
Role of Helicobacter pylori Infection in Pathogenesis, Evolution, and Complication of Atherosclerotic Plaque

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
3. Pathogenesis of Atherosclerosis: The Centrality of Endothelial Integrity
3.1. Immune Function of Endothelial Cells and Atherosclerosis
3.2. Inflammation of Endothelial Cells and Atherosclerosis
3.3. Platelets Aggregation and Clotting Formation in Atherosclerosis
4. H. pylori and Pathogenicity
4.1. Etiopathogenesis of H. pylori Colonization
- adhesion and colonization of the gastric mucosa,
- evasion of the immune response,
- induction of mucosal damage.
4.2. Role of Inflammation in H. pylori Infection: From Gastric Mucosa to Systemic Translocation
5. H. pylori and Atherosclerosis
- It induces the production of various pro-inflammatory factors (TNF-α, IFN-γ, IL-1, IL-2, IL-6, IL-8, IL-1).
- It indirectly causes the formation of immune complexes that mimic cross-antigenic reactions, common to autoimmune pathological manifestations.
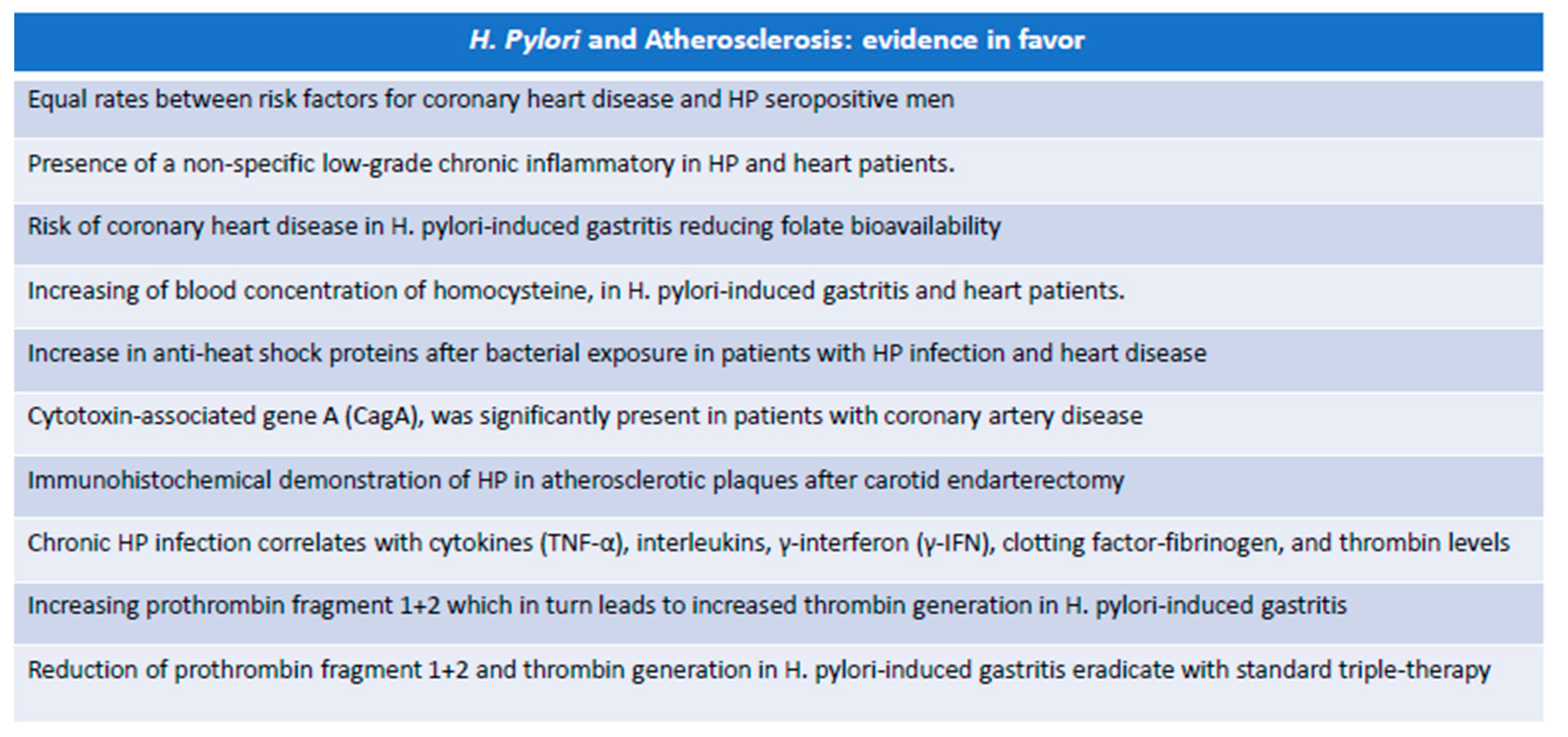
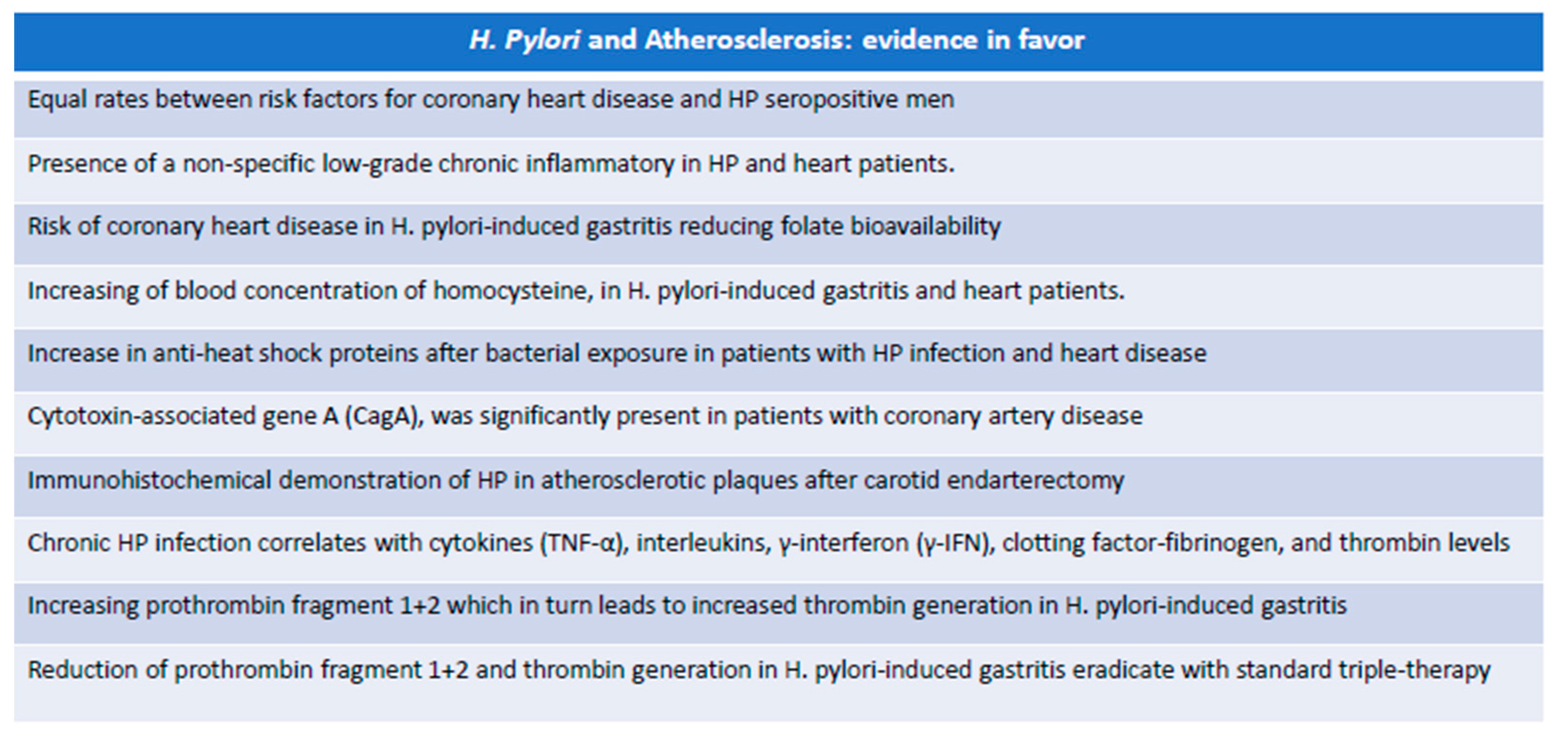
5.1. Experimental Evidence in Favor of the Role of H. pylori in Atherosclerosis
- First, it has been hypothesized that acute infection may precipitate ischemic events, especially in individuals with a significant presence of vascular risk factors. The key to this phenomenon is due to the transient imbalance of the blood coagulation system towards a prothrombotic state.
- Secondly, HP infection causes a chronic inflammatory state that affects atherosclerotic disease, supporting the hypothesis that the presence of microorganisms regardless of the site of primary colonization can lead to general inflammatory changes that weigh on the progression of atherosclerosis.
- Finally, it could be possible that some bacteria can influence atherogenesis due to direct colonization of the vascular wall or simply because parts of it act as immune complexes capable of activating an inflammatory state responsible for the evolution of the atherosclerotic plaque [116].
5.2. Experimental Evidence against the Possible Role of H. pylori in Atherosclerosis
6. Limits and Unanswered Questions
7. Conclusions
Funding
Conflicts of Interest
References
- Singh, S.S.; Pilkerton, C.S.; Shrader, C.D., Jr.; Frisbee, S.J. Subclinical atherosclerosis, cardiovascular health, and disease risk: Is there a case for the Cardiovascular Health Index in the primary prevention population? BMC Public Health 2018, 18, 429. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, G.; Muscoli, S.; De Rosa, S.; Cesaro, A.; Perrone, M.A.; Selvaggio, S.; Selvaggio, G.; Aimo, A.; Pedrinelli, R.; Mercuro, G.; et al. Evolving concepts in the pathophysiology of atherosclerosis: From endothelial dys-function to thrombus formation through multiple shades of inflammation. J. Cardiovasc. Med. 2023, 24 (Suppl. S2), e156–e167. [Google Scholar] [CrossRef] [PubMed]
- Barquera, S.; Pedroza-Tobías, A.; Medina, C.; Hernández-Barrera, L.; Bibbins-Domingo, K.; Lozano, R.; Moran, A.E. Global overview of the epidemiology of atherosclerotic cardiovascular disease. Arch. Med. Res. 2015, 46, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Gasbarrino, K.; Di Iorio, D.; Daskalopoulou, S.S. Importance of sex and gender in ischaemic stroke and carotid atherosclerotic disease. Eur. Heart J. 2021, 43, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Xian, X.; Wang, Z.; Bi, Y.; Chen, Q.; Han, X.; Tang, D.; Chen, R. Research Progress on the Relationship between Atherosclerosis and Inflammation. Biomolecules 2018, 8, 80. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, B.T.; Zhu, L.; Eckel, R.H.; Stafford, J.M. Sex differences in lipid and lipoprotein metabolism. Mol. Metab. 2018, 15, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.K.; Ober, J.C.; Krishnaswami, A.; Ferguson, J.J.; Anderson, H.V.; Golino, P.; Buja, L.M.; Willerson, J.T. Endogenous nitric oxide protects against platelet aggregation and cyclic flow variations in stenosed and endothelium-injured arteries. Circulation 1992, 86, 1302–1309. [Google Scholar] [CrossRef]
- Cohen, R.A. The role of nitric oxide and other endothelium-derived vasoactive substances in vascular disease. Prog. Cardiovasc. Dis. 1995, 38, 105–128. [Google Scholar] [CrossRef]
