
Hidradenitis Suppurativa: An Understanding of Genetic Factors and Treatment



Abstract
1. Introduction
2. Search Strategy
3. Genetics of Hidradenitis Suppurativa
3.1. γ-Secretase Gene
3.2. TNF-α
3.3. IL-17
3.4. IL-1β
3.5. IL-12/23
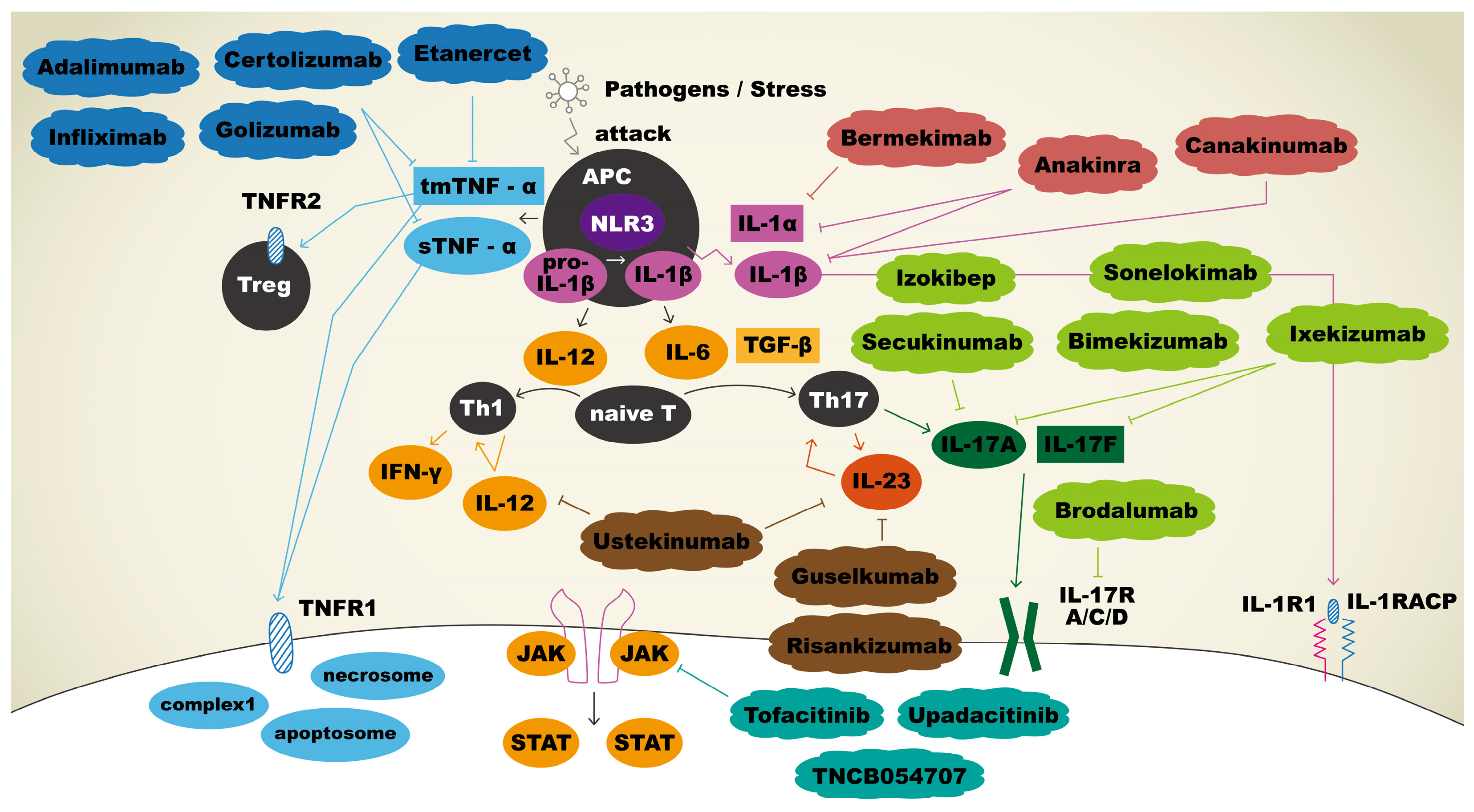
4. Current and Potential Therapeutic Agents Targeting Immune Mediators in Hidradenitis Suppurativa
4.1. Anti-TNF-α Therapy
4.2. Anti-IL-17 Therapy
4.3. Anti-IL-1 Therapy
4.4. Anti-IL-12/23 Therapy and Anti-IL-23 Therapy
4.5. Janus Kinase Inhibitors
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Choi, E.; Mir, S.A.; Ji, S.; Ooi, X.T.; Chua, E.W.L.; Yi Wei, Y.; Wenk, M.R.; Bendt, A.K.; Chandran, N.S. Understanding the systemic burden of disease in hidradenitis suppurativa from plasma lipidomic analysis. J. Dermatol. Sci. 2022, 107, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Pace, N.P.; Mintoff, D.; Borg, I. The Genomic Architecture of Hidradenitis Suppurativa-A Systematic Review. Front. Genet. 2022, 13, 861241. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.D.B.; Okoye, G.A.; Sokumbi, O. Histopathology of Hidradenitis Suppurativa: A Systematic Review. Dermatopathology 2022, 9, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Saunte, D.M.L.; Jemec, G.B.E. Hidradenitis Suppurativa: Advances in Diagnosis and Treatment. JAMA 2017, 318, 2019–2032. [Google Scholar] [CrossRef] [PubMed]
- Kadylak, D.; Baranska-Rybak, W. Botulinum toxin type A therapy for hidradenitis suppurativa: A case series. Dermatol. Sin. 2023, 41, 121–122. [Google Scholar] [CrossRef]
- Baranska-Rybak, W.; Lewandowski, M.; Swierczewska, Z. YouTube as a source of information for hidradenitis suppurativa treatment. Dermatol. Sin. 2022, 40, 156–161. [Google Scholar] [CrossRef]
- Atzori, L.; Zanniello, R.; Pilloni, L.; Rongioletti, F. Steatocystoma multiplex suppurativa associated with hidradenitis suppurativa successfully treated with adalimumab. J. Eur. Acad. Dermatol. Venereol. 2019, 33 (Suppl. S6), 42–44. [Google Scholar] [CrossRef]
- Revuz, J.E.; Canoui-Poitrine, F.; Wolkenstein, P.; Viallette, C.; Gabison, G.; Pouget, F.; Poli, F.; Faye, O.; Roujeau, J.C.; Bonnelye, G.; et al. Prevalence and factors associated with hidradenitis suppurativa: Results from two case-control studies. J. Am. Acad. Dermatol. 2008, 59, 596–601. [Google Scholar] [CrossRef]
- Jemec, G.B.; Heidenheim, M.; Nielsen, N.H. The prevalence of hidradenitis suppurativa and its potential precursor lesions. J. Am. Acad. Dermatol. 1996, 35, 191–194. [Google Scholar] [CrossRef]
- Zouboulis, C.C.; Benhadou, F.; Byrd, A.S.; Chandran, N.S.; Giamarellos-Bourboulis, E.J.; Fabbrocini, G.; Frew, J.W.; Fujita, H.; González-López, M.A.; Guillem, P.; et al. What causes hidradenitis suppurativa ?—15 years after. Exp. Dermatol. 2020, 29, 1154–1170. [Google Scholar] [CrossRef]
