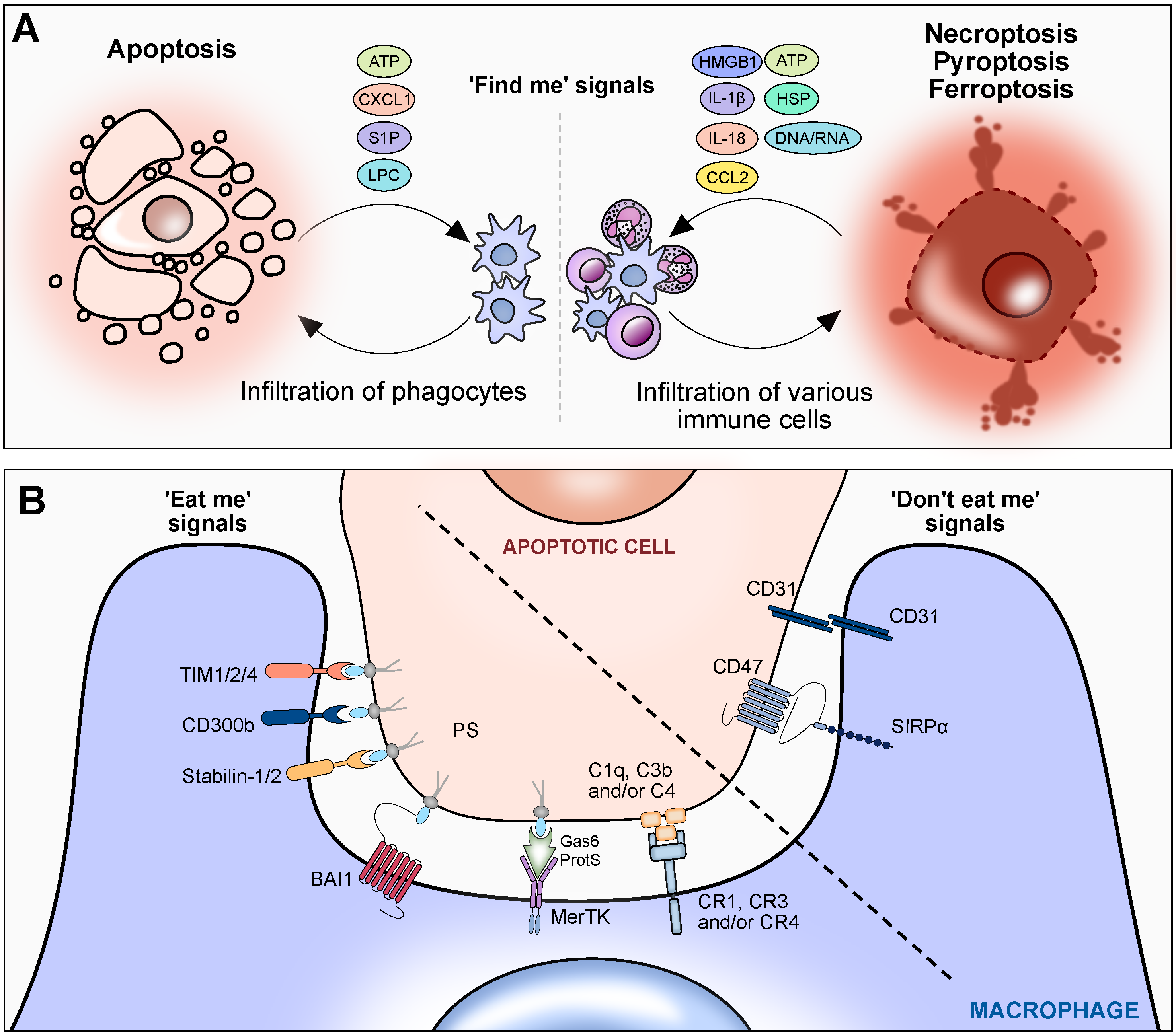
3. Cell Death Responses
3.1. ‘Find Me’ Signals
3.2. ‘Eat Me’ Signals
3.3. ‘Don’t Eat Me’ Signals
4. Cell Death in Liver Disease
4.1. Acetaminophen-Induced Liver Injury
4.1.1. Pyroptosis
4.1.2. Apoptosis
4.1.3. Necroptosis
4.1.4. Ferroptosis
4.1.5. Autophagy/Autophagy-Induced Cell Death
4.2. Alcohol-Associated Liver Disease
4.2.1. Pyroptosis
4.2.2. Apoptosis
4.2.3. Necroptosis
4.2.4. Ferroptosis
4.2.5. Autophagy/Autophagy-Induced Cell Death
4.3. Metabolic Dysfunction-Associated Steatotic Liver Disease
4.3.1. Pyroptosis
4.3.2. Apoptosis
4.3.3. Necroptosis
4.3.4. Ferroptosis
4.3.5. Autophagy/Autophagy-Induced Cell Death
4.4. Hepatocellular Carcinoma
4.4.1. Pyroptosis
4.4.2. Apoptosis
4.4.3. Necroptosis
4.4.4. Ferroptosis
4.4.5. Autophagy/Autophagy-Induced Cell Death
4.5. Tumor Microenvironment
4.5.1. Pyroptosis
4.5.2. Apoptosis
4.5.3. Necroptosis
4.5.4. Ferroptosis
4.5.5. Autophagy/Autophagy-Induced Cell Death
4.6. Liver Surgery/Ischemia-Reperfusion Injury
4.6.1. Pyroptosis
4.6.2. Apoptosis
4.6.3. Necroptosis
4.6.4. Ferroptosis
4.6.5. Autophagy/Autophagy-Induced Cell Death
5. Potential Treatment Options: Targeting Cell Death in Liver Diseases
5.1. Pyroptosis
5.2. Apoptosis
5.3. Necroptosis
5.4. Ferroptosis
5.5. Autophagy/Autophagy-Induced Cell Death
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Schwabe, R.F.; Luedde, T. Apoptosis and necroptosis in the liver: A matter of life and death. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 738–752. [Google Scholar] [CrossRef]
- Stoess, C.; Leszczynska, A.; Kui, L.; Feldstein, A.E. Pyroptosis and gasdermins—Emerging insights and therapeutic opportunities in metabolic dysfunction-associated steatohepatitis. Front. Cell Dev. Biol. 2023, 11, 1218807. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Vitale, I.; Pietrocola, F.; Guilbaud, E.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostini, M.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; et al. Apoptotic cell death in disease—Current understanding of the NCCD 2023. Cell Death Differ. 2023, 30, 1097–1154. [Google Scholar] [CrossRef]
- Knorr, J.; Wree, A.; Feldstein, A.E. Pyroptosis in Steatohepatitis and Liver Diseases. J. Mol. Biol. 2022, 434, 167271. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Alegre, K.; Deshpande, I.; Wu, S.; Lin, Z.; Kornfeld, O.S.; Lee, B.L.; Zhang, J.; Liu, J.; et al. Inhibiting membrane rupture with NINJ1 antibodies limits tissue injury. Nature 2023, 618, 1072–1077. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Kornfeld, O.S.; Lee, B.L.; Stowe, I.B.; O’Rourke, K.; Li, Q.; Sandoval, W.; Yan, D.; Kang, J.; Xu, M.; et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 2021, 591, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, K.E.; Khan, N.; Mildenhall, A.; Gerlic, M.; Croker, B.A.; D’Cruz, A.A.; Hall, C.; Kaur Spall, S.; Anderton, H.; Masters, S.L.; et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 2015, 6, 6282. [Google Scholar] [CrossRef]
- Kang, T.B.; Yang, S.H.; Toth, B.; Kovalenko, A.; Wallach, D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 2013, 38, 27–40. [Google Scholar] [CrossRef]
- Shojaie, L.; Iorga, A.; Dara, L. Cell Death in Liver Diseases: A Review. Int. J. Mol. Sci. 2020, 21, 9682. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783.e4. [Google Scholar] [CrossRef]
- Vercammen, D.; Beyaert, R.; Denecker, G.; Goossens, V.; Van Loo, G.; Declercq, W.; Grooten, J.; Fiers, W.; Vandenabeele, P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 1998, 187, 1477–1485. [Google Scholar] [CrossRef]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef]
- Lin, Y.; Devin, A.; Rodriguez, Y.; Liu, Z.G. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999, 13, 2514–2526. [Google Scholar] [CrossRef] [PubMed]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, K.; Chan, F.K. Regulation of RIPK3- and RHIM-dependent Necroptosis by the Proteasome. J. Biol. Chem. 2016, 291, 5948–5959. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Chan, F.K.; Kroemer, G. Necroptosis: Mechanisms and Relevance to Disease. Annu. Rev. Pathol. 2017, 12, 103–130. [Google Scholar] [CrossRef] [PubMed]
- Quarato, G.; Guy, C.S.; Grace, C.R.; Llambi, F.; Nourse, A.; Rodriguez, D.A.; Wakefield, R.; Frase, S.; Moldoveanu, T.; Green, D.R. Sequential Engagement of Distinct MLKL Phosphatidylinositol-Binding Sites Executes Necroptosis. Mol. Cell 2016, 61, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Matsuo, N.; Ishikura, H.; Moriguchi, T.; Yamamoto, T.; Tanaka, T. [Platelet volumetry and functional significance in septic rat: Alterations in adenylate pool: Preliminary report]. Nihon Geka Gakkai Zasshi 1989, 90, 317. [Google Scholar] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Winter, G.E.; Musavi, L.S.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol. 2015, 10, 1604–1609. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021, 593, 586–590. [Google Scholar] [CrossRef]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Muller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kossl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Chung, S.W. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019, 10, 822. [Google Scholar] [CrossRef]
- Li, X.; Chen, J.; Yuan, S.; Zhuang, X.; Qiao, T. Activation of the P62-Keap1-NRF2 Pathway Protects against Ferroptosis in Radiation-Induced Lung Injury. Oxidative Med. Cell. Longev. 2022, 2022, 8973509. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, B.; Zhu, J.; Yu, J.; Hui, J.; Xia, S.; Ji, J. Emerging Mechanisms and Targeted Therapy of Ferroptosis in Neurological Diseases and Neuro-oncology. Int. J. Biol. Sci. 2022, 18, 4260–4274. [Google Scholar] [CrossRef]
- Cheng, X.W.; Li, J.; Zhang, L.; Hu, W.J.; Zong, L.; Xu, X.; Qiao, J.P.; Zheng, M.J.; Jiang, X.W.; Liang, Z.K.; et al. Identification of SARS-CoV-2 Variants and Their Clinical Significance in Hefei, China. Front. Med. 2021, 8, 784632. [Google Scholar] [CrossRef]
- Dang, Q.; Sun, Z.; Wang, Y.; Wang, L.; Liu, Z.; Han, X. Ferroptosis: A double-edged sword mediating immune tolerance of cancer. Cell Death Dis. 2022, 13, 925. [Google Scholar] [CrossRef] [PubMed]
- Poznanski, S.M.; Singh, K.; Ritchie, T.M.; Aguiar, J.A.; Fan, I.Y.; Portillo, A.L.; Rojas, E.A.; Vahedi, F.; El-Sayes, A.; Xing, S.; et al. Metabolic flexibility determines human NK cell functional fate in the tumor microenvironment. Cell Metab. 2021, 33, 1205–1220.e5. [Google Scholar] [CrossRef]
- Liu, Y.; Duan, C.; Dai, R.; Zeng, Y. Ferroptosis-mediated Crosstalk in the Tumor Microenvironment Implicated in Cancer Progression and Therapy. Front. Cell Dev. Biol. 2021, 9, 739392. [Google Scholar] [CrossRef]
- Qian, H.; Chao, X.; Williams, J.; Fulte, S.; Li, T.; Yang, L.; Ding, W.X. Autophagy in liver diseases: A review. Mol. Asp. Med. 2021, 82, 100973. [Google Scholar] [CrossRef]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar]
- Takahashi, Y.; He, H.; Tang, Z.; Hattori, T.; Liu, Y.; Young, M.M.; Serfass, J.M.; Chen, L.; Gebru, M.; Chen, C.; et al. An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure. Nat. Commun. 2018, 9, 2855. [Google Scholar] [CrossRef]
- Eskelinen, E.L. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol. Asp. Med. 2006, 27, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Chao, X.; Qian, H.; Wang, S.; Fulte, S.; Ding, W.X. Autophagy and liver cancer. Clin. Mol. Hepatol. 2020, 26, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Boada-Romero, E.; Martinez, J.; Heckmann, B.L.; Green, D.R. The clearance of dead cells by efferocytosis. Nat. Rev. Mol. Cell Biol. 2020, 21, 398–414. [Google Scholar] [CrossRef]
- Lauber, K.; Blumenthal, S.G.; Waibel, M.; Wesselborg, S. Clearance of apoptotic cells: Getting rid of the corpses. Mol. Cell 2004, 14, 277–287. [Google Scholar] [CrossRef]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317. [Google Scholar] [CrossRef]
- Lauber, K.; Bohn, E.; Krober, S.M.; Xiao, Y.J.; Blumenthal, S.G.; Lindemann, R.K.; Marini, P.; Wiedig, C.; Zobywalski, A.; Baksh, S.; et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 2003, 113, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Gude, D.R.; Alvarez, S.E.; Paugh, S.W.; Mitra, P.; Yu, J.; Griffiths, R.; Barbour, S.E.; Milstien, S.; Spiegel, S. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J. 2008, 22, 2629–2638. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nunez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef]