- Seki, J.; Nishio, M.; Kato, Y.; Motoyama, Y.; Yoshida, K. FK409, a new nitric-oxide donor, suppresses smooth muscle proliferation in the rat model of balloon angioplasty. Atherosclerosis 1995, 117, 97–106. [Google Scholar] [CrossRef]
- Ridker, P.M.; Cushman, M.; Stampfer, M.J.; Tracy, R.P.; Hennekens, C.H. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N. Engl. J. Med. 1997, 336, 973–979. [Google Scholar] [CrossRef]
- Ridker, P.M.; Hennekens, C.H.; Buring, J.E.; Rifai, N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N. Engl. J. Med. 2000, 342, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; He, Q.; Jin, D.; Ma, B.; Yao, K.; Zou, X. Association between helicobacter pylori infection and subclinical atherosclerosis: A systematic review and meta-analysis. Medicine 2021, 100, e27840. [Google Scholar] [CrossRef] [PubMed]
- Consolazio, A.; Borgia, M.C.; Ferro, D.; Iacopini, F.; Paoluzi, O.A.; Crispino, P.; Nardi, F.; Rivera, M.; Paoluzi, P. Increased thrombin generation and circulating levels of tumour necrosis factor-α in patients with chronic Helicobacter pylori-positive gastritis. Aliment. Pharmacol. Ther. 2004, 20, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Longo-Mbenza, B.; Nkondi; Mokondjimobe, E.; Gombet, T.; Ibara, J.R.; Mbolla, E.; Vangu, D.N.; Fuele, S.M. Helicobacter pylori infection is identified as a cardiovascular risk factor in Central Africans. Vasc. Health Risk Manag. 2012, 8, 455–461. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, Q.; Liu, Y.; Cui, R.; Lu, K.; Zhao, Y. Association between Helicobacter pylori infection and carotid atherosclerosis in patients with vascular dementia. J. Neurol. Sci. 2016, 362, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global prevalence of Helicobacter pylori infection: Systematic review and meta-Analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhou, W.; Luo, L.; Xu, W. Helicobacter pylori infection is not related to increased carotid intima-media thickness in general population. Sci. Rep. 2018, 8, 14180. [Google Scholar] [CrossRef]
- Xu, Z.; Li, J.; Wang, H.; Xu, G. Helicobacter pylori infection and atherosclerosis: Is there a causal relationship? Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 2293–2301. [Google Scholar] [CrossRef]
- Du, L.; Liu, J.; Jin, C.; Ma, Y.; Yin, L.; Man, S.; Li, S.; Li, L.; Ning, Y.; Zhang, X. Association between Helicobacter pylori infection and carotid atherosclerosis in Chinese adults. Atheroscler. Plus 2021, 44, 25–30. [Google Scholar] [CrossRef]
- Viles-Gonzalez, J.F.; Fuster, V.; Badimon, J.J. Atherothrombosis: A widespread disease with unpredictable and life-threatening consequences. Eur. Heart J. 2004, 25, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Torzewski, M. The Initial Human Atherosclerotic Lesion and Lipoprotein Modification—A Deep Connection. Int. J. Mol. Sci. 2021, 22, 11488. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Li, L.; Shen, X.; Li, Q.; Xu, W.; Wang, X.; Tao, Y.; Yin, H. An update on lipid oxidation and inflammation in cardiovascular diseases. Free. Radic. Biol. Med. 2019, 144, 266–278. [Google Scholar] [CrossRef]
- Shatila, M.; Thomas, A.S. Current and Future Perspectives in the Diagnosis and Management of Helicobacter pylori Infection. J. Clin. Med. 2022, 11, 5086. [Google Scholar] [CrossRef]
- Ciarambino, T.; Crispino, P.; Giordano, M. Hyperuricemia and Endothelial Function: Is It a Simple Association or Do Gender Differences Play a Role in This Binomial? Biomedicines 2022, 10, 3067. [Google Scholar] [CrossRef]
- Kajikawa, M.; Higashi, Y. Obesity and Endothelial Function. Biomedicines 2022, 10, 1745. [Google Scholar] [CrossRef]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Papežíková, I.; Pekarová, M.; Kolářová, H.; Klinke, A.; Lau, D.; Baldus, S.; Lojek, A.; Kubala, L. Uric acid modulates vascular endothelial function through the down regulation of nitric oxide production. Free. Radic. Res. 2012, 47, 82–88. [Google Scholar] [CrossRef]
- Kotlyarov, S. Immune Function of Endothelial Cells: Evolutionary Aspects, Molecular Biology and Role in Atherogenesis. Int. J. Mol. Sci. 2022, 23, 9770. [Google Scholar] [CrossRef]