- Moltrasio, C.; Tricarico, P.M.; Romagnuolo, M.; Marzano, A.V.; Crovella, S. Hidradenitis Suppurativa: A Perspective on Genetic Factors Involved in the Disease. Biomedicines 2022, 10, 2039. [Google Scholar] [CrossRef]
- Jørgensen, A.R.; Brøgger-Mikkelsen, M.; Ring, H.C.; Thomsen, S.F. Patients with a familial predisposition to hidradenitis suppurativa have a distinct clinical phenotype. J. Am. Acad. Dermatol. 2020, 83, 1809–1811. [Google Scholar] [CrossRef]
- Tricarico, P.M.; Moltrasio, C.; Gradišek, A.; Marzano, A.V.; Flacher, V.; Boufenghour, W.; von Stebut, E.; Schmuth, M.; Jaschke, W.; Gams, M.; et al. Holistic health record for Hidradenitis suppurativa patients. Sci. Rep. 2022, 12, 8415. [Google Scholar] [CrossRef] [PubMed]
- Poli, F.; Jemec, G.B.E.; Revuz, J. Clinical Presentation. In Hidradenitis Suppurativa; Jemec, G.B.E., Revuz, J., Leyden, J.J., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 11–24. [Google Scholar]
- Jemec, G.B. Clinical practice. Hidradenitis suppurativa. N. Engl. J. Med. 2012, 366, 158–164. [Google Scholar] [CrossRef]
- Almuhanna, N.; Wortsman, X.; Wohlmuth-Wieser, I.; Kinoshita-Ise, M.; Alhusayen, R. Overview of Ultrasound Imaging Applications in Dermatology. J. Cutan. Med. Surg. 2021, 25, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Revuz, J.E.; Jemec, G.B. Diagnosing Hidradenitis Suppurativa. Dermatol. Clin. 2016, 34, 1–5. [Google Scholar] [CrossRef]
- Wieczorek, M.; Walecka, I. Hidradenitis suppurativa—Known and unknown disease. Reumatologia 2018, 56, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Sabat, R.; Jemec, G.B.E.; Matusiak, Ł.; Kimball, A.B.; Prens, E.; Wolk, K. Hidradenitis suppurativa. Nat. Rev. Dis. Primers 2020, 6, 18. [Google Scholar] [CrossRef]
- Teng, Y.; Tao, X.; Lu, W.; Huang, Y.; Xu, D.; Li, M.; Fan, Y. Identification of Hidradenitis Suppurativa-Related mRNA Expression Patterns through Analysis of Gene Expression Omnibus. Dose Response 2020, 18, 1559325820942646. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimmons, J.S.; Guilbert, P.R.; Fitzsimmons, E.M. Evidence of genetic factors in hidradenitis suppurativa. Br. J. Dermatol. 1985, 113, 1–8. [Google Scholar] [CrossRef]
- Gao, M.; Wang, P.G.; Cui, Y.; Yang, S.; Zhang, Y.H.; Lin, D.; Zhang, K.Y.; Liang, Y.H.; Sun, L.D.; Yan, K.L.; et al. Inversa acne (hidradenitis suppurativa): A case report and identification of the locus at chromosome 1p21.1–1q25.3. J. Investig. Dermatol. 2006, 126, 1302–1306. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gao, M.; Lv, Y.M.; Yang, X.; Ren, Y.Q.; Jiang, T.; Zhang, X.; Guo, B.R.; Li, M.; Zhang, Q.; et al. Confirmation by exome sequencing of the pathogenic role of NCSTN mutations in acne inversa (hidradenitis suppurativa). J. Investig. Dermatol. 2011, 131, 1570–1572. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Davis, J.W.; Idler, K.B.; Mostafa, N.M.; Okun, M.M.; Waring, J.F. Genetic analysis of NCSTN for potential association with hidradenitis suppurativa in familial and nonfamilial patients. Br. J. Dermatol. 2016, 175, 414–416. [Google Scholar] [CrossRef] [PubMed]