- Wang, J.; Kubes, P. A Reservoir of Mature Cavity Macrophages that Can Rapidly Invade Visceral Organs to Affect Tissue Repair. Cell 2016, 165, 668–678. [Google Scholar] [CrossRef]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- McDonald, B.; Pittman, K.; Menezes, G.B.; Hirota, S.A.; Slaba, I.; Waterhouse, C.C.; Beck, P.L.; Muruve, D.A.; Kubes, P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010, 330, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Marques, P.E.; Amaral, S.S.; Pires, D.A.; Nogueira, L.L.; Soriani, F.M.; Lima, B.H.; Lopes, G.A.; Russo, R.C.; Avila, T.V.; Melgaco, J.G.; et al. Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology 2012, 56, 1971–1982. [Google Scholar] [CrossRef]
- Antoniades, C.G.; Quaglia, A.; Taams, L.S.; Mitry, R.R.; Hussain, M.; Abeles, R.; Possamai, L.A.; Bruce, M.; McPhail, M.; Starling, C.; et al. Source and characterization of hepatic macrophages in acetaminophen-induced acute liver failure in humans. Hepatology 2012, 56, 735–746. [Google Scholar] [CrossRef]
- Dal-Secco, D.; Wang, J.; Zeng, Z.; Kolaczkowska, E.; Wong, C.H.; Petri, B.; Ransohoff, R.M.; Charo, I.F.; Jenne, C.N.; Kubes, P. A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J. Exp. Med. 2015, 212, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Fadok, V.A.; Voelker, D.R.; Campbell, P.A.; Cohen, J.J.; Bratton, D.L.; Henson, P.M. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 1992, 148, 2207–2216. [Google Scholar] [CrossRef] [PubMed]
- Segawa, K.; Yanagihashi, Y.; Yamada, K.; Suzuki, C.; Uchiyama, Y.; Nagata, S. Phospholipid flippases enable precursor B cells to flee engulfment by macrophages. Proc. Natl. Acad. Sci. USA 2018, 115, 12212–12217. [Google Scholar] [CrossRef]
- Zargarian, S.; Shlomovitz, I.; Erlich, Z.; Hourizadeh, A.; Ofir-Birin, Y.; Croker, B.A.; Regev-Rudzki, N.; Edry-Botzer, L.; Gerlic, M. Phosphatidylserine externalization, “necroptotic bodies” release, and phagocytosis during necroptosis. PLoS Biol. 2017, 15, e2002711. [Google Scholar] [CrossRef] [PubMed]
- Ousingsawat, J.; Schreiber, R.; Kunzelmann, K. TMEM16F/Anoctamin 6 in Ferroptotic Cell Death. Cancers 2019, 11, 625. [Google Scholar] [CrossRef]
- Wang, Q.; Imamura, R.; Motani, K.; Kushiyama, H.; Nagata, S.; Suda, T. Pyroptotic cells externalize eat-me and release find-me signals and are efficiently engulfed by macrophages. Int. Immunol. 2013, 25, 363–372. [Google Scholar] [CrossRef]
- Westman, J.; Grinstein, S.; Marques, P.E. Phagocytosis of Necrotic Debris at Sites of Injury and Inflammation. Front. Immunol. 2019, 10, 3030. [Google Scholar] [CrossRef] [PubMed]
- Geng, K.; Kumar, S.; Kimani, S.G.; Kholodovych, V.; Kasikara, C.; Mizuno, K.; Sandiford, O.; Rameshwar, P.; Kotenko, S.V.; Birge, R.B. Requirement of Gamma-Carboxyglutamic Acid Modification and Phosphatidylserine Binding for the Activation of Tyro3, Axl, and Mertk Receptors by Growth Arrest-Specific 6. Front. Immunol. 2017, 8, 1521. [Google Scholar] [CrossRef]
- Llacuna, L.; Barcena, C.; Bellido-Martin, L.; Fernandez, L.; Stefanovic, M.; Mari, M.; Garcia-Ruiz, C.; Fernandez-Checa, J.C.; Garcia de Frutos, P.; Morales, A. Growth arrest-specific protein 6 is hepatoprotective against murine ischemia/reperfusion injury. Hepatology 2010, 52, 1371–1379. [Google Scholar] [CrossRef]
- Li, Z.; Weinman, S.A. Regulation of Hepatic Inflammation via Macrophage Cell Death. Semin. Liver Dis. 2018, 38, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Bellan, M.; Cittone, M.G.; Tonello, S.; Rigamonti, C.; Castello, L.M.; Gavelli, F.; Pirisi, M.; Sainaghi, P.P. Gas6/TAM System: A Key Modulator of the Interplay between Inflammation and Fibrosis. Int. J. Mol. Sci. 2019, 20, 5070. [Google Scholar] [CrossRef] [PubMed]
- Rantakari, P.; Patten, D.A.; Valtonen, J.; Karikoski, M.; Gerke, H.; Dawes, H.; Laurila, J.; Ohlmeier, S.; Elima, K.; Hubscher, S.G.; et al. Stabilin-1 expression defines a subset of macrophages that mediate tissue homeostasis and prevent fibrosis in chronic liver injury. Proc. Natl. Acad. Sci. USA 2016, 113, 9298–9303. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Huang, M.; Yao, Y.M. Efferocytosis and Its Role in Inflammatory Disorders. Front. Cell Dev. Biol. 2022, 10, 839248. [Google Scholar] [CrossRef] [PubMed]
- Oldenborg, P.A.; Zheleznyak, A.; Fang, Y.F.; Lagenaur, C.F.; Gresham, H.D.; Lindberg, F.P. Role of CD47 as a marker of self on red blood cells. Science 2000, 288, 2051–2054. [Google Scholar] [CrossRef]
- Poon, I.K.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef]
- Elward, K.; Griffiths, M.; Mizuno, M.; Harris, C.L.; Neal, J.W.; Morgan, B.P.; Gasque, P. CD46 plays a key role in tailoring innate immune recognition of apoptotic and necrotic cells. J. Biol. Chem. 2005, 280, 36342–36354. [Google Scholar] [CrossRef] [PubMed]
- Lerbs, T.; Cui, L.; King, M.E.; Chai, T.; Muscat, C.; Chung, L.; Brown, R.; Rieger, K.; Shibata, T.; Wernig, G. CD47 prevents the elimination of diseased fibroblasts in scleroderma. JCI Insight 2020, 5, e140458. [Google Scholar] [CrossRef]
- Shi, H.; Wang, X.; Li, F.; Gerlach, B.D.; Yurdagul, A., Jr.; Moore, M.P.; Zeldin, S.; Zhang, H.; Cai, B.; Zheng, Z.; et al. CD47-SIRPalpha axis blockade in NASH promotes necroptotic hepatocyte clearance by liver macrophages and decreases hepatic fibrosis. Sci. Transl. Med. 2022, 14, eabp8309. [Google Scholar] [CrossRef]
- Park, Y.J.; Liu, G.; Lorne, E.F.; Zhao, X.; Wang, J.; Tsuruta, Y.; Zmijewski, J.; Abraham, E. PAI-1 inhibits neutrophil efferocytosis. Proc. Natl. Acad. Sci. USA 2008, 105, 11784–11789. [Google Scholar] [CrossRef]
- Chidiac, A.S.; Buckley, N.A.; Noghrehchi, F.; Cairns, R. Paracetamol (acetaminophen) overdose and hepatotoxicity: Mechanism, treatment, prevention measures, and estimates of burden of disease. Expert Opin. Drug Metab. Toxicol. 2023, 19, 297–317. [Google Scholar] [CrossRef]
- Reuben, A.; Tillman, H.; Fontana, R.J.; Davern, T.; McGuire, B.; Stravitz, R.T.; Durkalski, V.; Larson, A.M.; Liou, I.; Fix, O.; et al. Outcomes in Adults With Acute Liver Failure Between 1998 and 2013: An Observational Cohort Study. Ann. Intern. Med. 2016, 164, 724–732. [Google Scholar] [CrossRef]
- Andrade, R.J.; Chalasani, N.; Bjornsson, E.S.; Suzuki, A.; Kullak-Ublick, G.A.; Watkins, P.B.; Devarbhavi, H.; Merz, M.; Lucena, M.I.; Kaplowitz, N.; et al. Drug-induced liver injury. Nat. Rev. Dis. Primers 2019, 5, 58. [Google Scholar] [CrossRef] [PubMed]
- Iorga, A.; Dara, L.; Kaplowitz, N. Drug-Induced Liver Injury: Cascade of Events Leading to Cell Death, Apoptosis or Necrosis. Int. J. Mol. Sci. 2017, 18, 1018. [Google Scholar] [CrossRef]
- Dara, L.; Johnson, H.; Suda, J.; Win, S.; Gaarde, W.; Han, D.; Kaplowitz, N. Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology 2015, 62, 1847–1857. [Google Scholar] [CrossRef]
- Kon, K.; Kim, J.S.; Jaeschke, H.; Lemasters, J.J. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology 2004, 40, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Shen, Y.; Huang, J.; Lu, L.; Zhang, Q. MCC950 Ameliorates Acute Liver Injury Through Modulating Macrophage Polarization and Myeloid-Derived Suppressor Cells Function. Front. Med. 2021, 8, 752223. [Google Scholar] [CrossRef]
- Li, Z.; Wang, H.; Zhu, J.; Nan, N.; Lin, Y.; Zhuang, X.; Li, L.; Zhang, Y.; Huang, P. Inhibition of TWEAK/Tnfrsf12a axis protects against acute liver failure by suppressing RIPK1-dependent apoptosis. Cell Death Discov. 2022, 8, 328. [Google Scholar] [CrossRef]
- Yang, C.; Sun, P.; Deng, M.; Loughran, P.; Li, W.; Yi, Z.; Li, S.; Zhang, X.; Fan, J.; Billiar, T.R.; et al. Gasdermin D protects against noninfectious liver injury by regulating apoptosis and necroptosis. Cell Death Dis. 2019, 10, 481. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.R.; Sharpe, M.R.; Williams, C.D.; Taha, M.; Curry, S.C.; Jaeschke, H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J. Clin. Investig. 2012, 122, 1574–1583. [Google Scholar] [CrossRef]
- McGill, M.R.; Yan, H.M.; Ramachandran, A.; Murray, G.J.; Rollins, D.E.; Jaeschke, H. HepaRG cells: A human model to study mechanisms of acetaminophen hepatotoxicity. Hepatology 2011, 53, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, X.; Anstaett, M.; Hadem, J.; Stiefel, P.; Bahr, M.J.; Lehner, F.; Manns, M.P.; Schulze-Osthoff, K.; Bantel, H. Caspase activation is associated with spontaneous recovery from acute liver failure. Hepatology 2008, 47, 1624–1633. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; McGill, M.R.; Dorko, K.; Kumer, S.C.; Schmitt, T.M.; Forster, J.; Jaeschke, H. Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicol. Appl. Pharmacol. 2014, 279, 266–274. [Google Scholar] [CrossRef]