- Linton, M.F.; Yancey, P.G.; Davies, S.S.; Jerome, W.G.; Linton, E.F.; Song, W.L.; Doran, A.C.; Vickers, K.C. Il ruolo dei lipidi e delle lipoproteine nell’aterosclerosi. In Endotesto; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Mai, J.; Virtue, A.; Shen, J.; Wang, H.; Yang, X.-F. An evolving new paradigm: Endothelial cells—Conditional innate immune cells. J. Hematol. Oncol. 2013, 6, 61. [Google Scholar] [CrossRef]
- Murdoch, C.; Monk, P.N.; Finn, A. Cxc chemokine receptor expression on human endothelial cells. Cytokine 1999, 11, 704–712. [Google Scholar] [CrossRef]
- Qu, D.; Wang, L.; Huo, M.; Song, W.; Lau, C.-W.; Xu, J.; Xu, A.; Yao, X.; Chiu, J.-J.; Tian, X.Y.; et al. Focal TLR4 activation mediates disturbed flow-induced endothelial inflammation. Cardiovasc. Res. 2019, 116, 226–236. [Google Scholar] [CrossRef]
- Li, X.; Wang, L.; Fang, P.; Sun, Y.; Jiang, X.; Wang, H.; Yang, X.-F. Lysophospholipids induce innate immune transdifferentiation of endothelial cells, resulting in prolonged endothelial activation. J. Biol. Chem. 2018, 293, 11033–11045. [Google Scholar] [CrossRef]
- Bai, B.; Yang, Y.; Wang, Q.; Li, M.; Tian, C.; Liu, Y.; Aung, L.H.H.; Li, P.-F.; Yu, T.; Chu, X.-M. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 2020, 11, 776. [Google Scholar] [CrossRef]
- Zhang, B.; Cao, G.L.; Cross, A.; Domachowske, J.B.; Rosen, G.M. Differential antibacterial activity of nitric oxide from the immunological isozyme of nitric oxide synthase transduced into endothelial cells. Nitric Oxide 2002, 7, 42–49. [Google Scholar] [CrossRef]
- Gao, C.; Xie, R.; Li, W.; Zhou, J.; Liu, S.; Cao, F.; Liu, Y.; Ma, R.; Si, Y.; Liu, Y.; et al. Endothelial cell phagocytosis of senescent neutrophils decreases procoagulant activity. Thromb. Haemost. 2013, 109, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Alva-Murillo, N.; Téllez-Pérez, A.D.; Sagrero-Cisneros, E.; López-Meza, J.E.; Ochoa-Zarzosa, A. Expression of antimicrobial peptides by bovine endothelial cells. Cell. Immunol. 2012, 280, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Campinho, P.; Vilfan, A.; Vermot, J. Blood Flow Forces in Shaping the Vascular System: A Focus on Endothelial Cell Behavior. Front. Physiol. 2020, 11, 552. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.D.E.; Hansen, A.J.; Lundbæk, J.A. Regulation of endothelial cell migration by amphiphiles—Are changes in cell membrane physical properties involved? Angiogenesis 2007, 10, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Järvisalo, M.J.; Juonala, M.; Raitakari, O.T. Assessment of inflammatory markers and endothelial function. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Gliozzi, M.; Scicchitano, M.; Bosco, F.; Musolino, V.; Carresi, C.; Scarano, F.; Maiuolo, J.; Nucera, S.; Maretta, A.; Paone, S.; et al. Modulation of Nitric Oxide Synthases by Oxidized LDLs: Role in Vascular Inflammation and Atherosclerosis Development. Int. J. Mol. Sci. 2019, 20, 3294. [Google Scholar] [CrossRef]
- Eligini, S.; Colli, S.; Habib, A.; Aldini, G.; Altomare, A.; Banfi, C. Cyclooxygenase-2 Glycosylation Is Affected by Peroxynitrite in Endothelial Cells: Impact on Enzyme Activity and Degradation. Antioxidants 2021, 10, 496. [Google Scholar] [CrossRef] [PubMed]
- Sabe, S.A.; Feng, J.; Sellke, F.W.; Abid, M.R. Mechanisms and clinical implications of endothelium-dependent vasomotor dysfunction in coronary microvasculature. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H819–H841. [Google Scholar] [CrossRef] [PubMed]
- Kotlyarov, S.; Kotlyarova, A. Involvement of Fatty Acids and Their Metabolites in the Development of Inflammation in Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 1308. [Google Scholar] [CrossRef] [PubMed]
- Salvemini, D.; Misko, T.P.; Masferrer, J.L.; Seibert, K.; Currie, M.G.; Needleman, P. Nitric oxide activates cyclooxygenase enzymes. Proc. Natl. Acad. Sci. USA 1993, 90, 7240–7244. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.F. The role of nitric oxide in prostaglandin biology; update. Nitric Oxide 2011, 25, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Mollace, V.; Muscoli, C.; Masini, E.; Cuzzocrea, S.; Salvemini, D. Modulation of prostaglandin biosynthesis by nitric oxide and nitric oxide donors. Pharmacol. Rev. 2005, 57, 217–252. [Google Scholar] [CrossRef] [PubMed]