- Matusiak, L.; Bieniek, A.; Szepietowski, J.C. Increased serum tumour necrosis factor-alpha in hidradenitis suppurativa patients: Is there a basis for treatment with anti-tumour necrosis factor-alpha agents? Acta Derm. Venereol. 2009, 89, 601–603. [Google Scholar] [CrossRef] [PubMed]
- van der Zee, H.H.; de Ruiter, L.; van den Broecke, D.G.; Dik, W.A.; Laman, J.D.; Prens, E.P. Elevated levels of tumour necrosis factor (TNF)-α, interleukin (IL)-1β and IL-10 in hidradenitis suppurativa skin: A rationale for targeting TNF-α and IL-1β. Br. J. Dermatol. 2011, 164, 1292–1298. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.M.; Moran, B.; Petrasca, A.; Smith, C.M. IL-17 in inflammatory skin diseases psoriasis and hidradenitis suppurativa. Clin. Exp. Immunol. 2020, 201, 121–134. [Google Scholar] [CrossRef]
- Kurayev, A.; Ashkar, H.; Saraiya, A.; Gottlieb, A.B. Hidradenitis Suppurativa: Review of the Pathogenesis and Treatment. J. Drugs Dermatol. 2016, 15, 1017–1022. [Google Scholar]
- Schlapbach, C.; Hänni, T.; Yawalkar, N.; Hunger, R.E. Expression of the IL-23/Th17 pathway in lesions of hidradenitis suppurativa. J. Am. Acad. Dermatol. 2011, 65, 790–798. [Google Scholar] [CrossRef]
- Pink, A.E.; Simpson, M.A.; Desai, N.; Dafou, D.; Hills, A.; Mortimer, P.; Smith, C.H.; Trembath, R.C.; Barker, J.N.W. Mutations in the γ-secretase genes NCSTN, PSENEN, and PSEN1 underlie rare forms of hidradenitis suppurativa (acne inversa). J. Investig. Dermatol. 2012, 132, 2459–2461. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Y.; Xu, H.; Zhang, Y.W. The γ-secretase complex: From structure to function. Front. Cell Neurosci. 2014, 8, 427. [Google Scholar] [CrossRef]
- Chataigner, L.M.P.; Leloup, N.; Janssen, B.J.C. Structural Perspectives on Extracellular Recognition and Conformational Changes of Several Type-I Transmembrane Receptors. Front. Mol. Biosci. 2020, 7, 129. [Google Scholar] [CrossRef]
- Dumbrava, E.E.I.; Mills, G.B.; Yap, T.A. Targeting gamma secretase: Has progress moved up a Notch? Ann. Oncol. 2018, 29, 1889–1891. [Google Scholar] [CrossRef]
- Kopan, R. Notch signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011213. [Google Scholar] [CrossRef]
- Jfri, A.; O’Brien, E.; Litvinov, I.; Alavi, A.; Netchiporouk, E. Hidradenitis Suppurativa: Comprehensive Review of Predisposing Genetic Mutations and Changes. J. Cutan. Med. Surg. 2019, 23, 1203475419852049. [Google Scholar] [CrossRef]
