

SRPK1 Promotes Glioma Proliferation, Migration, and Invasion through Activation of Wnt/β-Catenin and JAK-2/STAT-3 Signaling Pathways

Abstract
1. Introduction
2. Methods
2.1. Materials
2.2. Cell Culture
2.3. Neuroglial Tumor Tissue
2.4. Study Design
2.5. Study Setting
2.6. Sample Size Determination
2.7. Immunofluorescence Microscopy
2.8. Transfection of Cells with siRNA or Overexpression (OE) of Plasmid
2.9. Immunohistochemistry
2.10. RNA Extraction and qPCR
2.11. Cell Proliferation Assay
2.12. RNA-Seq
2.13. Flow Cytometry
2.14. Western Blot
2.15. Transwell Infiltration Experiment
2.16. Monomer Red Fluorescent Protein (mRFP)—Green Fluorescent Protein (GFP)—LC3 Tandem Fluorescence Protein Quenching Test
2.17. In Vivo Experiments
2.18. Statistical Analysis
3. Results
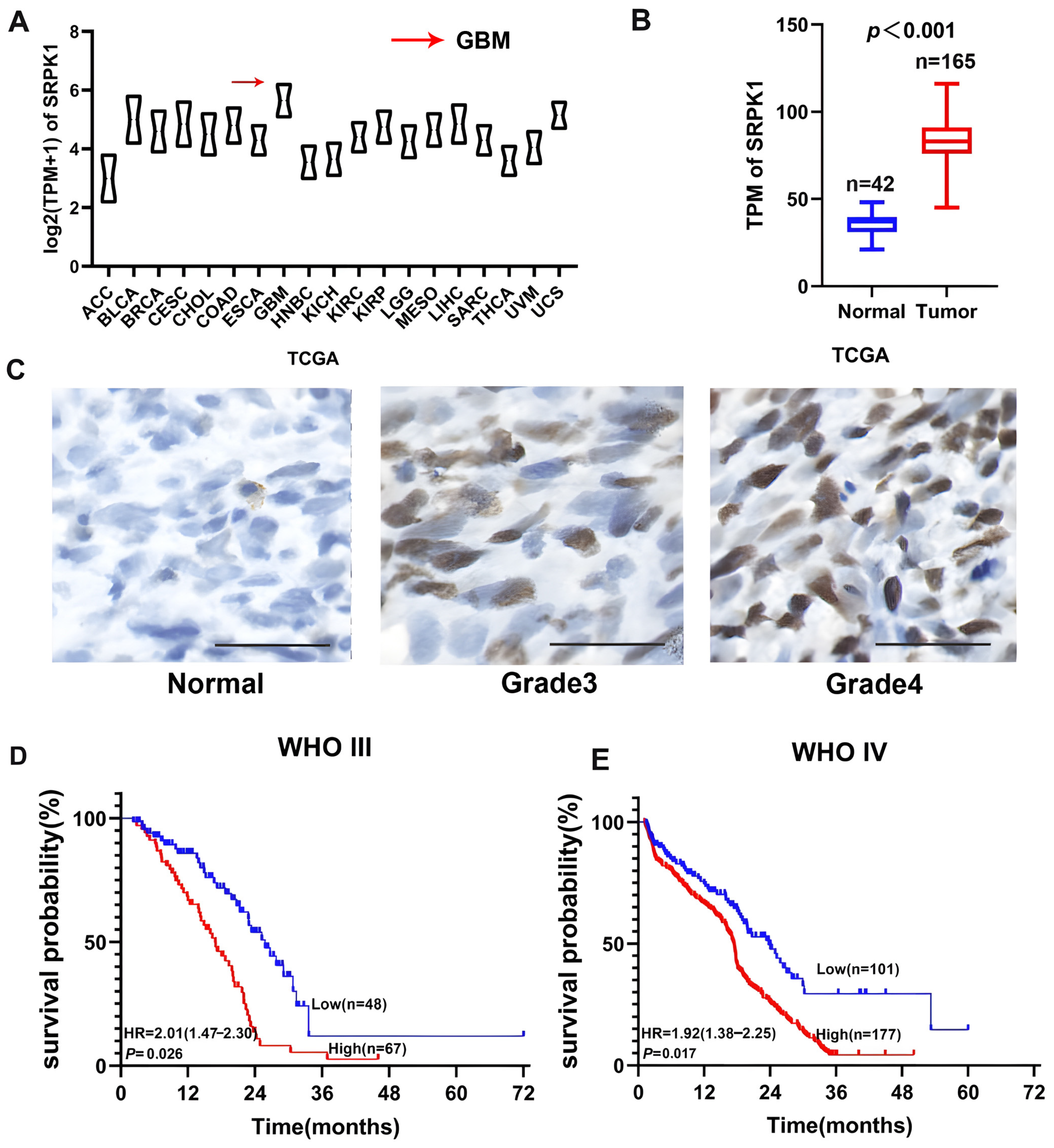
3.1. SRPK1 Overexpression Was Positively Correlated with Glioma Grade and Negatively Correlated with Patient Prognosis
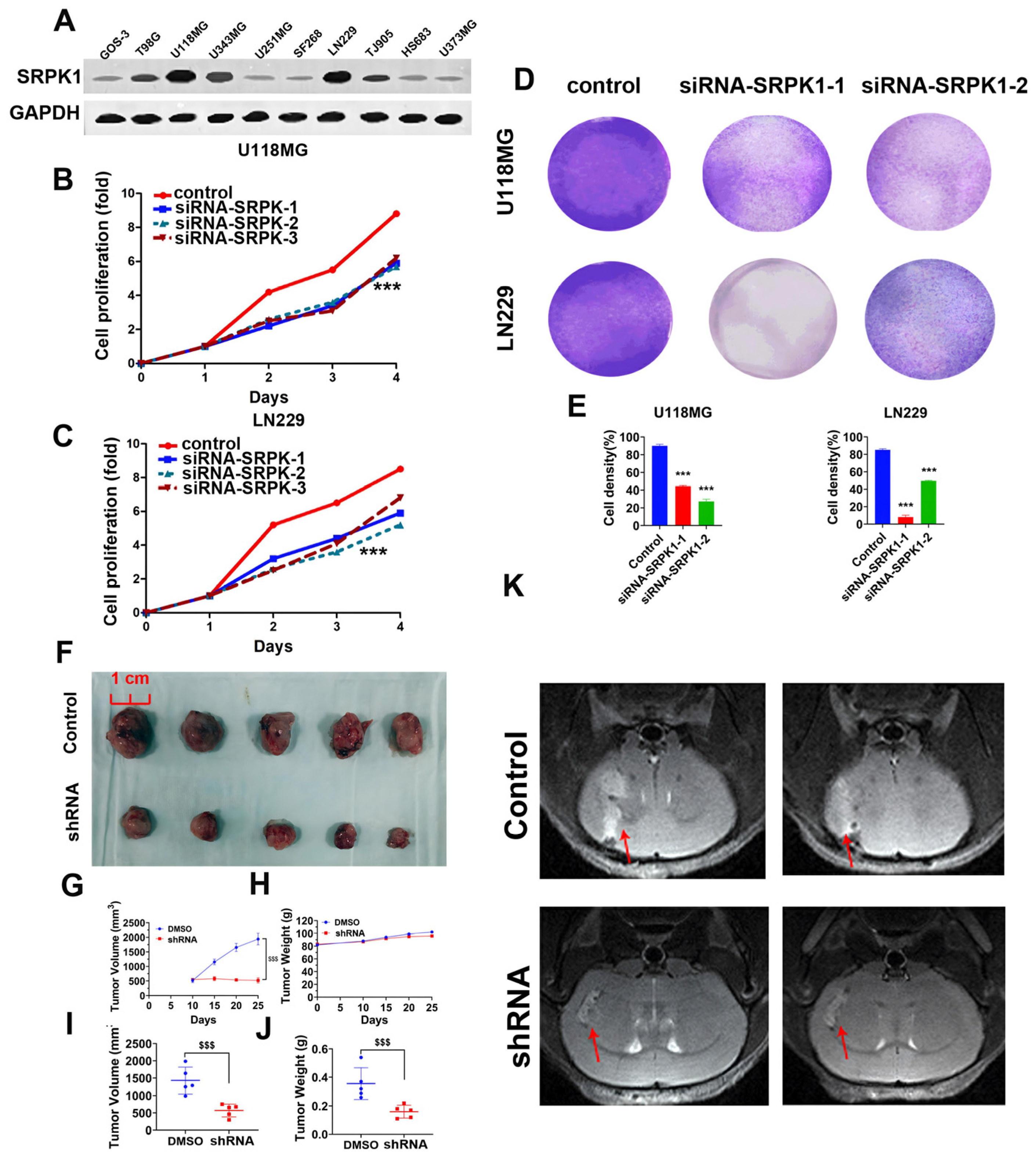
3.2. Low Expression of SRPK1 Inhibits Glioma Cell Viability and Proliferation
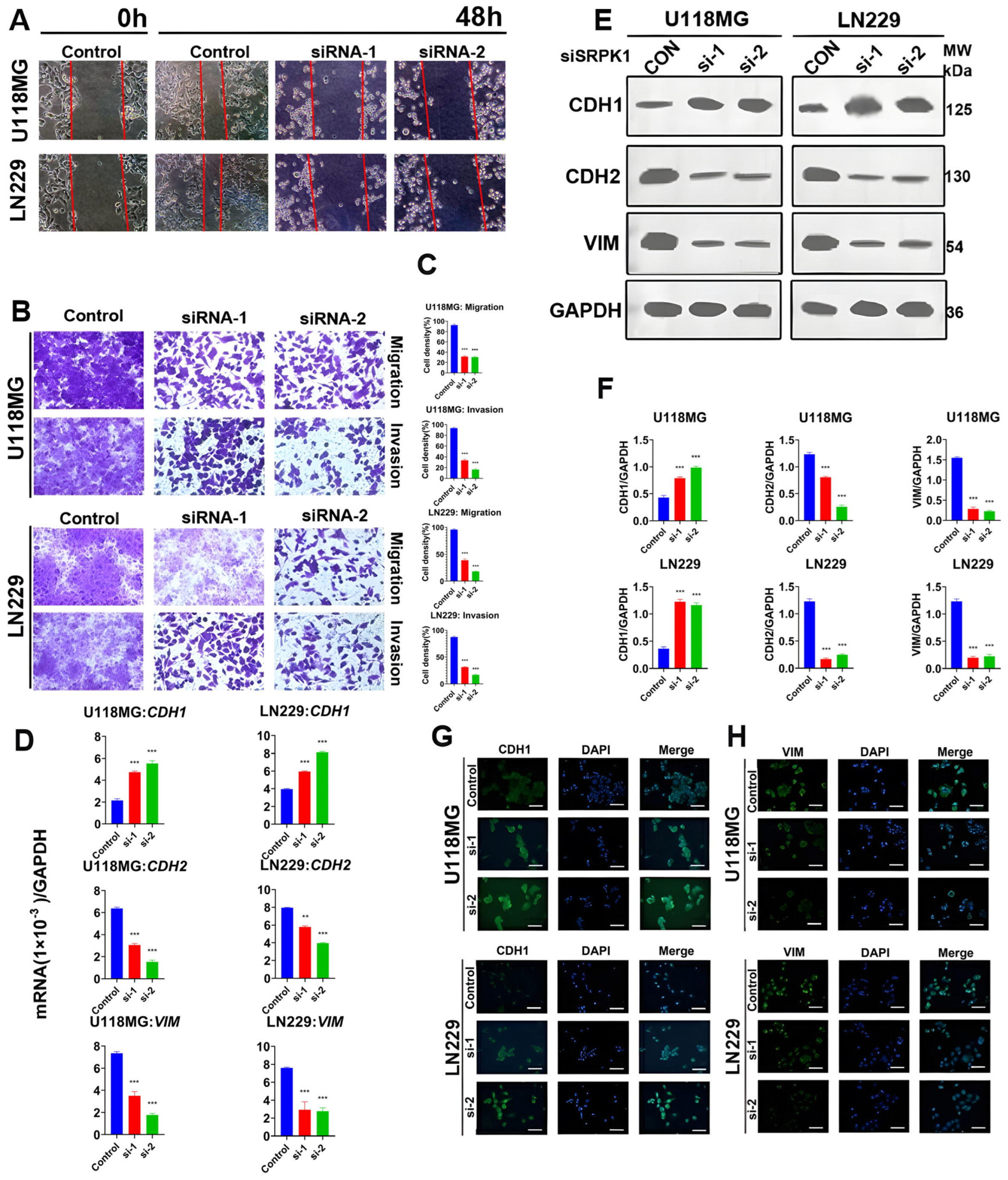
3.3. Silencing SRPK1 will Inhibit Glioma Cell Migration and Invasion
3.4. Lack of SRPK1 Expression Induces Apoptosis and G2/M Phase Blockade
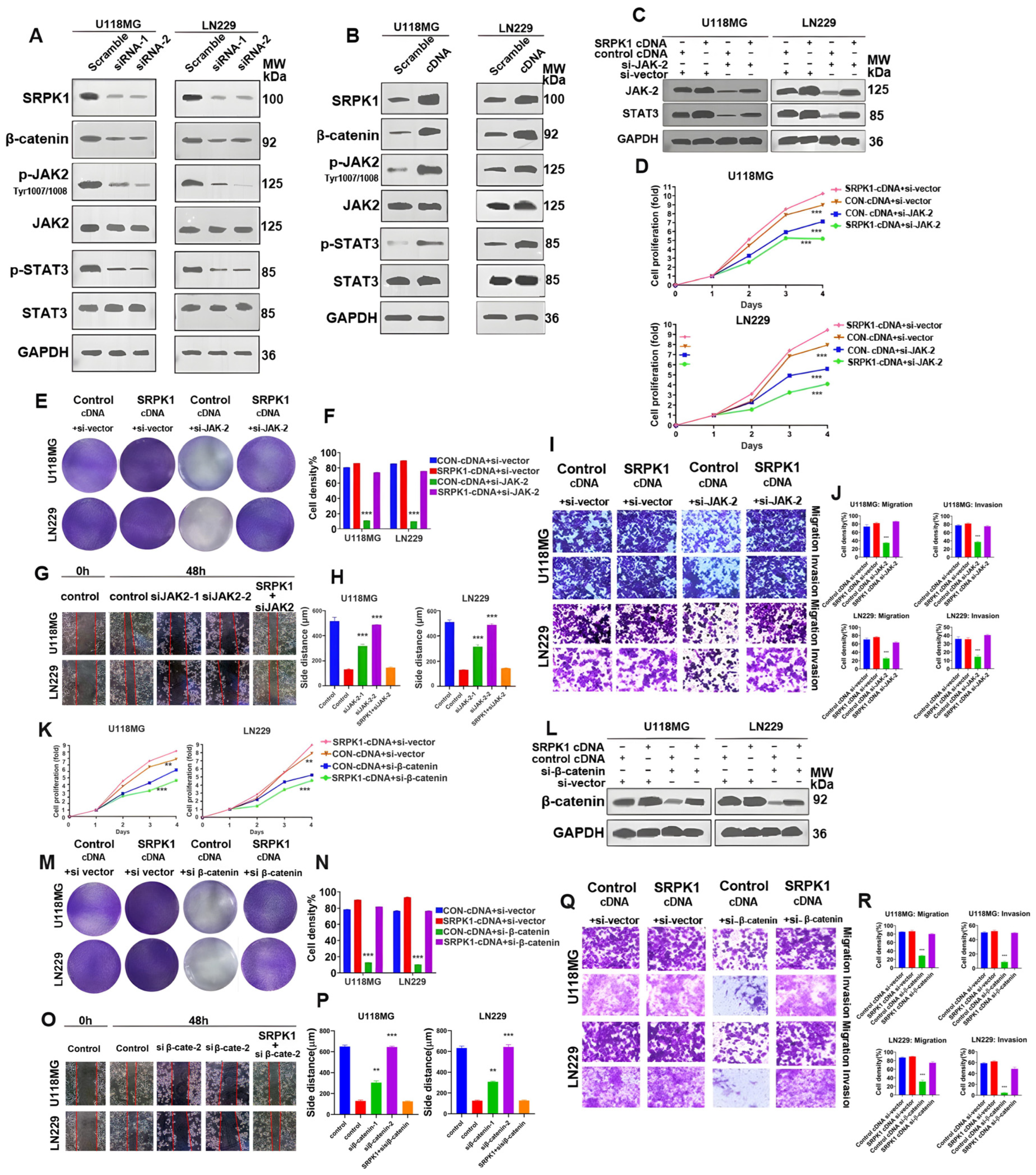
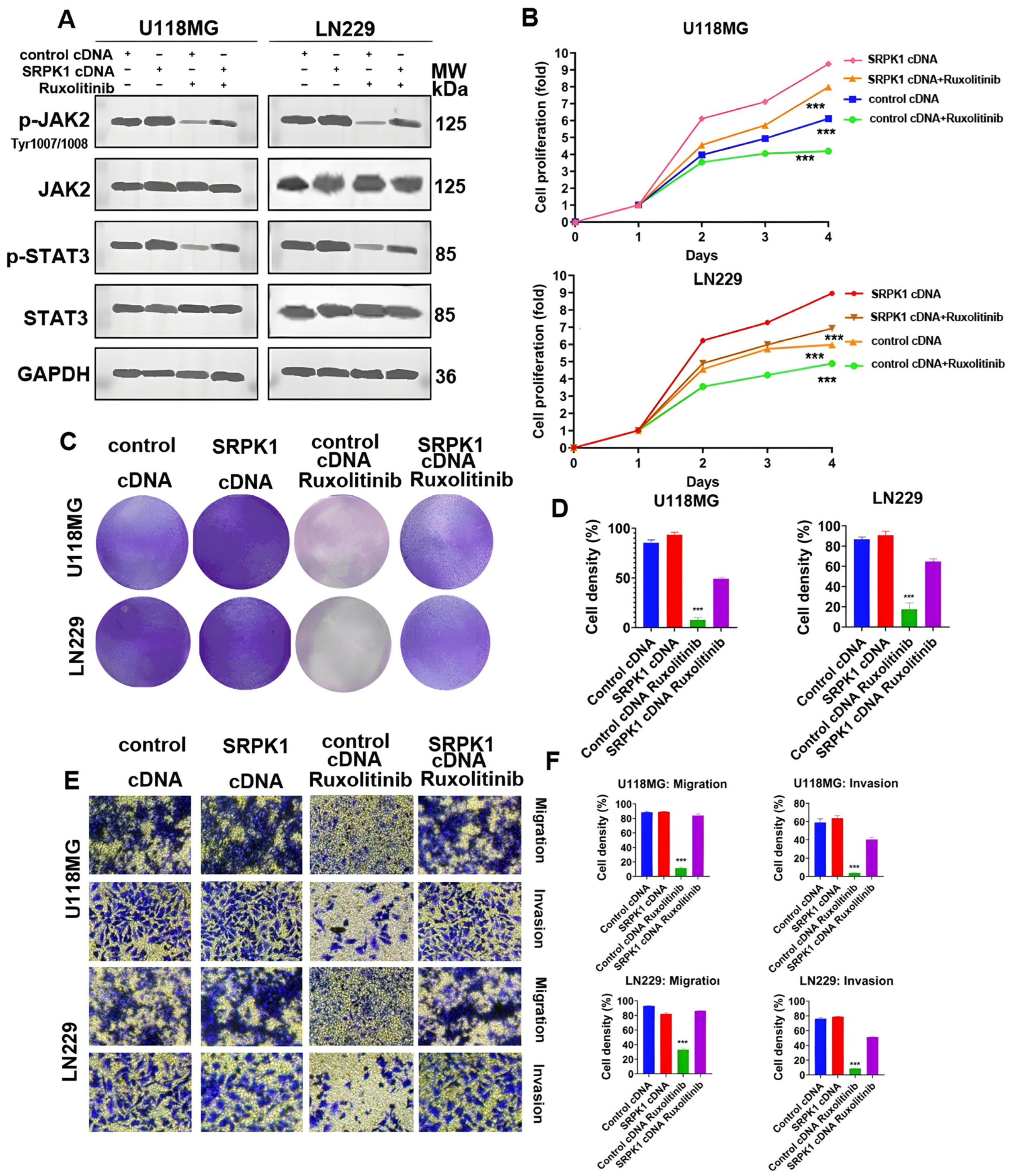
3.5. SRPK1 Promotes Glioma Proliferation and Migration through Activation of Wnt/β-Catenin and JAK-2/STAT-3 Signaling Pathways, Invasion
3.6. The Expression Levels of SRPK1 and Related Molecules in Subcutaneous Graft and Glioma Tissues of Nude Mice Were Positively Correlated