- Martí i Líndez, A.-A.; Reith, W. Arginine-dependent immune responses. Cell. Mol. Life Sci. 2021, 78, 5303–5324. [Google Scholar] [CrossRef]
- Ley, K. M1 Means Kill; M2 Means Heal. J. Immunol. 2017, 199, 2191–2193. [Google Scholar] [CrossRef]
- Zhu, C.; Yu, Y.; Montani, J.-P.; Ming, X.-F.; Yang, Z. Arginase-I enhances vascular endothelial inflammation and senescence through eNOS-uncoupling. BMC Res. Notes 2017, 10, 82. [Google Scholar] [CrossRef]
- Wu, J.M.F.; Cheng, Y.Y.; Tang, T.W.H.; Shih, C.; Chen, J.H.; Hsieh, P.C.H. Prostaglandin E2 Receptor 2 Modulates Macrophage Activity for Cardiac Repair. J. Am. Heart Assoc. 2018, 7, e009216. [Google Scholar] [CrossRef]
- Jiang, J.-X.; Zhang, S.-J.; Liu, Y.-N.; Lin, X.-X.; Sun, Y.-H.; Shen, H.-J.; Yan, X.-F.; Xie, Q.-M. EETs alleviate ox-LDL-induced inflammation by inhibiting LOX-1 receptor expression in rat pulmonary arterial endothelial cells. Eur. J. Pharmacol. 2014, 727, 43–51. [Google Scholar] [CrossRef]
- Kozak, W.; Aronoff, D.M.; Boutaud, O.; Kozak, A. 11,12-epoxyeicosatrienoic acid attenuates synthesis of prostaglandin E2 in rat monocytes stimulated with lipopolysaccharide. Exp. Biol. Med. 2003, 228, 786–794. [Google Scholar] [CrossRef]
- Nappi, F.; Fiore, A.; Masiglat, J.; Cavuoti, T.; Romandini, M.; Nappi, P.; Singh, S.S.A.; Couetil, J.-P. Endothelium-Derived Relaxing Factors and Endothelial Function: A Systematic Review. Biomedicines 2022, 10, 2884. [Google Scholar] [CrossRef]
- Kowalczyk, A.; Kleniewska, P.; Kolodziejczyk, M.; Skibska, B.; Goraca, A. The role of endothelin-1 and endothelin receptor antagonists in inflammatory response and sepsis. Arch. Immunol. Ther. Exp. 2014, 63, 41–52. [Google Scholar] [CrossRef]
- Lordan, R.; Tsoupras, A.; Zabetakis, I. Platelet Activation and Prothrombotic Mediators at the Nexus of Inflammation and Atherosclerosis: Potential Role of Antiplatelet Agents. Blood Rev. 2020, 45, 100694. [Google Scholar] [CrossRef] [PubMed]
- Estevez, B.; Du, X. New Concepts and Mechanisms of Platelet Activation Signaling. Physiology 2017, 32, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Huilcaman, R.; Venturini, W.; Fuenzalida, L.; Cayo, A.; Segovia, R.; Valenzuela, C.; Brown, N.; Moore-Carrasco, R. Platelets, a Key Cell in Inflammation and Atherosclerosis Progression. Cells 2022, 11, 1014. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, B.; Varga-Szabo, D.; Elvers, M. Integrins in Platelet Activation. J. Thromb. Haemost. 2009, 7, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Shattil, S.J.; Newman, P.J. Integrins: Dynamic Scaffolds for Adhesion and Signaling in Platelets. Blood 2004, 104, 1606–1615. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Huang, A.L.; Kyaw, T.S.; Bobik, A.; Peter, K. Atherosclerotic Plaque Rupture: Identifying the Straw That Breaks the Camel’s Back. Arter. Thromb. Vasc. Biol. 2016, 36, e63–e72. [Google Scholar] [CrossRef]
- Koenen, R.R.; von Hundelshausen, P.; Nesmelova, I.V.; Zernecke, A.; Liehn, E.A.; Sarabi, A.; Kramp, B.K.; Piccinini, A.M.; Paludan, S.R.; Kowalska, M.A.; et al. Disrupting Functional Interactions between Platelet Chemokines Inhibits Atherosclerosis in Hyperlipidemic mice. Nat. Med. 2009, 15, 97–103. [Google Scholar] [CrossRef]
- Gencer, S.; Evans, B.R.; van der Vorst, E.P.C.; Döring, Y.; Weber, C. Inflammatory Chemokines in Atherosclerosis. Cells 2021, 10, 226. [Google Scholar] [CrossRef]
- Gonzalez, J.; Donoso, W.; Díaz, N.; Albornoz, M.E.; Huilcaman, R.; Morales, E.; Moore-Carrasco, R. High Fat Diet Induces Adhesion of Platelets to Endothelium in Two Models of Dyslipidemia. J. Obes. 2014, 2014, 591270. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.A.C.; Serbulea, V.; Baylis, R.A.; Shankman, L.S.; Bradley, X.; Alencar, G.F.; Owsiany, K.; Deaton, R.A.; Karnewar, S.; Shamsuzzaman, S.; et al. Multiple Cell Types Contribute to the Atherosclerotic Lesion Fibrous Cap by PDGFRβ and Bioenergetic Mechanisms. Nat. Metab. 2021, 3, 166–181. [Google Scholar] [CrossRef] [PubMed]