- Yi, Y.; Zhang, W.; Tao, L.; Shao, Q.; Xu, Q.; Chen, Y.; Zhang, H.; Zhang, J.; Weng, D. RIP1 kinase inactivation protects against acetaminophen-induced acute liver injury in mice. Free Radic. Biol. Med. 2021, 174, 57–65. [Google Scholar] [CrossRef]
- Hameed, H.; Farooq, M.; Piquet-Pellorce, C.; Hamon, A.; Samson, M.; Le Seyec, J. Questioning the RIPK1 kinase activity involvement in acetaminophen-induced hepatotoxicity in mouse. Free Radic. Biol. Med. 2022, 178, 243–245. [Google Scholar] [CrossRef]
- Yamada, N.; Karasawa, T.; Kimura, H.; Watanabe, S.; Komada, T.; Kamata, R.; Sampilvanjil, A.; Ito, J.; Nakagawa, K.; Kuwata, H.; et al. Ferroptosis driven by radical oxidation of n-6 polyunsaturated fatty acids mediates acetaminophen-induced acute liver failure. Cell Death Dis. 2020, 11, 144. [Google Scholar] [CrossRef]
- Lorincz, T.; Jemnitz, K.; Kardon, T.; Mandl, J.; Szarka, A. Ferroptosis is Involved in Acetaminophen Induced Cell Death. Pathol. Oncol. Res. 2015, 21, 1115–1121. [Google Scholar] [CrossRef]
- Shan, X.; Li, J.; Liu, J.; Feng, B.; Zhang, T.; Liu, Q.; Ma, H.; Wu, H.; Wu, H. Targeting ferroptosis by poly(acrylic) acid coated Mn3O4 nanoparticles alleviates acute liver injury. Nat. Commun. 2023, 14, 7598. [Google Scholar] [CrossRef]
- Ni, H.M.; Bockus, A.; Boggess, N.; Jaeschke, H.; Ding, W.X. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 2012, 55, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Ye, H.; Chen, C.; Wu, H.; Zheng, K.; Martin-Adrados, B.; Caparros, E.; Frances, R.; Nelson, L.J.; Gomez Del Moral, M.; Asensio, I.; et al. Genetic and pharmacological inhibition of XBP1 protects against APAP hepatotoxicity through the activation of autophagy. Cell Death Dis. 2022, 13, 143. [Google Scholar] [CrossRef]
- Huang, D.Q.; Mathurin, P.; Cortez-Pinto, H.; Loomba, R. Global epidemiology of alcohol-associated cirrhosis and HCC: Trends, projections and risk factors. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 37–49. [Google Scholar] [CrossRef]
- Miyata, T.; Nagy, L.E. Programmed cell death in alcohol-associated liver disease. Clin. Mol. Hepatol. 2020, 26, 618–625. [Google Scholar] [CrossRef]
- Khanova, E.; Wu, R.; Wang, W.; Yan, R.; Chen, Y.; French, S.W.; Llorente, C.; Pan, S.Q.; Yang, Q.; Li, Y.; et al. Pyroptosis by caspase11/4-gasdermin-D pathway in alcoholic hepatitis in mice and patients. Hepatology 2018, 67, 1737–1753. [Google Scholar] [CrossRef] [PubMed]
- Petrasek, J.; Iracheta-Vellve, A.; Saha, B.; Satishchandran, A.; Kodys, K.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Szabo, G. Metabolic danger signals, uric acid and ATP, mediate inflammatory cross-talk between hepatocytes and immune cells in alcoholic liver disease. J. Leukoc. Biol. 2015, 98, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Iracheta-Vellve, A.; Petrasek, J.; Satishchandran, A.; Gyongyosi, B.; Saha, B.; Kodys, K.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Szabo, G. Inhibition of sterile danger signals, uric acid and ATP, prevents inflammasome activation and protects from alcoholic steatohepatitis in mice. J. Hepatol. 2015, 63, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Heo, M.J.; Kim, T.H.; You, J.S.; Blaya, D.; Sancho-Bru, P.; Kim, S.G. Alcohol dysregulates miR-148a in hepatocytes through FoxO1, facilitating pyroptosis via TXNIP overexpression. Gut 2019, 68, 708–720. [Google Scholar] [CrossRef]
- Nagata, S.; Tanaka, M. Programmed cell death and the immune system. Nat. Rev. Immunol. 2017, 17, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, P.; Szabo, G. Signalling pathways in alcohol-induced liver inflammation. J. Hepatol. 2009, 50, 1258–1266. [Google Scholar] [CrossRef]
- Pan, X.S.; Li, B.W.; Wang, L.L.; Li, N.; Lin, H.M.; Zhang, J.; Du, N.; Zhu, Y.Q.; Wu, X.; Hu, C.M.; et al. Kupffer cell pyroptosis mediated by METTL3 contributes to the progression of alcoholic steatohepatitis. FASEB J. 2023, 37, e22965. [Google Scholar] [CrossRef]
- Contreras-Zentella, M.L.; Villalobos-Garcia, D.; Hernandez-Munoz, R. Ethanol Metabolism in the Liver, the Induction of Oxidant Stress, and the Antioxidant Defense System. Antioxidants 2022, 11, 1258. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, A.; Shiva, S.; Wigley, A.; Ulasova, E.; Chhieng, D.; Bailey, S.M.; Darley-Usmar, V.M. The role of iNOS in alcohol-dependent hepatotoxicity and mitochondrial dysfunction in mice. Hepatology 2004, 40, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Ji, C. New Insights into the Pathogenesis of Alcohol-Induced ER Stress and Liver Diseases. Int. J. Hepatol. 2014, 2014, 513787. [Google Scholar] [CrossRef] [PubMed]
- Brentnall, M.; Rodriguez-Menocal, L.; De Guevara, R.L.; Cepero, E.; Boise, L.H. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Zhang, C.L.; Xiao, M.; Yang, R.; Xie, K.Q. Critical Roles of Kupffer Cells in the Pathogenesis of Alcoholic Liver Disease: From Basic Science to Clinical Trials. Front. Immunol. 2016, 7, 538. [Google Scholar] [CrossRef]
- Keshavarzian, A.; Farhadi, A.; Forsyth, C.B.; Rangan, J.; Jakate, S.; Shaikh, M.; Banan, A.; Fields, J.Z. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J. Hepatol. 2009, 50, 538–547. [Google Scholar] [CrossRef]
- Kishore, R.; Hill, J.R.; McMullen, M.R.; Frenkel, J.; Nagy, L.E. ERK1/2 and Egr-1 contribute to increased TNF-alpha production in rat Kupffer cells after chronic ethanol feeding. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G6–G15. [Google Scholar] [CrossRef]
- Kolios, G.; Valatas, V.; Kouroumalis, E. Role of Kupffer cells in the pathogenesis of liver disease. World J. Gastroenterol. 2006, 12, 7413–7420. [Google Scholar] [CrossRef]
- Parlesak, A.; Schafer, C.; Schutz, T.; Bode, J.C.; Bode, C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J. Hepatol. 2000, 32, 742–747. [Google Scholar] [CrossRef]
- Roychowdhury, S.; Chiang, D.J.; Mandal, P.; McMullen, M.R.; Liu, X.; Cohen, J.I.; Pollard, J.; Feldstein, A.E.; Nagy, L.E. Inhibition of apoptosis protects mice from ethanol-mediated acceleration of early markers of CCl4-induced fibrosis but not steatosis or inflammation. Alcohol. Clin. Exp. Res. 2012, 36, 1139–1147. [Google Scholar] [CrossRef]
- Roychowdhury, S.; McMullen, M.R.; Pisano, S.G.; Liu, X.; Nagy, L.E. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology 2013, 57, 1773–1783. [Google Scholar] [CrossRef]
- Miyata, T.; Wu, X.; Fan, X.; Huang, E.; Sanz-Garcia, C.; Ross, C.K.C.; Roychowdhury, S.; Bellar, A.; McMullen, M.R.; Dasarathy, J.; et al. Differential role of MLKL in alcohol-associated and non-alcohol-associated fatty liver diseases in mice and humans. JCI Insight 2021, 6, e140180. [Google Scholar] [CrossRef]
- Wu, X.; Fan, X.; McMullen, M.R.; Miyata, T.; Kim, A.; Pathak, V.; Wu, J.; Day, L.Z.; Hardesty, J.E.; Welch, N.; et al. Macrophage-derived MLKL in alcohol-associated liver disease: Regulation of phagocytosis. Hepatology 2023, 77, 902–919. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Wang, M.; Yu, H.M.; Han, F.X.; Wu, Q.S.; Cai, X.J.; Kurihara, H.; Chen, Y.X.; Li, Y.F.; He, R.R. Ferroptosis is involved in alcohol-induced cell death in vivo and in vitro. Biosci. Biotechnol. Biochem. 2020, 84, 1621–1628. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, S.; Fu, Y.; Yan, L.; Feng, Y.; Chen, Y.; Wu, Y.; Deng, Y.; Zhang, G.; Chen, Z.; et al. Computational repositioning of dimethyl fumarate for treating alcoholic liver disease. Cell Death Dis. 2020, 11, 641. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ye, T.J.; DeCaro, E.; Buehler, B.; Stahl, Z.; Bonavita, G.; Daniels, M.; You, M. Intestinal SIRT1 Deficiency Protects Mice from Ethanol-Induced Liver Injury by Mitigating Ferroptosis. Am. J. Pathol. 2020, 190, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ye, T.J.; Bonavita, G.; Daniels, M.; Kainrad, N.; Jogasuria, A.; You, M. Adipose-Specific Lipin-1 Overexpression Renders Hepatic Ferroptosis and Exacerbates Alcoholic Steatohepatitis in Mice. Hepatol. Commun. 2019, 3, 656–669. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Li, M.; Chen, X.; Ni, H.M.; Lin, C.W.; Gao, W.; Lu, B.; Stolz, D.B.; Clemens, D.L.; Yin, X.M. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 2010, 139, 1740–1752. [Google Scholar] [CrossRef] [PubMed]