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, K.; Wu, Z.; Zhang, H.; Zhang, N.; Wu, W.; Wang, Z.; Dai, Z.; Zhang, X.; Zhang, L.; Peng, Y.; et al. Glioma targeted therapy: Insight into future of molecular approaches. Mol. Cancer 2022, 21, 39. [Google Scholar] [CrossRef]
- Ratti, S.; Marvi, M.V.; Mongiorgi, S.; Obeng, E.O.; Rusciano, I.; Ramazzotti, G.; Morandi, L.; Asioli, S.; Zoli, M.; Mazzatenta, D.; et al. Impact of phospholipase C β1 in glioblastoma: A study on the main mechanisms of tumor aggres-siveness. Cell Mol. Life Sci. 2022, 79, 195. [Google Scholar] [CrossRef]
- Wang, G.; Wang, J.; Niu, C.; Zhao, Y.; Wu, P. Neutrophils: New Critical Regulators of Glioma. Front. Immunol. 2022, 13, 927233. [Google Scholar] [CrossRef]
- Rahme, G.J.; Javed, N.M.; Puorro, K.L.; Xin, S.; Hovestadt, V.; Johnstone, S.E.; Bernstein, B.E. Modeling epigenetic lesions that cause gliomas. Cell 2023, 186, 3674–3685.e14. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Meng, J.; Zhu, L.; Peng, Y. Exosomal noncoding RNAs in Glioma: Biological functions and potential clinical applications. Mol. Cancer 2020, 19, 66. [Google Scholar] [CrossRef] [PubMed]
- Van den Bent, M.J.; Geurts, M.; French, P.J.; Smits, M.; Capper, D.; Bromberg, J.E.C.; Chang, S.M. Primary brain tumours in adults. Lancet 2023, 402, 1564–1579. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Blumcke, I.; Budday, S.; Poduri, A.; Lal, D.; Kobow, K.; Baulac, S. Neocortical development and epilepsy: Insights from focal cor-tical dysplasia and brain tumours. Lancet Neurol. 