- Groh, L.; Keating, S.T.; Joosten, L.A.B.; Netea, M.G.; Riksen, N.P. Monocyte and Macrophage Immunometabolism in Atherosclerosis. In Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 2018; Volume 40, pp. 203–214. [Google Scholar]
- Oggero, S.; de Gaetano, M.; Marcone, S.; Fitzsimons, S.; Pinto, A.L.; Ikramova, D.; Barry, M.; Burke, D.; Montero-Melendez, T.; Cooper, D.; et al. Extracellular Vesicles from Monocyte/Platelet Aggregates Modulate Human Atherosclerotic Plaque Reactivity. J. Extracell. Vesicles 2021, 10, 12084. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Doran, J. The Many Faces of Cytokine Release Syndrome-Related Coagulopathy. Clin. Hematol. Int. 2021, 3, 3–12. [Google Scholar] [CrossRef]
- Vajen, T.; Benedikter, B.J.; Heinzmann, A.C.A.; Vasina, E.M.; Henskens, Y.; Parsons, M.; Maguire, P.B.; Stassen, F.R.; Heemskerk, J.W.M.; Schurgers, L.J.; et al. Platelet Extracellular Vesicles Induce a Pro-Inflammatory Smooth Muscle cell Phenotype. J. Extracell. Vesicles 2017, 6, 1322454. [Google Scholar] [CrossRef]
- Bakogiannis, C.; Sachse, M.; Stamatelopoulos, K.; Stellos, K. Platelet-Derived Chemokines in Inflammation and Atherosclerosis. Cytokine 2019, 122, 154157. [Google Scholar] [CrossRef]
- Kaczor, D.M.; Kramann, R.; Hackeng, T.M.; Schurgers, L.J.; Koenen, R.R. Differential Effects of Platelet Factor 4 (CXCL4) and Its Non-Allelic Variant (CXCL4L1) on Cultured Human Vascular Smooth Muscle Cells. Int. J. Mol. Sci. 2022, 23, 580. [Google Scholar] [CrossRef]
- Trostchansky, A.; Moore-Carrasco, R.; Fuentes, E. Oxidative Pathways of Arachidonic Acid as Targets for Regulation of Platelet Activation. Prostaglandins Other Lipid Mediat. 2019, 145, 106382. [Google Scholar] [CrossRef]
- Markus, H.S.; Risley, P.; Mendall, M.A.; Steinmetz, H.; Sitzer, M. Helicobacter pylori infection, the cytotoxin gene a strain, and carotid artery intima-media thickness. J. Cardiovasc. Risk 2002, 9, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Manolakis, A.; Kapsoritakis, A.N.; Potamianos, S.P. A review of the postulated mechanisms concerning the association of Helicobacter pylori with ischemic heart disease. Helicobacter 2007, 12, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; He, Q.; Song, D.; Wang, Y.; Liu, X.; Ding, G.; Xing, W. Association of Helicobacter pylori Infection With Carotid Atherosclerosis in a Northern Chinese Population: A Cross-Sectional Study. Front. Cardiovasc. Med. 2022, 8, 795795. [Google Scholar] [CrossRef] [PubMed]
- Thorell, K.; Yahara, K.; Berthenet, E.; Lawson, D.J.; Mikhail, J.; Kato, I.; Mendez, A.; Rizzato, C.; Bravo, M.M.; Suzuki, R.; et al. Correction: Rapid evolution of distinct Helicobacter pylori subpopulations in the Americas. PLoS Genet. 2017, 13, e1006730. [Google Scholar] [CrossRef] [PubMed]
- Bravo, D.; Hoare, A.; Soto, C.; Valenzuela, M.A.; Quest, A.F. Helicobacter pylori in human health and disease: Mechanisms for local gastric and systemic effects. World J. Gastroenterol. 2018, 24, 3071–3089. [Google Scholar] [CrossRef] [PubMed]
- Denic, M.; Touati, E.; De Reuse, H. Review: Pathogenesis of Helicobacter pylori infection. Helicobacter 2020, 25, e12736. [Google Scholar] [CrossRef] [PubMed]
- Hanyu, H.; Engevik, K.A.; Matthis, A.L.; Ottemann, K.M.; Montrose, M.H.; Aihara, E. Helicobacter pylori Uses the TlpB Receptor to Sense Sites of Gastric Injury. Infect. Immun. 2019, 87, e00202-19. [Google Scholar] [CrossRef] [PubMed]
- Idowu, S.; Bertrand, P.P.; Walduck, A.K. Gastric organoids: Advancing the study of H. pylori pathogenesis and inflammation. Helicobacter 2022, 27, e12891. [Google Scholar] [CrossRef]
- Soejima, M.; Koda, Y. Molecular mechanisms of Lewis antigen expression. Leg. Med. 2005, 7, 266–269. [Google Scholar] [CrossRef]
- Odenbreit, S.; Faller, G.; Haas, R. Role of the AlpAB proteins and lipopolysaccharide in adhesion of Helicobacter pylori to human gastric tissue. Int. J. Med. Microbiol. 2002, 292, 247–256. [Google Scholar] [CrossRef]
- Alm, R.A.; Bina, J.; Andrews, B.M.; Doig, P.; Hancock, R.E.W.; Trust, T.J. Comparative Genomics of Helicobacter pylori: Analysis of the Outer Membrane Protein Families. Infect. Immun. 2000, 68, 4155–4168. [Google Scholar] [CrossRef]
- Oleastro, M.; Ménard, A. The Role of Helicobacter pylori Outer Membrane Proteins in Adherence and Pathogenesis. Biology 2013, 2, 1110–1134. [Google Scholar] [CrossRef]