- Rautou, P.E.; Mansouri, A.; Lebrec, D.; Durand, F.; Valla, D.; Moreau, R. Autophagy in liver diseases. J. Hepatol. 2010, 53, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Thomes, P.G.; Trambly, C.S.; Fox, H.S.; Tuma, D.J.; Donohue, T.M., Jr. Acute and Chronic Ethanol Administration Differentially Modulate Hepatic Autophagy and Transcription Factor EB. Alcohol. Clin. Exp. Res. 2015, 39, 2354–2363. [Google Scholar] [CrossRef] [PubMed]
- Chao, X.; Wang, S.; Zhao, K.; Li, Y.; Williams, J.A.; Li, T.; Chavan, H.; Krishnamurthy, P.; He, X.C.; Li, L.; et al. Impaired TFEB-Mediated Lysosome Biogenesis and Autophagy Promote Chronic Ethanol-Induced Liver Injury and Steatosis in Mice. Gastroenterology 2018, 155, 865–879.e12. [Google Scholar] [CrossRef]
- Kucukoglu, O.; Guldiken, N.; Chen, Y.; Usachov, V.; El-Heliebi, A.; Haybaeck, J.; Denk, H.; Trautwein, C.; Strnad, P. High-fat diet triggers Mallory-Denk body formation through misfolding and crosslinking of excess keratin 8. Hepatology 2014, 60, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Sohail, M.A.; Gomes, D.A.; Hashmi, A.; Nagata, J.; Sutterwala, F.S.; Mahmood, S.; Jhandier, M.N.; Shi, Y.; Flavell, R.A.; et al. Inflammasome-mediated regulation of hepatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G1248–G1257. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Lukens, J.R.; Kanneganti, T.D. Mitochondria: Diversity in the regulation of the NLRP3 inflammasome. Trends Mol. Med. 2015, 21, 193–201. [Google Scholar] [CrossRef]
- Gaul, S.; Leszczynska, A.; Alegre, F.; Kaufmann, B.; Johnson, C.D.; Adams, L.A.; Wree, A.; Damm, G.; Seehofer, D.; Calvente, C.J.; et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J. Hepatol. 2021, 74, 156–167. [Google Scholar] [CrossRef]
- Xu, B.; Jiang, M.; Chu, Y.; Wang, W.; Chen, D.; Li, X.; Zhang, Z.; Zhang, D.; Fan, D.; Nie, Y.; et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 2018, 68, 773–782. [Google Scholar] [CrossRef]
- Kaufmann, B.; Kui, L.; Reca, A.; Leszczynska, A.; Kim, A.D.; Booshehri, L.M.; Wree, A.; Friess, H.; Hartmann, D.; Broderick, L.; et al. Cell-specific Deletion of NLRP3 Inflammasome Identifies Myeloid Cells as Key Drivers of Liver Inflammation and Fibrosis in Murine Steatohepatitis. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 751–767. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Ferreira, D.M.; Castro, R.E.; Machado, M.V.; Evangelista, T.; Silvestre, A.; Costa, A.; Coutinho, J.; Carepa, F.; Cortez-Pinto, H.; Rodrigues, C.M. Apoptosis and insulin resistance in liver and peripheral tissues of morbidly obese patients is associated with different stages of non-alcoholic fatty liver disease. Diabetologia 2011, 54, 1788–1798. [Google Scholar] [CrossRef]
- Tenney, J.R.; Rozhkov, L.; Horn, P.; Miles, L.; Miles, M.V. Cerebral glucose hypometabolism is associated with mitochondrial dysfunction in patients with intractable epilepsy and cortical dysplasia. Epilepsia 2014, 55, 1415–1422. [Google Scholar] [CrossRef]
- Machado, M.V.; Michelotti, G.A.; Pereira, T.d.A.; Boursier, J.; Kruger, L.; Swiderska-Syn, M.; Karaca, G.; Xie, G.; Guy, C.D.; Bohinc, B.; et al. Reduced lipoapoptosis, hedgehog pathway activation and fibrosis in caspase-2 deficient mice with non-alcoholic steatohepatitis. Gut 2015, 64, 1148–1157. [Google Scholar] [CrossRef]
- Thapaliya, S.; Wree, A.; Povero, D.; Inzaugarat, M.E.; Berk, M.; Dixon, L.; Papouchado, B.G.; Feldstein, A.E. Caspase 3 inactivation protects against hepatic cell death and ameliorates fibrogenesis in a diet-induced NASH model. Dig. Dis. Sci. 2014, 59, 1197–1206. [Google Scholar] [CrossRef]
- Zhao, P.; Sun, X.; Chaggan, C.; Liao, Z.; In Wong, K.; He, F.; Singh, S.; Loomba, R.; Karin, M.; Witztum, J.L.; et al. An AMPK–caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science 2020, 367, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Goodman, Z.; Jabbar, A.; Vemulapalli, R.; Younes, Z.H.; Freilich, B.; Sheikh, M.Y.; Schattenberg, J.M.; Kayali, Z.; Zivony, A.; et al. A randomized, placebo-controlled trial of emricasan in patients with NASH and F1-F3 fibrosis. J. Hepatol. 2020, 72, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Tiegs, G.; Horst, A.K. TNF in the liver: Targeting a central player in inflammation. Semin. Immunopathol. 2022, 44, 445–459. [Google Scholar] [CrossRef]
- Afonso, M.B.; Rodrigues, P.M.; Mateus-Pinheiro, M.; Simao, A.L.; Gaspar, M.M.; Majdi, A.; Arretxe, E.; Alonso, C.; Santos-Laso, A.; Jimenez-Aguero, R.; et al. RIPK3 acts as a lipid metabolism regulator contributing to inflammation and carcinogenesis in non-alcoholic fatty liver disease. Gut 2021, 70, 2359–2372. [Google Scholar] [CrossRef] [PubMed]
- Chenxu, G.; Minxuan, X.; Yuting, Q.; Tingting, G.; Jing, F.; Jinxiao, L.; Sujun, W.; Yongjie, M.; Deshuai, L.; Qiang, L.; et al. Loss of RIP3 initiates annihilation of high-fat diet initialized nonalcoholic hepatosteatosis: A mechanism involving Toll-like receptor 4 and oxidative stress. Free Radic. Biol. Med. 2019, 134, 23–41. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Su, Y.; Sun, L.; He, S.; Meng, L.; Liao, D.; Liu, X.; Ma, Y.; Liu, C.; Li, S.; et al. Discovery of a Highly Potent, Selective, and Metabolically Stable Inhibitor of Receptor-Interacting Protein 1 (RIP1) for the Treatment of Systemic Inflammatory Response Syndrome. J. Med. Chem. 2017, 60, 972–986. [Google Scholar] [CrossRef]
- Wu, X.; Poulsen, K.L.; Sanz-Garcia, C.; Huang, E.; McMullen, M.R.; Roychowdhury, S.; Dasarathy, S.; Nagy, L.E. MLKL-dependent signaling regulates autophagic flux in a murine model of non-alcohol-associated fatty liver and steatohepatitis. J. Hepatol. 2020, 73, 616–627. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Belt, P.; Wilson, L.A.; Yeh, M.M.; Neuschwander-Tetri, B.A.; Chalasani, N.; Sanyal, A.J.; Nelson, J.E.; the NASH Clinical Research Network. Serum ferritin is an independent predictor of histologic severity and advanced fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2012, 55, 77–85. [Google Scholar] [CrossRef]
- Gao, G.; Xie, Z.; Li, E.W.; Yuan, Y.; Fu, Y.; Wang, P.; Zhang, X.; Qiao, Y.; Xu, J.; Holscher, C.; et al. Dehydroabietic acid improves nonalcoholic fatty liver disease through activating the Keap1/Nrf2-ARE signaling pathway to reduce ferroptosis. J. Nat. Med. 2021, 75, 540–552. [Google Scholar] [CrossRef]
- Loguercio, C.; De Girolamo, V.; de Sio, I.; Tuccillo, C.; Ascione, A.; Baldi, F.; Budillon, G.; Cimino, L.; Di Carlo, A.; Di Marino, M.P.; et al. Non-alcoholic fatty liver disease in an area of southern Italy: Main clinical, histological, and pathophysiological aspects. J. Hepatol. 2001, 35, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Qiu, T.; Wang, N.; Yao, X.; Jiang, L.; Jia, X.; Tao, Y.; Zhang, J.; Zhu, Y.; Yang, G.; et al. Ferroptosis mediated by the interaction between Mfn2 and IREalpha promotes arsenic-induced nonalcoholic steatohepatitis. Environ. Res. 2020, 188, 109824. [Google Scholar] [CrossRef]
- Qi, J.; Kim, J.W.; Zhou, Z.; Lim, C.W.; Kim, B. Ferroptosis Affects the Progression of Nonalcoholic Steatohepatitis via the Modulation of Lipid Peroxidation-Mediated Cell Death in Mice. Am. J. Pathol. 2020, 190, 68–81. [Google Scholar] [CrossRef]
- Li, X.; Wang, T.X.; Huang, X.; Li, Y.; Sun, T.; Zang, S.; Guan, K.L.; Xiong, Y.; Liu, J.; Yuan, H.X. Targeting ferroptosis alleviates methionine-choline deficient (MCD)-diet induced NASH by suppressing liver lipotoxicity. Liver Int. 2020, 40, 1378–1394. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, A.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillon, J.; Lo Iacono, O.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef]
- Rodriguez-Navarro, J.A.; Kaushik, S.; Koga, H.; Dall’Armi, C.; Shui, G.; Wenk, M.R.; Di Paolo, G.; Cuervo, A.M. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2012, 109, E705–E714. [Google Scholar] [CrossRef]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010, 11, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.P.; Lin, J.; Yang, Y.C.; Tsai, M.K.; Tsao, C.K.; Etzel, C.; Huang, M.; Hsu, C.Y.; Ye, Y.; Mishra, L.; et al. Hepatocellular carcinoma risk prediction model for the general population: The predictive power of transaminases. J. Natl. Cancer Inst. 2012, 104, 1599–1611. [Google Scholar] [CrossRef]