2021, 20, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Wei, Y.; Wang, X.; Zhang, Z.; Yin, J.; Li, W.; Chen, L.; Lyu, X.; Shi, Z.; Yan, W.; et al. DNA-methylation-mediated activating of lncRNA SNHG12 promotes temozolomide resistance in glioblastoma. Mol. Cancer 2020, 19, 28. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Liu, Y.; Chou, F.-J.; Yang, C. Genotoxic therapy and resistance mechanism in gliomas. Pharmacol. Ther. 2021, 228, 107922. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Gong, Z.; Li, K.; Zhang, Q.; Xu, Z.; Xu, Y. SRPK1/2 and PP1α exert opposite functions by modulating SRSF1-guided MKNK2 alternative splicing in colon adenocarcinoma. J. Exp. Clin. Cancer Res. 2021, 40, 75. [Google Scholar] [CrossRef]
- Huang, J.Q.; Li, H.F.; Zhu, J.; Song, J.W.; Zhang, X.B.; Gong, P.; Liu, Q.Y.; Zhou, C.H.; Wang, L.; Gong, L.Y. SRPK1/AKT axis promotes oxaliplatin-induced anti-apoptosis via NF-κB activation in colon cancer. J. Transl. Med. 2021, 19, 280. [Google Scholar] [CrossRef] [PubMed]
- Gou, L.-T.; Lim, D.-H.; Ma, W.; Aubol, B.E.; Hao, Y.; Wang, X.; Zhao, J.; Liang, Z.; Shao, C.; Zhang, X.; et al. Initiation of Parental Genome Reprogramming in Fertilized Oocyte by Splicing Kinase SRPK1-Catalyzed Protamine Phosphorylation. Cell 2020, 180, 1212–1227.e14. [Google Scholar] [CrossRef]
- Li, Y.; Xu, J.; Lu, Y.; Bian, H.; Yang, L.; Wu, H.; Zhang, X.; Zhang, B.; Xiong, M.; Chang, Y.; et al. DRAK2 aggravates nonalcoholic fatty liver disease progression through SRSF6-associated RNA alternative splicing. Cell Metab. 2021, 33, 2004–2020.e9. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-Q.; Duan, L.-X.; Liu, Q.-Y.; Li, H.-F.; Hu, A.-P.; Song, J.-W.; Lin, C.; Huang, B.; Yao, D.; Peng, B.; et al. Serine-arginine protein kinase 1 (SRPK1) promotes EGFR-TKI resistance by enhancing GSK3β Ser9 autophosphorylation independent of its kinase activity in non-small-cell lung cancer. Oncogene 2023, 42, 1233–1246. [Google Scholar] [CrossRef]