- Xu, C.; Soyfoo, D.M.; Wu, Y.; Xu, S. Virulence of Helicobacter pylori outer membrane proteins: An updated review. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Doohan, D.; Rezkitha, Y.A.A.; Waskito, L.A.; Yamaoka, Y.; Miftahussurur, M. Helicobacter pylori BabA–SabA Key Roles in the Adherence Phase: The Synergic Mechanism for Successful Colonization and Disease Development. Toxins 2021, 13, 485. [Google Scholar] [CrossRef] [PubMed]
- Unemo, M.; Aspholm-Hurtig, M.; Ilver, D.; Bergström, J.; Borén, T.; Danielsson, D.; Teneberg, S. The Sialic Acid Binding SabA Adhesin of Helicobacter pylori Is Essential for Nonopsonic Activation of Human Neutrophils. J. Biol. Chem. 2005, 280, 15390–15397. [Google Scholar] [CrossRef] [PubMed]
- Giraldi, L.; Michelazzo, M.B.; Arzani, D.; Persiani, R.; Pastorino, R.; Boccia, S. MUC1, MUC5AC, and MUC6 polymorphisms, Helicobacter pylori infection, and gastric cancer: A systematic review and meta-analysis. Eur. J. Cancer Prev. 2018, 27, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Tegtmeyer, N. Type IV Secretion and Signal Transduction of Helicobacter pylori CagA through Interactions with Host Cell Receptors. Toxins 2017, 9, 115. [Google Scholar] [CrossRef] [PubMed]
- Baj, J.; Forma, A.; Sitarz, M.; Portincasa, P.; Garruti, G.; Krasowska, D.; Maciejewski, R. Helicobacter pylori Virulence Factors—Mechanisms of Bacterial Pathogenicity in the Gastric Microenvironment. Cells 2020, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, N.; Tay, A.C.Y.; Marshall, B.J.; Jain, U. Helicobacter pylori VacA, a distinct toxin exerts diverse functionalities in numerous cells: An overview. Helicobacter 2018, 24, e12544. [Google Scholar] [CrossRef] [PubMed]
- Sharndama, H.C.; Mba, I.E. Helicobacter pylori: An up-to-date overview on the virulence and pathogenesis mechanisms. Braz. J. Microbiol. 2022, 53, 33–50. [Google Scholar] [CrossRef]
- Sharma, V.; Aggarwal, A. Helicobacter pylori: Does it add to risk of coronary artery disease. World J. Cardiol. 2015, 7, 19–25. [Google Scholar] [CrossRef]
- Nejati, S.; Karkhah, A.; Darvish, H.; Validi, M.; Ebrahimpour, S.; Nouri, H.R. Influence of Helicobacter pylori virulence factors CagA and VacA on pathogenesis of gastrointestinal disorders. Microb. Pathog. 2018, 117, 43–48. [Google Scholar] [CrossRef]
- Lina, T.T.; Alzahrani, S.; Gonzalez, J.; Pinchuk, I.V.; Beswick, E.J.; Reyes, V.E. Immune evasion strategies used by Helicobacter pylori. World J. Gastroenterol. 2014, 20, 12753–12766. [Google Scholar] [CrossRef]
- Mejías-Luque, R.; Gerhard, M. Immune Evasion Strategies and Persistence of Helicobacter pylori. In Molecular Pathogenesis and Signal Transduction by Helicobacter pylori; Current Topics in Microbiology and Immunology; Springer: Cham, Switzerland, 2017; Volume 400, pp. 53–71. [Google Scholar] [CrossRef]
- Morey, P.; Pfannkuch, L.; Pang, E.; Boccellato, F.; Sigal, M.; Imai-Matsushima, A.; Dyer, V.; Koch, M.; Mollenkopf, H.J.; Schlaermann, P.; et al. Helicobacter pylori Depletes Cholesterol in Gastric Glands to Prevent Interferon Gamma Signaling and Escape the Inflammatory Response. Gastroenterology 2018, 154, 1391–1404.e9. [Google Scholar] [CrossRef]
- Chen, Y.; Tang, Z.; Fu, L.; Liu, R.; Yang, L.; Wang, B. Inhibitory and Injury-Protection Effects of O-Glycan on Gastric Epithelial Cells Infected with Helicobacter pylori. Infect. Immun. 2022, 90, e0039322. [Google Scholar] [CrossRef]
- Stent, A.; Every, A.L.; Chionh, Y.T.; Ng, G.Z.; Sutton, P. Superoxide dismutase from Helicobacter pylori suppresses the production of pro-inflammatory cytokines during in vivo infection. Helicobacter 2017, 23, e12459. [Google Scholar] [CrossRef]
- Tran, L.S.; Chonwerawong, M.; Ferrero, R.L. Regulation and functions of inflammasome-mediated cytokines in Helicobacter pylori infection. Microbes Infect. 2017, 19, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Parsons, B.N.; Ijaz, U.Z.; D’Amore, R.; Burkitt, M.D.; Eccles, R.; Lenzi, L.; Duckworth, C.A.; Moore, A.R.; Tiszlavicz, L.; Varro, A.; et al. Comparison of the human gastric microbiota in hypochlorhydric states arising as a result of Helicobacter pylori-induced atrophic gastritis, autoimmune atrophic gastritis and proton pump inhibitor use. PLoS Pathog. 2017, 13, e1006653. [Google Scholar] [CrossRef] [PubMed]