- Moossavi, M.; Parsamanesh, N.; Bahrami, A.; Atkin, S.L.; Sahebkar, A. Role of the NLRP3 inflammasome in cancer. Mol. Cancer 2018, 17, 158. [Google Scholar] [CrossRef]
- Jiang, M.; Qi, L.; Li, L.; Li, Y. The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer. Cell Death Discov. 2020, 6, 112. [Google Scholar] [CrossRef]
- Xia, X.; Wang, X.; Cheng, Z.; Qin, W.; Lei, L.; Jiang, J.; Hu, J. The role of pyroptosis in cancer: Pro-cancer or pro-”host”? Cell Death Dis. 2019, 10, 650. [Google Scholar] [CrossRef]
- Fang, G.; Zhang, Q.; Fan, J.; Li, H.; Ding, Z.; Fu, J.; Wu, Y.; Zeng, Y.; Liu, J. Pyroptosis related genes signature predicts prognosis and immune infiltration of tumor microenvironment in hepatocellular carcinoma. BMC Cancer 2022, 22, 999. [Google Scholar] [CrossRef]
- Li, Y.; Li, Y.; Zhang, X.; Duan, X.; Feng, H.; Yu, Z.; Gao, Y. A novel association of pyroptosis-related gene signature with the prognosis of hepatocellular carcinoma. Front. Oncol. 2022, 12, 986827. [Google Scholar] [CrossRef]
- Chu, Q.; Jiang, Y.; Zhang, W.; Xu, C.; Du, W.; Tuguzbaeva, G.; Qin, Y.; Li, A.; Zhang, L.; Sun, G.; et al. Pyroptosis is involved in the pathogenesis of human hepatocellular carcinoma. Oncotarget 2016, 7, 84658–84665. [Google Scholar] [CrossRef]
- Wei, Q.; Mu, K.; Li, T.; Zhang, Y.; Yang, Z.; Jia, X.; Zhao, W.; Huai, W.; Guo, P.; Han, L. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab. Investig. 2014, 94, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Da, Q.; Li, Z.; Lin, Q.; Yi, J.; Su, Y.; Yu, G.; Ren, Q.; Liu, X.; Lin, Z.; et al. Inhibition of NEK7 Suppressed Hepatocellular Carcinoma Progression by Mediating Cancer Cell Pyroptosis. Front. Oncol. 2022, 12, 812655. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Boger, R.; Vick, B.; Urbanik, T.; Haybaeck, J.; Zoller, S.; Teufel, A.; Krammer, P.H.; Opferman, J.T.; Galle, P.R.; et al. Hepatocyte-specific deletion of the antiapoptotic protein myeloid cell leukemia-1 triggers proliferation and hepatocarcinogenesis in mice. Hepatology 2010, 51, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Hikita, H.; Kodama, T.; Shimizu, S.; Li, W.; Shigekawa, M.; Tanaka, S.; Hosui, A.; Miyagi, T.; Tatsumi, T.; Kanto, T.; et al. Bak deficiency inhibits liver carcinogenesis: A causal link between apoptosis and carcinogenesis. J. Hepatol. 2012, 57, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.T.; Gautheron, J.; Feoktistova, M.; Roderburg, C.; Loosen, S.H.; Roy, S.; Benz, F.; Schemmer, P.; Buchler, M.W.; Nachbur, U.; et al. RIPK1 Suppresses a TRAF2-Dependent Pathway to Liver Cancer. Cancer Cell 2017, 31, 94–109. [Google Scholar] [CrossRef]
- Tan, S.; Zhao, J.; Sun, Z.; Cao, S.; Niu, K.; Zhong, Y.; Wang, H.; Shi, L.; Pan, H.; Hu, J.; et al. Hepatocyte-specific TAK1 deficiency drives RIPK1 kinase-dependent inflammation to promote liver fibrosis and hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2020, 117, 14231–14242. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.; Thadathil, N.; Ohene-Marfo, P.; Tran, A.L.; Van Der Veldt, M.; Georgescu, C.; Oh, S.; Nicklas, E.H.; Wang, D.; Haritha, N.H.; et al. Absence of Either Ripk3 or Mlkl Reduces Incidence of Hepatocellular Carcinoma Independent of Liver Fibrosis. Mol. Cancer Res. 2023, 21, 933–946. [Google Scholar] [CrossRef]
- Vucur, M.; Ghallab, A.; Schneider, A.T.; Adili, A.; Cheng, M.; Castoldi, M.; Singer, M.T.; Buttner, V.; Keysberg, L.S.; Kusgens, L.; et al. Sublethal necroptosis signaling promotes inflammation and liver cancer. Immunity 2023, 56, 1578–1595.e8. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tang, M.; Ke, R.X.; Li, P.L.; Sheng, Z.G.; Zhu, B.Z. The anti-cancer drug candidate CBL0137 induced necroptosis via forming left-handed Z-DNA and its binding protein ZBP1 in liver cells. Toxicol. Appl. Pharmacol. 2024, 482, 116765. [Google Scholar] [CrossRef]
- Tang, B.; Zhu, J.; Li, J.; Fan, K.; Gao, Y.; Cheng, S.; Kong, C.; Zheng, L.; Wu, F.; Weng, Q.; et al. The ferroptosis and iron-metabolism signature robustly predicts clinical diagnosis, prognosis and immune microenvironment for hepatocellular carcinoma. Cell Commun. Signal. 2020, 18, 174. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.Y.; Wang, D.S.; Lin, H.C.; Chen, X.X.; Yang, H.; Zheng, Y.; Li, Y.H. A Novel Ferroptosis-related Gene Signature for Overall Survival Prediction in Patients with Hepatocellular Carcinoma. Int. J. Biol. Sci. 2020, 16, 2430–2441. [Google Scholar] [CrossRef]
- He, F.; Zhang, P.; Liu, J.; Wang, R.; Kaufman, R.J.; Yaden, B.C.; Karin, M. ATF4 suppresses hepatocarcinogenesis by inducing SLC7A11 (xCT) to block stress-related ferroptosis. J. Hepatol. 2023, 79, 362–377. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Wu, K.; Yan, M.; Liu, T.; Wang, Z.; Duan, Y.; Xia, Y.; Ji, G.; Shen, Y.; Wang, L.; Li, L.; et al. Creatine kinase B suppresses ferroptosis by phosphorylating GPX4 through a moonlighting function. Nat. Cell Biol. 2023, 25, 714–725. [Google Scholar] [CrossRef]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef]
- Zatloukal, K.; Stumptner, C.; Fuchsbichler, A.; Heid, H.; Schnoelzer, M.; Kenner, L.; Kleinert, R.; Prinz, M.; Aguzzi, A.; Denk, H. p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am. J. Pathol. 2002, 160, 255–263. [Google Scholar] [CrossRef]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef]
- Desideri, E.; Castelli, S.; Dorard, C.; Toifl, S.; Grazi, G.L.; Ciriolo, M.R.; Baccarini, M. Impaired degradation of YAP1 and IL6ST by chaperone-mediated autophagy promotes proliferation and migration of normal and hepatocellular carcinoma cells. Autophagy 2023, 19, 152–162. [Google Scholar] [CrossRef]
- Ueno, T.; Sato, W.; Horie, Y.; Komatsu, M.; Tanida, I.; Yoshida, M.; Ohshima, S.; Mak, T.W.; Watanabe, S.; Kominami, E. Loss of Pten, a tumor suppressor, causes the strong inhibition of autophagy without affecting LC3 lipidation. Autophagy 2008, 4, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Thoen, L.F.; Guimaraes, E.L.; Dolle, L.; Mannaerts, I.; Najimi, M.; Sokal, E.; van Grunsven, L.A. A role for autophagy during hepatic stellate cell activation. J. Hepatol. 2011, 55, 1353–1360. [Google Scholar] [CrossRef]
- Myojin, Y.; Hikita, H.; Sugiyama, M.; Sasaki, Y.; Fukumoto, K.; Sakane, S.; Makino, Y.; Takemura, N.; Yamada, R.; Shigekawa, M.; et al. Hepatic Stellate Cells in Hepatocellular Carcinoma Promote Tumor Growth Via Growth Differentiation Factor 15 Production. Gastroenterology 2021, 160, 1741–1754.e16. [Google Scholar] [CrossRef] [PubMed]
- Duran, A.; Hernandez, E.D.; Reina-Campos, M.; Castilla, E.A.; Subramaniam, S.; Raghunandan, S.; Roberts, L.R.; Kisseleva, T.; Karin, M.; Diaz-Meco, M.T.; et al. p62/SQSTM1 by Binding to Vitamin D Receptor Inhibits Hepatic Stellate Cell Activity, Fibrosis, and Liver Cancer. Cancer Cell 2016, 30, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Donne, R.; Lujambio, A. The liver cancer immune microenvironment: Therapeutic implications for hepatocellular carcinoma. Hepatology 2023, 77, 1773–1796. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.Y.; Hsieh, C.H.; Lin, P.H.; Chen, Y.T.; Hsu, D.S.; Tai, S.K.; Chu, P.Y.; Yang, M.H. Snail-regulated exosomal microRNA-21 suppresses NLRP3 inflammasome activity to enhance cisplatin resistance. J. Immunother. Cancer 2022, 10, e004832. [Google Scholar] [CrossRef] [PubMed]
- Ershaid, N.; Sharon, Y.; Doron, H.; Raz, Y.; Shani, O.; Cohen, N.; Monteran, L.; Leider-Trejo, L.; Ben-Shmuel, A.; Yassin, M.; et al. NLRP3 inflammasome in fibroblasts links tissue damage with inflammation in breast cancer progression and metastasis. Nat. Commun. 2019, 10, 4375. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, D.; Hong, M.; Liu, J.; Li, Y.; Hao, J.; Lu, L.; Yin, Z.; Wu, Y. Apoptosis, Pyroptosis, and Ferroptosis Conspiringly Induce Immunosuppressive Hepatocellular Carcinoma Microenvironment and gammadelta T-Cell Imbalance. Front. Immunol. 2022, 13, 845974. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Kim, N.; Heo, J.; Shum, D.; Heo, T.; Seo, H.R. Inhibition of DNMT3B expression in activated hepatic stellate cells overcomes chemoresistance in the tumor microenvironment of hepatocellular carcinoma. Sci. Rep. 2024, 14, 115. [Google Scholar] [CrossRef]