- Kurimchak, A.M.; Kumar, V.; Herrera-Montávez, C.; Johnson, K.J.; Srivastava, N.; Davarajan, K.; Peri, S.; Cai, K.Q.; Mantia-Smaldone, G.M.; Duncan, J.S. Kinome Profiling of Primary Endometrial Tumors Using Multiplexed Inhibitor Beads and Mass Spec-trometry Identifies SRPK1 as Candidate Therapeutic Target. Mol. Cell Proteom. 2020, 19, 2068–2090. [Google Scholar] [CrossRef]
- Yaron, T.M.; Heaton, B.E.; Levy, T.M.; Johnson, J.L.; Jordan, T.X.; Cohen, B.M.; Kerelsky, A.; Lin, T.Y.; Liberatore, K.M.; Bulaon, D.K.; et al. Host protein kinases required for SARS-CoV-2 nucleocapsid phosphorylation and viral replication. Sci. Signal. 2022, 15, eabm0808. [Google Scholar] [CrossRef]
- Neal, M.E.H.; Birkeland, A.C.; Bhangale, A.D.; Zhai, J.; Kulkarni, A.; Foltin, S.K.; Jewell, B.M.; Ludwig, M.L.; Pinatti, L.; Jiang, H.; et al. Genetic analysis of sinonasal undifferentiated carcinoma discovers recurrent SWI/SNF alterations and a novel PGAP3-SRPK1 fusion gene. BMC Cancer 2021, 21, 636. [Google Scholar]
- Nicholson, J.G.; Fine, H.A. Diffuse Glioma Heterogeneity and Its Therapeutic Implications. Cancer Discov. 2021, 11, 575–590. [Google Scholar] [CrossRef]
- Liu, T.; Zhu, C.; Chen, X.; Guan, G.; Zou, C.; Shen, S.; Wu, J.; Wang, Y.; Lin, Z.; Chen, L.; et al. Ferroptosis, as the most enriched programmed cell death process in glioma, induces immunosuppression and immunotherapy resistance. Neuro-Oncology 2022, 24, 1113–1125. [Google Scholar] [CrossRef]