- Fakharian, F.; Asgari, B.; Nabavi-Rad, A.; Sadeghi, A.; Soleimani, N.; Yadegar, A.; Zali, M.R. The interplay between Helicobacter pylori and the gut microbiota: An emerging driver influencing the immune system homeostasis and gastric carcinogenesis. Front. Cell. Infect. Microbiol. 2022, 12, 953718. [Google Scholar] [CrossRef] [PubMed]
- Chahal, G.; Padra, M.; Erhardsson, M.; Jin, C.; Quintana-Hayashi, M.; Venkatakrishnan, V.; Padra, J.T.; Stenbäck, H.; Thorell, A.; Karlsson, N.G.; et al. A Complex Connection Between the Diversity of Human Gastric Mucin O-Glycans, Helicobacter pylori Binding, Helicobacter Infection and Fucosylation. Mol. Cell. Proteom. 2022, 21, 100421. [Google Scholar] [CrossRef]
- Brawner, K.M.; Kumar, R.; Serrano, C.A.; Ptacek, T.; Lefkowitz, E.; Morrow, C.D.; Zhi, D.; Kyanam-Kabir-Baig, K.R.; Smythies, L.E.; Harris, P.R.; et al. Helicobacter pylori infection is associated with an altered gastric microbiota in children. Mucosal Immunol. 2017, 10, 1169–1177. [Google Scholar] [CrossRef]
- Xia, X.; Zhang, L.; Chi, J.; Li, H.; Liu, X.; Hu, T.; Li, R.; Guo, Y.; Zhang, X.; Wang, H.; et al. Helicobacter pylori Infection Impairs Endothelial Function Through an Exosome-Mediated Mechanism. J. Am. Heart Assoc. 2020, 9, e014120. [Google Scholar] [CrossRef]
- He, J.; Liu, Y.; Ouyang, Q.; Li, R.; Li, J.; Chen, W.; Hu, W.; He, L.; Bao, Q.; Li, P.; et al. Helicobacter pylori and unignorable extragastric diseases: Mechanism and implications. Front. Microbiol. 2022, 13, 972777. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, F.; Shi, S. Helicobacter pylori Infection Increase the Risk of Myocardial Infarction: A Meta-Analysis of 26 Studies Involving more than 20,000 Participants. Helicobacter 2015, 20, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, Z.; Xia, X.; Chi, J.; Li, H.; Liu, X.; Li, R.; Li, Y.; Liu, D.; Tian, D.; et al. Helicobacter pylori infection selectively increases the risk for carotid atherosclerosis in young males. Atherosclerosis 2019, 291, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.-Y.; Hu, K.-C.; Liu, C.-J.; Hung, C.-L.; Bair, M.-J.; Chen, M.-J.; Wang, H.-Y.; Wu, M.-S.; Shih, S.-C.; Liu, C.-C. Helicobacter pylori infection combined with non-alcoholic fatty liver disease increase the risk of atherosclerosis: Focus in carotid artery plaque. Medicine 2019, 98, e14672. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Mendall, M.A.; Carrington, D.; Strachan, D.P.; Leatham, E.; Molineaux, N.; Levy, J.; Blakeston, C.; A Seymour, C.; Camm, A.J.; et al. Association of Helicobacter pylori and Chlamydia pneumoniae infections with coronary heart disease and cardiovascular risk factors. BMJ 1995, 311, 711–714, Erratum in BMJ 1995, 311, 985. [Google Scholar] [CrossRef] [PubMed]
- Markle, H.V. Coronary artery disease associated with Helicobacter pylori infection is at least partially due to inadequate folate status. Med. Hypotheses 1997, 49, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Birnie, D.H.; Holme, E.R.; McKay, I.C.; Hood, S.; McColl, K.E.; Hillis, W.S. Association between antibodies to heat shock protein 65 and coronary atherosclerosis. Possible mechanism of action of Helicobacter pylori and other bacterial infections in increasing cardiovascular risk. Eur. Heart J. 1998, 19, 387–394. [Google Scholar] [CrossRef]
- Pasceri, V.; Cammarota, G.; Patti, G.; Cuoco, L.; Gasbarrini, A.; Grillo, R.L.; Fedeli, G.; Gasbarrini, G.; Maseri, A. Association of virulent Helicobacter pylori strains with ischemic heart disease. Circulation 1998, 97, 1675–1679. [Google Scholar] [CrossRef]
- Ameriso, S.F.; Fridman, E.A.; Leiguarda, R.C.; Sevlever, G.E. Detection of Helicobacter pylori in human carotid atherosclerotic plaques. Stroke 2001, 32, 385–391. [Google Scholar] [CrossRef]
- Huang, G.; Fang, N.; Kuang, M.Q.; Huang, Y.Q.; Zhang, K.H. Establishment of a risk assessment system for peptic ulcer recurrence and its value in individualized intervention. Am. J. Transl. Res. 2021, 13, 2969–2975. [Google Scholar]