- Wang, C.; Chen, C.; Hu, W.; Tao, L.; Chen, J. Revealing the role of necroptosis microenvironment: FCGBP + tumor-associated macrophages drive primary liver cancer differentiation towards cHCC-CCA or iCCA. Apoptosis, 2023; ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Seehawer, M.; Heinzmann, F.; D’Artista, L.; Harbig, J.; Roux, P.F.; Hoenicke, L.; Dang, H.; Klotz, S.; Robinson, L.; Dore, G.; et al. Necroptosis microenvironment directs lineage commitment in liver cancer. Nature 2018, 562, 69–75. [Google Scholar] [CrossRef]
- Conche, C.; Finkelmeier, F.; Pesic, M.; Nicolas, A.M.; Bottger, T.W.; Kennel, K.B.; Denk, D.; Ceteci, F.; Mohs, K.; Engel, E.; et al. Combining ferroptosis induction with MDSC blockade renders primary tumours and metastases in liver sensitive to immune checkpoint blockade. Gut 2023, 72, 1774–1782. [Google Scholar] [CrossRef]
- Song, Y.J.; Zhang, S.S.; Guo, X.L.; Sun, K.; Han, Z.P.; Li, R.; Zhao, Q.D.; Deng, W.J.; Xie, X.Q.; Zhang, J.W.; et al. Autophagy contributes to the survival of CD133+ liver cancer stem cells in the hypoxic and nutrient-deprived tumor microenvironment. Cancer Lett. 2013, 339, 70–81. [Google Scholar] [CrossRef]
- Lin, C.W.; Chen, Y.S.; Lin, C.C.; Lee, P.H.; Lo, G.H.; Hsu, C.C.; Hsieh, P.M.; Koh, K.W.; Chou, T.C.; Dai, C.Y.; et al. Autophagy-related gene LC3 expression in tumor and liver microenvironments significantly predicts recurrence of hepatocellular carcinoma after surgical resection. Clin. Transl. Gastroenterol. 2018, 9, e166. [Google Scholar] [CrossRef]
- Dar, W.A.; Sullivan, E.; Bynon, J.S.; Eltzschig, H.; Ju, C. Ischaemia reperfusion injury in liver transplantation: Cellular and molecular mechanisms. Liver Int. 2019, 39, 788–801. [Google Scholar] [CrossRef]
- Peralta, C.; Jimenez-Castro, M.B.; Gracia-Sancho, J. Hepatic ischemia and reperfusion injury: Effects on the liver sinusoidal milieu. J. Hepatol. 2013, 59, 1094–1106. [Google Scholar] [CrossRef] [PubMed]
- Bahde, R.; Spiegel, H.U. Hepatic ischaemia-reperfusion injury from bench to bedside. Br. J. Surg. 2010, 97, 1461–1475. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.; Ou, Z.; Wang, Y.; Liu, Y.; Sun, K.; Zhang, J.; Zhang, J.; Deng, W.; Zeng, W.; Xia, R.; et al. XBP1 modulates endoplasmic reticulum and mitochondria crosstalk via regulating NLRP3 in renal ischemia/reperfusion injury. Cell Death Discov. 2023, 9, 69. [Google Scholar] [CrossRef]
- Xu, L.; Zeng, Z.; Niu, C.; Liu, D.; Lin, S.; Liu, X.; Szabo, G.; Lu, J.; Zheng, S.; Zhou, P. Normothermic ex vivo heart perfusion with NLRP3 inflammasome inhibitor Mcc950 treatment improves cardiac function of circulatory death hearts after transplantation. Front. Cardiovasc. Med. 2023, 10, 1126391. [Google Scholar] [CrossRef]
- Sadowsky, D.; Zamora, R.; Barclay, D.; Yin, J.; Fontes, P.; Vodovotz, Y. Machine Perfusion of Porcine Livers with Oxygen-Carrying Solution Results in Reprogramming of Dynamic Inflammation Networks. Front. Pharmacol. 2016, 7, 413. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Ye, S.; Zeng, C.; Xue, S.; Hu, X.; Zhang, X.; Gao, S.; Xiong, Y.; He, X.; Vivalda, S.; et al. Hypothermic oxygenated perfusion (HOPE) attenuates ischemia/reperfusion injury in the liver through inhibition of the TXNIP/NLRP3 inflammasome pathway in a rat model of donation after cardiac death. FASEB J. 2018, 32, 6212–6227. [Google Scholar] [CrossRef] [PubMed]
- Scheuermann, U.; Zhu, M.; Song, M.; Yerxa, J.; Gao, Q.; Davis, R.P.; Zhang, M.; Parker, W.; Hartwig, M.G.; Kwun, J.; et al. Damage-Associated Molecular Patterns Induce Inflammatory Injury During Machine Preservation of the Liver: Potential Targets to Enhance a Promising Technology. Liver Transplant. 2019, 25, 610–626. [Google Scholar] [CrossRef]
- Yu, Y.; Cheng, Y.; Pan, Q.; Zhang, Y.J.; Jia, D.G.; Liu, Y.F. Effect of the Selective NLRP3 Inflammasome Inhibitor mcc950 on Transplantation Outcome in a Pig Liver Transplantation Model With Organs From Donors After Circulatory Death Preserved by Hypothermic Machine Perfusion. Transplantation 2019, 103, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, S.; Zhang, G.; Cai, J. NOD1 induces pyroptotic cell death to aggravate liver ischemia-reperfusion injury in mice. MedComm 2022, 3, e170. [Google Scholar] [CrossRef]
- Kolachala, V.L.; Lopez, C.; Shen, M.; Shayakhmetov, D.; Gupta, N.A. Ischemia reperfusion injury induces pyroptosis and mediates injury in steatotic liver thorough Caspase 1 activation. Apoptosis 2021, 26, 361–370. [Google Scholar] [CrossRef]
- Hu, Y.; Zhan, F.; Wang, Y.; Wang, D.; Lu, H.; Wu, C.; Xia, Y.; Meng, L.; Zhang, F.; Wang, X.; et al. The Ninj1/Dusp1 Axis Contributes to Liver Ischemia Reperfusion Injury by Regulating Macrophage Activation and Neutrophil Infiltration. Cell Mol. Gastroenterol. Hepatol. 2023, 15, 1071–1084. [Google Scholar] [CrossRef]
- Gao, W.; Bentley, R.C.; Madden, J.F.; Clavien, P.A. Apoptosis of sinusoidal endothelial cells is a critical mechanism of preservation injury in rat liver transplantation. Hepatology 1998, 27, 1652–1660. [Google Scholar] [CrossRef]
- Sasaki, H.; Matsuno, T.; Tanaka, N.; Orita, K. Activation of apoptosis during the reperfusion phase after rat liver ischemia. Transplant. Proc. 1996, 28, 1908–1909. [Google Scholar]
- Ye, L.; He, S.; Mao, X.; Zhang, Y.; Cai, Y.; Li, S. Effect of Hepatic Macrophage Polarization and Apoptosis on Liver Ischemia and Reperfusion Injury During Liver Transplantation. Front. Immunol. 2020, 11, 1193. [Google Scholar] [CrossRef]
- Martin-Villalba, A.; Llorens-Bobadilla, E.; Wollny, D. CD95 in cancer: Tool or target? Trends Mol. Med. 2013, 19, 329–335. [Google Scholar] [CrossRef]
- Al-Saeedi, M.; Steinebrunner, N.; Kudsi, H.; Halama, N.; Mogler, C.; Buchler, M.W.; Krammer, P.H.; Schemmer, P.; Muller, M. Neutralization of CD95 ligand protects the liver against ischemia-reperfusion injury and prevents acute liver failure. Cell Death Dis. 2018, 9, 132. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Yang, J.; Sun, G.; Hu, J.; Zhang, Q.; Cai, J.; Yuan, D.; Li, H.; Hei, Z.; Yao, W. Macrophage extracellular traps aggravate iron overload-related liver ischaemia/reperfusion injury. Br. J. Pharmacol. 2021, 178, 3783–3796. [Google Scholar] [CrossRef] [PubMed]
- Saeed, W.K.; Jun, D.W.; Jang, K.; Chae, Y.J.; Lee, J.S.; Kang, H.T. Does necroptosis have a crucial role in hepatic ischemia-reperfusion injury? PLoS ONE 2017, 12, e0184752. [Google Scholar] [CrossRef] [PubMed]
- Rosentreter, D.; Funken, D.; Reifart, J.; Mende, K.; Rentsch, M.; Khandoga, A. RIP1-Dependent Programmed Necrosis is Negatively Regulated by Caspases During Hepatic Ischemia-Reperfusion. Shock 2015, 44, 72–76. [Google Scholar] [CrossRef]
- Shi, S.; Bonaccorsi-Riani, E.; Schurink, I.; van den Bosch, T.; Doukas, M.; Lila, K.A.; Roest, H.P.; Xhema, D.; Gianello, P.; de Jonge, J.; et al. Liver Ischemia and Reperfusion Induce Periportal Expression of Necroptosis Executor pMLKL Which Is Associated With Early Allograft Dysfunction After Transplantation. Front. Immunol. 2022, 13, 890353. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.M.; Chao, X.; Kaseff, J.; Deng, F.; Wang, S.; Shi, Y.H.; Li, T.; Ding, W.X.; Jaeschke, H. Receptor-Interacting Serine/Threonine-Protein Kinase 3 (RIPK3)-Mixed Lineage Kinase Domain-Like Protein (MLKL)-Mediated Necroptosis Contributes to Ischemia-Reperfusion Injury of Steatotic Livers. Am. J. Pathol. 2019, 189, 1363–1374. [Google Scholar] [CrossRef]
- Baidya, R.; Gautheron, J.; Crawford, D.H.G.; Wang, H.; Bridle, K.R. Inhibition of MLKL Attenuates Necroptotic Cell Death in a Murine Cell Model of Ischaemia Injury. J. Clin. Med. 2021, 10, 212. [Google Scholar] [CrossRef]
- Zhong, W.; Wang, X.; Rao, Z.; Pan, X.; Sun, Y.; Jiang, T.; Wang, P.; Zhou, H.; Wang, X. Aging aggravated liver ischemia and reperfusion injury by promoting hepatocyte necroptosis in an endoplasmic reticulum stress-dependent manner. Ann. Transl. Med. 2020, 8, 869. [Google Scholar] [CrossRef]
- Xu, J.; Wu, D.; Zhou, S.; Hu, H.; Li, F.; Guan, Z.; Zhan, X.; Gao, Y.; Wang, P.; Rao, Z. MLKL deficiency attenuated hepatocyte oxidative DNA damage by activating mitophagy to suppress macrophage cGAS-STING signaling during liver ischemia and reperfusion injury. Cell Death Discov. 2023, 9, 58. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Qu, X.; Tian, Y.; Jie, Z.; Xi, Z.; Xue, F.; Ma, X.; Zhu, J.; Xia, Q. Macrophage Notch1 inhibits TAK1 function and RIPK3-mediated hepatocyte necroptosis through activation of beta-catenin signaling in liver ischemia and reperfusion injury. Cell Commun. Signal 2022, 20, 144. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Song, Z.; Yu, J.; Li, C.; Jin, C.; Duan, W.; Liu, X.; Liu, Y.; Huang, S.; Tuo, Y.; et al. Hepatocyte-specific TMEM16A deficiency alleviates hepatic ischemia/reperfusion injury via suppressing GPX4-mediated ferroptosis. Cell Death Dis. 2022, 13, 1072. [Google Scholar] [CrossRef]