- Duggan, W.P.; O’connell, E.; Prehn, J.H.M.; Burke, J.P. Serine-Arginine Protein Kinase 1 (SRPK1): A systematic review of its multimodal role in oncogenesis. Mol. Cell. Biochem. 2022, 477, 2451–2467. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Chang, Y.; Zhang, L.; Zhang, Y.; Tian, T.; Feng, G.; Zhou, S.; Zheng, Q.; Han, F.; Huang, F. SRPK1 Dissimilarly Impacts on the Growth, Metastasis, Chemosensitivity and Angiogenesis of Glioma in Normoxic and Hypoxic Conditions. J. Cancer 2013, 4, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Larsson, C.; Xu, D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: Old actors and new players. Oncogene 2019, 38, 6172–6183. [Google Scholar] [CrossRef]
- Pogacar, Z.; Groot, K.; Jochems, F.; Dos Santos Días, M.; Mulero-Sánchez, A.; Morris, B.; Roosen, M.; Wardak, L.; De Conti, G.; Velds, A.; et al. Genetic and compound screens uncover factors modulating cancer cell response to indisulam. Life Sci. Alliance 2022, 5, e202101348. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-W.; Sahm, F.; Steffl, B.; Arrillaga-Romany, I.; Cahill, D.; Monje, M.; Herold-Mende, C.; Wick, W.; Turcan, Ş. TERT and DNMT1 expression predict sensitivity to decitabine in gliomas. Neuro-Oncology 2021, 23, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary brain tumours in adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yin, L.; Ma, L.; Sun, H. Artificial Intelligence Applications in Glioma With 1p/19q Co-Deletion: A Systematic Review. J. Magn. Reson. Imaging 2023, 58, 1338–1352. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Ananda, H.; Singh, N.; Qamar, I. Identification of a novel and potent small molecule inhibitor of SRPK1: Mech-anism of dual inhibition of SRPK1 for the inhibition of cancer progression. Aging 2020, 13, 163–180. [Google Scholar] [CrossRef]
- Belali, T.; Wodi, C.; Clark, B.; Cheung, M.K.; Craig, T.J.; Wheway, G.; Wagner, N.; Wagner, K.D.; Roberts, S.; Porazinski, S.; et al. WT1 activates transcription of the splice factor kinase SRPK1 gene in PC3 and K562 cancer cells in the absence of core-pressor BASP1. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194642. [Google Scholar] [CrossRef]
- Zhao, X.; Li, J.; Liu, Z.; Powers, S. Combinatorial CRISPR/Cas9 Screening Reveals Epistatic Networks of Interacting Tumor Suppressor Genes and Therapeutic Targets in Human Breast Cancer. Cancer Res. 2021, 81, 6090–6105. [Google Scholar] [CrossRef]
- Malvi, P.; Janostiak, R.; Chava, S.; Manrai, P.; Yoon, E.; Singh, K.; Harigopal, M.; Gupta, R.; Wajapeyee, N. LIMK2 promotes the metastatic progression of triple-negative breast cancer by activating SRPK1. Oncogenesis 2020, 9, 77. [Google Scholar] [CrossRef] [PubMed]
- Akhoundova, D.; Hussung, S.; Sivakumar, S.; Töpfer, A.; Rechsteiner, M.; Kahraman, A.; Arnold, F.; Angst, F.; Britschgi, C.; Zoche, M.; et al. ROS1 genomic rearrangements are rare actionable drivers in microsatellite stable colorectal cancer. Int. J. Cancer 2022, 151, 2161–2171. [Google Scholar] [CrossRef] [PubMed]