- Crabtree, J.E.; Shallcross, T.M.; Heatley, R.V.; Wyatt, J.I. Mucosal tumour necrosis factor alpha and interleukin-6 in patients with Helicobacter pylori associated gastritis. Gut 1991, 32, 1473–1477. [Google Scholar] [CrossRef] [PubMed]
- Bauer, K.A.; Rosenberg, R.D. The pathophysiology of the prethrombotic state in humans: Insights gained from studies using markers of hemostatic system activation. Blood 1987, 70, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zheng, H.; Qiu, M.; Hao, S.; Liu, X.; Zhu, X.; Cai, X.; Huang, Y. Helicobacter pylori infection and risk of cardiovascular disease. Helicobacter 2023, 28, e12967. [Google Scholar] [CrossRef] [PubMed]
- Malnick, S.D.; Goland, S.; Kaftoury, A.; Schwarz, H.; Pasik, S.; Mashiach, A.; Sthoeger, Z. Evaluation of carotid arterial plaques after endarterectomy for Helicobacter pylori infection. Am. J. Cardiol. 1999, 83, 1586–1587. [Google Scholar] [CrossRef] [PubMed]
- Blasi, F.; Denti, F.; Erba, M.; Cosentini, R.; Raccanelli, R.; Rinaldi, A.; Fagetti, L.; Esposito, G.; Ruberti, U.; Allegra, L. Detection of Chlamydia pneumoniae but not Helicobacter pylori in atherosclerotic plaques of aortic aneurysms. J. Clin. Microbiol. 1996, 34, 2766–2769. [Google Scholar] [CrossRef] [PubMed]
- Blasi, F.; Boman, J.; Esposito, G.; Melissano, G.; Chiesa, R.; Cosentini, R.; Tarsia, P.; Tshomba, Y.; Betti, M.; Alessi, M.; et al. Chlamydia pneumoniae DNA detection in peripheral blood mononuclear cells is predictive of vascular infection. J. Infect. Dis. 1999, 180, 2074–2076. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.J.; Perng, C.L.; Shyu, R.Y.; Chen, C.H.; Lou, Q.; Chong, S.K.F.; Lee, C.H. Comparison of five PCR methods for detection of Helicobacter pylori DNA in gastric tissues. J. Clin. Microbiol. 1999, 37, 772–774. [Google Scholar] [CrossRef] [PubMed]
- Akyön, Y.; Pinar, A.; Farsak, B.; Böke, E.; Günalp, A. Helicobacter pylori and Chlamydia pneumoniae DNA found in atherosclerotic plaques by polymerase chain reaction. Gut 1999, 45, A89. [Google Scholar]
- Laek, B.; Szklo, M.; McClelland, R.L.; Ding, J.; Tsai, M.Y.; Bluemke, D.A.; Tracy, R.; Matsushita, K. The prospective association of Chlamydia pneumoniae and four other pathogens with development of coronary artery calcium: The multi-ethnic study of atherosclerosis (MESA). Atherosclerosis 2013, 230, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Schöttker, B.; Adamu, M.A.; Weck, M.N.; Müller, H.; Brenner, H. Helicobacter pylori infection, chronic atrophic gastritis and major cardiovascular events: A population-based cohort study. Atherosclerosis 2012, 220, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Li, Y.; Dong, C.; Si, G.; Xu, Y.; Peng, M.; Li, Y. Helicobacter pylori infection and the progression of atherosclerosis: A systematic review and meta-analysis. Helicobacter 2021, 27, e12865. [Google Scholar] [CrossRef]
- Li, B.; Xia, Y.; Hu, B. Infection and atherosclerosis: TLR-dependent pathways. Cell. Mol. Life Sci. 2020, 77, 2751–2769. [Google Scholar] [CrossRef]
- Tobias, P.; Curtiss, L.K. Thematic review series: The immune system and atherogenesis. Paying the price for pathogen protection: Toll receptors in atherogenesis. J. Lipid Res. 2005, 46, 404–411. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciarambino, T.; Crispino, P.; Minervini, G.; Giordano, M. Role of Helicobacter pylori Infection in Pathogenesis, Evolution, and Complication of Atherosclerotic Plaque. Biomedicines 2024, 12, 400. https://doi.org/10.3390/biomedicines12020400
Ciarambino T, Crispino P, Minervini G, Giordano M. Role of Helicobacter pylori Infection in Pathogenesis, Evolution, and Complication of Atherosclerotic Plaque. Biomedicines. 2024; 12(2):400. https://doi.org/10.3390/biomedicines12020400
Chicago/Turabian StyleCiarambino, Tiziana, Pietro Crispino, Giovanni Minervini, and Mauro Giordano. 2024. "Role of Helicobacter pylori Infection in Pathogenesis, Evolution, and Complication of Atherosclerotic Plaque" Biomedicines 12, no. 2: 400. https://doi.org/10.3390/biomedicines12020400
APA StyleCiarambino, T., Crispino, P., Minervini, G., & Giordano, M. (2024). Role of Helicobacter pylori Infection in Pathogenesis, Evolution, and Complication of Atherosclerotic Plaque. Biomedicines, 12(2), 400. https://doi.org/10.3390/biomedicines12020400