- Morishita, H.; Mizushima, N. Diverse Cellular Roles of Autophagy. Annu. Rev. Cell Dev. Biol. 2019, 35, 453–475. [Google Scholar] [CrossRef]
- Liu, A.; Wang, W.; Lu, Z.; Liu, Z.; Zhou, W.; Zhong, Z.; Ye, Q. Mild hypothermia pretreatment extenuates liver ischemia-reperfusion injury through Rab7-mediated autophagosomes-lysosomes fusion. Biochem. Biophys. Res. Commun. 2021, 550, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.N.; Li, W.; Bai, C.; Dong, Y.; Wu, Y.; An, W. Augmenter of liver regeneration-mediated mitophagy protects against hepatic ischemia/reperfusion injury. Am. J. Transplant. 2022, 22, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Witek, R.P.; Stone, W.C.; Karaca, F.G.; Syn, W.K.; Pereira, T.A.; Agboola, K.M.; Omenetti, A.; Jung, Y.; Teaberry, V.; Choi, S.S.; et al. Pan-caspase inhibitor VX-166 reduces fibrosis in an animal model of nonalcoholic steatohepatitis. Hepatology 2009, 50, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Concas, D.; Kudo, H.; Levene, A.; Pollard, J.; Charlton, P.; Thomas, H.C.; Thursz, M.R.; Goldin, R.D. Impact of pan-caspase inhibition in animal models of established steatosis and non-alcoholic steatohepatitis. J. Hepatol. 2010, 53, 542–550. [Google Scholar] [CrossRef]
- Barreyro, F.J.; Holod, S.; Finocchietto, P.V.; Camino, A.M.; Aquino, J.B.; Avagnina, A.; Carreras, M.C.; Poderoso, J.J.; Gores, G.J. The pan-caspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis. Liver Int. 2015, 35, 953–966. [Google Scholar] [CrossRef]
- Frenette, C.T.; Morelli, G.; Shiffman, M.L.; Frederick, R.T.; Rubin, R.A.; Fallon, M.B.; Cheng, J.T.; Cave, M.; Khaderi, S.A.; Massoud, O.; et al. Emricasan Improves Liver Function in Patients with Cirrhosis and High Model for End-Stage Liver Disease Scores Compared With Placebo. Clin. Gastroenterol. Hepatol. 2019, 17, 774–783.e4. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Sheikh, M.Y.; Sanyal, A.J.; Lim, J.K.; Conjeevaram, H.; Chalasani, N.; Abdelmalek, M.; Bakken, A.; Renou, C.; Palmer, M.; et al. A phase 2, randomized, double-blind, placebo-controlled study of GS-9450 in subjects with nonalcoholic steatohepatitis. Hepatology 2012, 55, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Tsao, G.; Bosch, J.; Kayali, Z.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.; Satapathy, S.K.; Ghabril, M.; Shiffman, M.L.; Younes, Z.H.; et al. Randomized placebo-controlled trial of emricasan for non-alcoholic steatohepatitis-related cirrhosis with severe portal hypertension. J. Hepatol. 2020, 72, 885–895. [Google Scholar] [CrossRef]
- Safadi, R.; Konikoff, F.M.; Mahamid, M.; Zelber-Sagi, S.; Halpern, M.; Gilat, T.; Oren, R.; FLORA Group. The fatty acid-bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2014, 12, 2085–2091.e1. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Wang, J.; Yu, F.; Tang, Y.; Ding, G.; Yang, Z.; Sun, Y. Protective Effect of Meretrix meretrix Oligopeptides on High-Fat-Diet-Induced Non-Alcoholic Fatty Liver Disease in Mice. Mar. Drugs 2018, 16, 39. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Johnson, J.D.; Lee, J.J.; Song, J.; Matthews, M.; Hellerstein, M.K.; McWherter, C.A. Seladelpar combined with complementary therapies improves fibrosis, inflammation, and liver injury in a mouse model of nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2024, 326, G120–G132. [Google Scholar] [CrossRef]
- Hajighasem, A.; Farzanegi, P.; Mazaheri, Z. Effects of combined therapy with resveratrol, continuous and interval exercises on apoptosis, oxidative stress, and inflammatory biomarkers in the liver of old rats with non-alcoholic fatty liver disease. Arch. Physiol. Biochem. 2019, 125, 142–149. [Google Scholar] [CrossRef]
- Sharma, R.S.; Harrison, D.J.; Kisielewski, D.; Cassidy, D.M.; McNeilly, A.D.; Gallagher, J.R.; Walsh, S.V.; Honda, T.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; et al. Experimental Nonalcoholic Steatohepatitis and Liver Fibrosis Are Ameliorated by Pharmacologic Activation of Nrf2 (NF-E2 p45-Related Factor 2). Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 367–398. [Google Scholar] [CrossRef]
- Park, H.S.; Song, J.W.; Park, J.H.; Lim, B.K.; Moon, O.S.; Son, H.Y.; Lee, J.H.; Gao, B.; Won, Y.S.; Kwon, H.J. TXNIP/VDUP1 attenuates steatohepatitis via autophagy and fatty acid oxidation. Autophagy 2021, 17, 2549–2564. [Google Scholar] [CrossRef] [PubMed]
- Love, S.; Mudasir, M.A.; Bhardwaj, S.C.; Singh, G.; Tasduq, S.A. Long-term administration of tacrolimus and everolimus prevents high cholesterol-high fructose-induced steatosis in C57BL/6J mice by inhibiting de-novo lipogenesis. Oncotarget 2017, 8, 113403–113417. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.G.; Jeng, L.-B.; Saliba, F.; Soin, A.S.; Lee, W.-C.; De Simone, P.; Nevens, F.; Suh, K.-S.; Fischer, L.; Joo, D.J.; et al. Efficacy and Safety of Everolimus With Reduced Tacrolimus in Liver Transplant Recipients: 24-month Results From the Pooled Analysis of 2 Randomized Controlled Trials. Transplantation 2021, 105, 1564–1575. [Google Scholar] [CrossRef]
- Zhang, D.; Ma, Y.; Liu, J.; Deng, Y.; Zhou, B.; Wen, Y.; Li, M.; Wen, D.; Ying, Y.; Luo, S.; et al. Metformin Alleviates Hepatic Steatosis and Insulin Resistance in a Mouse Model of High-Fat Diet-Induced Nonalcoholic Fatty Liver Disease by Promoting Transcription Factor EB-Dependent Autophagy. Front. Pharmacol. 2021, 12, 689111. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Xuan, W.; Li, J.; Yao, H.; Huang, C.; Li, J. AMPK protects against alcohol-induced liver injury through UQCRC2 to up-regulate mitophagy. Autophagy 2021, 17, 3622–3643. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, G.; Han, D.H.; Lee, M.; Kim, I.; Kim, B.; Kim, K.H.; Song, Y.M.; Yoo, J.E.; Wang, H.J.; et al. Ezetimibe ameliorates steatohepatitis via AMP activated protein kinase-TFEB-mediated activation of autophagy and NLRP3 inflammasome inhibition. Autophagy 2017, 13, 1767–1781. [Google Scholar] [CrossRef]
- Loomba, R.; Sanyal, A.J.; Kowdley, K.V.; Bhatt, D.L.; Alkhouri, N.; Frias, J.P.; Bedossa, P.; Harrison, S.A.; Lazas, D.; Barish, R.; et al. Randomized, Controlled Trial of the FGF21 Analogue Pegozafermin in NASH. N. Engl. J. Med. 2023, 389, 998–1008. [Google Scholar] [CrossRef]
- Zeng, X.; Yang, J.; Hu, O.; Huang, J.; Ran, L.; Chen, M.; Zhang, Y.; Zhou, X.; Zhu, J.; Zhang, Q.; et al. Dihydromyricetin Ameliorates Nonalcoholic Fatty Liver Disease by Improving Mitochondrial Respiratory Capacity and Redox Homeostasis Through Modulation of SIRT3 Signaling. Antioxid. Redox Signal. 2019, 30, 163–183. [Google Scholar] [CrossRef]
- Xu, X.; Zhu, X.P.; Bai, J.Y.; Xia, P.; Li, Y.; Lu, Y.; Li, X.Y.; Gao, X. Berberine alleviates nonalcoholic fatty liver induced by a high-fat diet in mice by activating SIRT3. FASEB J. 2019, 33, 7289–7300. [Google Scholar] [CrossRef]
- Yan, C.; Zhang, Y.; Zhang, X.; Aa, J.; Wang, G.; Xie, Y. Curcumin regulates endogenous and exogenous metabolism via Nrf2-FXR-LXR pathway in NAFLD mice. Biomed. Pharmacother. 2018, 105, 274–281. [Google Scholar] [CrossRef]
- Wang, X.; Chang, X.; Zhan, H.; Zhang, Q.; Li, C.; Gao, Q.; Yang, M.; Luo, Z.; Li, S.; Sun, Y. Curcumin and Baicalin ameliorate ethanol-induced liver oxidative damage via the Nrf2/HO-1 pathway. J. Food Biochem. 2020, 44, e13425. [Google Scholar] [CrossRef] [PubMed]
- Hammoutene, A.; Laouirem, S.; Albuquerque, M.; Colnot, N.; Brzustowski, A.; Valla, D.; Provost, N.; Delerive, P.; Paradis, V.; QUID-NASH Research Group. A new NRF2 activator for the treatment of human metabolic dysfunction-associated fatty liver disease. JHEP Rep. 2023, 5, 100845. [Google Scholar] [CrossRef]
- Yue, S.; Zhou, H.; Wang, X.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Zhai, Y. Prolonged Ischemia Triggers Necrotic Depletion of Tissue-Resident Macrophages To Facilitate Inflammatory Immune Activation in Liver Ischemia Reperfusion Injury. J. Immunol. 2017, 198, 3588–3595. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.F.; He, W.; Zhang, C.; Liu, X.J.; Lu, Y.; Wang, H.; Zhang, Z.H.; Chen, X.; Xu, D.X. Role of receptor interacting protein (RIP)1 on apoptosis-inducing factor-mediated necroptosis during acetaminophen-evoked acute liver failure in mice. Toxicol. Lett. 2014, 225, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Majdi, A.; Aoudjehane, L.; Ratziu, V.; Islam, T.; Afonso, M.B.; Conti, F.; Mestiri, T.; Lagouge, M.; Foufelle, F.; Ballenghien, F.; et al. Inhibition of receptor-interacting protein kinase 1 improves experimental non-alcoholic fatty liver disease. J. Hepatol. 2020, 72, 627–635. [Google Scholar] [CrossRef]