- Jia, T.; Jacquet, T.; Dalonneau, F.; Coudert, P.; Vaganay, E.; Exbrayat-Héritier, C.; Vollaire, J.; Josserand, V.; Ruggiero, F.; Coll, J.L.; et al. FGF-2 promotes angiogenesis through a SRSF1/SRSF3/SRPK1-dependent axis that controls VEGFR1 splicing in endo-thelial cells. BMC Biol. 2021, 19, 173. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Chang, X.; Xie, L.; Wang, Y.; Hou, Y. Increased expression of SRPK1 (serine/arginine-rich protein-specific kinase 1) is associated with progression and unfavorable prognosis in cervical squamous cell carcinoma. Bioengineered 2022, 13, 6100–6112. [Google Scholar] [CrossRef]
- Schunk, S.J.; Floege, J.; Fliser, D.; Speer, T. WNT-β-catenin signalling—A versatile player in kidney injury and repair. Nat. Rev. Nephrol. 2021, 17, 172–184. [Google Scholar] [CrossRef]
- Choi, B.Y. Targeting Wnt/β-Catenin Pathway for Developing Therapies for Hair Loss. Int. J. Mol. Sci. 2020, 21, 4915. [Google Scholar] [CrossRef]
- El-Mokadem, B.M.; El-Abhar, H.S.; Abdallah, D.M.; Awad, A.S.; Soubh, A.A. Epac-1/Rap-1 signaling pathway orchestrates the reno-therapeutic effect of ticagrelor against renal ischemia/reperfusion model. Biomed. Pharmacother. 2021, 139, 111488. [Google Scholar] [CrossRef]
- Salem, H.A.; Abu-Elfotuh, K.; Alzahrani, S.; Rizk, N.I.; Ali, H.S.; Elsherbiny, N.; Aljohani, A.; Hamdan, A.M.E.; Chellasamy, P.; Abdou, N.S.; et al. Punicalagin’s Protective Effects on Parkinson’s Progression in Socially Isolated and Socialized Rats: Insights into Multifaceted Pathway. Pharmaceutics 2023, 15, 2420. [Google Scholar] [CrossRef]
- Lang, F.; Singh, R.K.; Pei, Y.; Zhang, S.; Sun, K.; Robertson, E.S. EBV epitranscriptome reprogramming by METTL14 is critical for viral-associated tumorigenesis. PLoS Pathog. 2019, 15, e1007796. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Uni HR | Multi HR | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p | HR | 95% CI | p | |
| Gender (male, female) | 0.93 | 0.75–1.35 | 0.765 | - | - | - |
| Age (<60, ≥60) | 0.97 | 0.85–1.25 | 0.151 | - | - | - |
| KPS (<70, ≥70) | 0.73 | 0.35–0.80 | 0.001 | 0.62 | 0.40–1.12 | 0.055 |
| Radio-chemotherapy (yes, no) | 0.27 | 0.18–0.35 | <0.001 | 0.33 | 0.22–0.39 | <0.001 |
| SRPK1 (high, low) | 1.86 | 1.25–2.50 | <0.001 | 1.55 | 1.05–2.54 | 0.005 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, M.; Sun, D.; Deng, L.; Liu, J.; Zhang, M.-J. SRPK1 Promotes Glioma Proliferation, Migration, and Invasion through Activation of Wnt/β-Catenin and JAK-2/STAT-3 Signaling Pathways. Biomedicines 2024, 12, 378. https://doi.org/10.3390/biomedicines12020378
Shi M, Sun D, Deng L, Liu J, Zhang M-J. SRPK1 Promotes Glioma Proliferation, Migration, and Invasion through Activation of Wnt/β-Catenin and JAK-2/STAT-3 Signaling Pathways. Biomedicines. 2024; 12(2):378. https://doi.org/10.3390/biomedicines12020378
Chicago/Turabian StyleShi, Mengna, Dan Sun, Lu Deng, Jing Liu, and Min-Jie Zhang. 2024. "SRPK1 Promotes Glioma Proliferation, Migration, and Invasion through Activation of Wnt/β-Catenin and JAK-2/STAT-3 Signaling Pathways" Biomedicines 12, no. 2: 378. https://doi.org/10.3390/biomedicines12020378
APA StyleShi, M., Sun, D., Deng, L., Liu, J., & Zhang, M.-J. (2024). SRPK1 Promotes Glioma Proliferation, Migration, and Invasion through Activation of Wnt/β-Catenin and JAK-2/STAT-3 Signaling Pathways. Biomedicines, 12(2), 378. https://doi.org/10.3390/biomedicines12020378