- Qian, X.; Jin, P.; Fan, K.; Pei, H.; He, Z.; Du, R.; Cao, C.; Yang, Y. Polystyrene microplastics exposure aggravates acute liver injury by promoting Kupffer cell pyroptosis. Int. Immunopharmacol. 2024, 126, 111307. [Google Scholar] [CrossRef]
- Coll, R.C.; Schroder, K.; Pelegrin, P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol. Sci. 2022, 43, 653–668. [Google Scholar] [CrossRef]
- Sun, K.; Wang, J.; Lan, Z.; Li, L.; Wang, Y.; Li, A.; Liu, S.; Li, Y. Sleeve Gastroplasty Combined with the NLRP3 Inflammasome Inhibitor CY-09 Reduces Body Weight, Improves Insulin Resistance and Alleviates Hepatic Steatosis in Mouse Model. Obes. Surg. 2020, 30, 3435–3443. [Google Scholar] [CrossRef]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- Marin-Aguilar, F.; Castejon-Vega, B.; Alcocer-Gomez, E.; Lendines-Cordero, D.; Cooper, M.A.; de la Cruz, P.; Andujar-Pulido, E.; Perez-Alegre, M.; Muntane, J.; Perez-Pulido, A.J.; et al. NLRP3 Inflammasome Inhibition by MCC950 in Aged Mice Improves Health via Enhanced Autophagy and PPARalpha Activity. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 1457–1464. [Google Scholar] [CrossRef]
- Yan, S.L.; Huang, C.S.; Mong, M.C.; Yin, M.C. Oridonin Attenuates the Effects of Chronic Alcohol Consumption Inducing Oxidative, Glycative and Inflammatory Injury in the Mouse Liver. In Vivo 2021, 35, 2141–2149. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Qin, H.; Yang, B.; Du, B.; Yun, X. Oridonin ameliorates carbon tetrachloride-induced liver fibrosis in mice through inhibition of the NLRP3 inflammasome. Drug Dev. Res. 2020, 81, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Vergis, N.; Patel, V.; Bogdanowicz, K.; Czyzewska-Khan, J.; Fiorentino, F.; Day, E.; Cross, M.; Foster, N.; Lord, E.; Goldin, R.; et al. IL-1 Signal Inhibition In Alcoholic Hepatitis (ISAIAH): A study protocol for a multicentre, randomised, placebo-controlled trial to explore the potential benefits of canakinumab in the treatment of alcoholic hepatitis. Trials 2021, 22, 792. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhang, X.; Yang, W.; Li, M.; Wang, G.; Luo, Q. Ferrostatin-1 Ameliorates Liver Dysfunction via Reducing Iron in Thioacetamide-induced Acute Liver Injury in Mice. Front. Pharmacol. 2022, 13, 869794. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Lan, X.T.; Zhang, Z.; Liu, Y.; Sun, D.Y.; Wang, X.J.; Ou-Yang, S.X.; Zhuang, C.L.; Shen, F.M.; Wang, P.; et al. Ferroptosis inhibitor liproxstatin-1 alleviates metabolic dysfunction-associated fatty liver disease in mice: Potential involvement of PANoptosis. Acta Pharmacol. Sin. 2023, 44, 1014–1028. [Google Scholar] [CrossRef]
- Li, S.; Zhuge, A.; Wang, K.; Xia, J.; Wang, Q.; Han, S.; Shen, J.; Li, L. Obeticholic acid and ferrostatin-1 differentially ameliorate non-alcoholic steatohepatitis in AMLN diet-fed ob/ob mice. Front. Pharmacol. 2022, 13, 1081553. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Feng, B.; Yu, J.; Yan, L.; Che, L.; Zhuo, Y.; Luo, Y.; Yu, B.; Wu, D.; Chen, D. Fibroblast growth factor 21 attenuates iron overload-induced liver injury and fibrosis by inhibiting ferroptosis. Redox Biol. 2021, 46, 102131. [Google Scholar] [CrossRef]
- Wang, X.; Sun, K.; Zhou, Y.; Wang, H.; Zhou, Y.; Liu, S.; Nie, Y.; Li, Y. NLRP3 inflammasome inhibitor CY-09 reduces hepatic steatosis in experimental NAFLD mice. Biochem. Biophys. Res. Commun. 2021, 534, 734–739. [Google Scholar] [CrossRef]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, H.; Zhang, J.; Ou, F.; Wang, J.; Liu, T.; Wu, J. Disulfiram alleviates acute lung injury and related intestinal mucosal barrier impairment by targeting GSDMD-dependent pyroptosis. J. Inflamm. 2022, 19, 17. [Google Scholar] [CrossRef]
- Cai, Q.; Sun, Z.; Xu, S.; Jiao, X.; Guo, S.; Li, Y.; Wu, H.; Yu, X. Disulfiram ameliorates ischemia/reperfusion-induced acute kidney injury by suppressing the caspase-11-GSDMD pathway. Ren. Fail. 2022, 44, 1169–1181. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Mifflin, L.; Ofengeim, D.; Yuan, J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat. Rev. Drug Discov. 2020, 19, 553–571. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.H.; Park, S.; Hong, E.; Choi, M.A.; Kwon, Y.M.; Park, J.W.; Lee, A.H.; Park, G.R.; Kim, H.Y.; Lee, S.M.; et al. Novel Inhibitor of Mixed-Lineage Kinase Domain-Like Protein: The Antifibrotic Effects of a Necroptosis Antagonist. ACS Pharmacol. Transl. Sci. 2023, 6, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Chen, D.; Zhu, Y.; Pan, T.; Xia, D.; Cai, T.; Lin, H.; Lin, J.; Jin, X.; Wu, F.; et al. GPX4-Regulated Ferroptosis Mediates S100-Induced Experimental Autoimmune Hepatitis Associated with the Nrf2/HO-1 Signaling Pathway. Oxid. Med. Cell Longev. 2021, 2021, 6551069. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.; Li, H.; Long, X.; Rye, K.A.; Ong, K.L. Fibroblast growth factor 21 in non-alcoholic fatty liver disease. Metabolism 2019, 101, 153994. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Cell Death | Drug | Stage | Target of Compound | Disease/Model | Ref. |
|---|---|---|---|---|---|
| Apoptosis | VX-166 | Preclinical | Pan-caspase | MASLD/MASH Cirrhosis | [135,220,221,222,223,224,225] |
| GS-9450 | Phase II, randomized, placebo-controlled trial | ||||
| Emricasan (IDN-6556) | Randomized, placebo-controlled trial | ||||
| Aramchol | Randomized, placebo-controlled trial | Stearoyl-CoA desaturase 1 | MASLD | [226] | |
| Meretrix oligopeptides | Preclinical | NF-κB | MASLD | [227] | |
| Seladelpar (MBX-8025) | Preclinical | Proliferator-activated receptor-delta | MASH | [228] | |
| Resveratrol | Preclinical | SIRT1 | MASLD | [229] | |
| TBE-31 | Preclinical | NRF2 | MASH/Fibrosis | [230] | |
| Autophagy | Rapamycin | Preclinical | mTORC1 | MASLD/MASH Liver transplantation with HCC | [231,232,233] |
| Everolimus | Randomized trial | ||||
| Metformin | Preclinical | AMPK | ALD MASLD | [234,235] | |
| Ezetimibe | Preclinical | AMPK, TFEB | MASH | [236] | |
| FGF21 | Randomized, placebo-controlled trial | AMPK | MASLD/MASH | [237] | |
| Dihydromyricetin Berberine | Preclinical | SIRT3 | MASLD | [238,239] | |
| Curcumin | Preclinical | NRF2/FXR/LXRα | MASLD ALD | [240,241] | |
| S217879 | Preclinical | NRF2 | MASLD | [242] | |
| Necroptosis | Necrostatin-1 | Preclinical | RIPK1 | Liver I/R injury Drug-induced acute liver injury | [243,244] |
| RIPA-56 | Preclinical | RIPK1 | MASLD/MASH | [245] | |
| Necrosulfonamide | Preclinical | MLKL | CCL4-induced acute injury | [246] | |
| Pyroptosis | CY-09, Tranilast, Oridonin MCC950 IFM-514 | Preclinical | NLRP3 | MASLD/MASH ALD Liver fibrosis | [247,248,249,250,251,252] |
| Canakinumab | Randomized, placebo-controlled trial | IL-1β | Alcoholic hepatitis | [253] | |
| Ferroptosis | Ferrostain-1, Liproxstatin-1 Vitamin E | Preclinical | Lipophilic antioxidation | TAA-induced ALI Autoimmune hepatitis MASLD/MASH ALD | [115,254,255,256] |
| Selenium | Preclinical | GPX4 | MASH | [145] | |
| FGF21 | Preclinical | HO-1 inhibition, NRF2 activation | Iron overload-induced liver damage and fibrosis | [257] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stoess, C.; Choi, Y.-K.; Onyuru, J.; Friess, H.; Hoffman, H.M.; Hartmann, D.; Feldstein, A.E. Cell Death in Liver Disease and Liver Surgery. Biomedicines 2024, 12, 559. https://doi.org/10.3390/biomedicines12030559
Stoess C, Choi Y-K, Onyuru J, Friess H, Hoffman HM, Hartmann D, Feldstein AE. Cell Death in Liver Disease and Liver Surgery. Biomedicines. 2024; 12(3):559. https://doi.org/10.3390/biomedicines12030559
Chicago/Turabian StyleStoess, Christian, Yeon-Kyung Choi, Janset Onyuru, Helmut Friess, Hal M. Hoffman, Daniel Hartmann, and Ariel E. Feldstein. 2024. "Cell Death in Liver Disease and Liver Surgery" Biomedicines 12, no. 3: 559. https://doi.org/10.3390/biomedicines12030559
APA StyleStoess, C., Choi, Y.-K., Onyuru, J., Friess, H., Hoffman, H. M., Hartmann, D., & Feldstein, A. E. (2024). Cell Death in Liver Disease and Liver Surgery. Biomedicines, 12(3), 559. https://doi.org/10.3390/biomedicines12030559