- He, Y.; Li, C.; Xu, H.; Duan, Z.; Liu, Y.; Zeng, R.; Li, M.; Wang, B. AKT-dependent hyperproliferation of keratinocytes in familial hidradenitis suppurativa with a NCSTN mutation: A potential role of defective miR-100-5p. Br. J. Dermatol. 2020, 182, 500–502. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xu, H.; Li, C.; Zhang, X.; Zhou, P.; Xiao, X.; Zhang, W.; Wu, Y.; Zeng, R.; Wang, B. Nicastrin/miR-30a-3p/RAB31 Axis Regulates Keratinocyte Differentiation by Impairing EGFR Signaling in Familial Acne Inversa. J. Investig. Dermatol. 2019, 139, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, T.; Mitoma, H.; Harashima, S.; Tsukamoto, H.; Shimoda, T. Transmembrane TNF-alpha: Structure, function and interaction with anti-TNF agents. Rheumatology 2010, 49, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.; McDermott, M. Tumour necrosis factor signalling in health and disease. F1000Res 2019, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Haas, T.L.; Emmerich, C.H.; Gerlach, B.; Schmukle, A.C.; Cordier, S.M.; Rieser, E.; Feltham, R.; Vince, J.; Warnken, U.; Wenger, T.; et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 2009, 36, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of tumour necrosis factor signalling: Live or let die. Nat. Rev. Immunol. 2015, 15, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Du, F.; Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef]
- Jang, D.I.; Lee, A.H.; Shin, H.Y.; Song, H.R.; Park, J.H.; Kang, T.B.; Lee, S.R.; Yang, S.H. The Role of Tumor Necrosis Factor Alpha (TNF-α) in Autoimmune Disease and Current TNF-α Inhibitors in Therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef]
- Kalliolias, G.D.; Ivashkiv, L.B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016, 12, 49–62. [Google Scholar] [CrossRef]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef]
- Venken, K.; Jacques, P.; Mortier, C.; Labadia, M.E.; Decruy, T.; Coudenys, J.; Hoyt, K.; Wayne, A.L.; Hughes, R.; Turner, M.; et al. RORγt inhibition selectively targets IL-17 producing iNKT and γδ-T cells enriched in Spondyloarthritis patients. Nat. Commun. 2019, 10, 9. [Google Scholar] [CrossRef]
- Liu, T.; Li, S.; Ying, S.; Tang, S.; Ding, Y.; Li, Y.; Qiao, J.; Fang, H. The IL-23/IL-17 Pathway in Inflammatory Skin Diseases: From Bench to Bedside. Front. Immunol. 2020, 11, 594735. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, C.; Qian, W.; Han, Y.; Li, X.; Deng, J. Structure of the unique SEFIR domain from human interleukin 17 receptor A reveals a composite ligand-binding site containing a conserved α-helix for Act1 binding and IL-17 signaling. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 1476–1483. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Liu, C.; Hartupee, J.; Altuntas, C.Z.; Gulen, M.F.; Jane-wit, D.; Xiao, J.; Lu, Y.; Giltiay, N.; Liu, J.; et al. The adaptor Act1 is required for interleukin 17–dependent signaling associated with autoimmune and inflammatory disease. Nat. Immunol. 2007, 8, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Chen, X.; Zhao, J.; Martin, B.; Zepp, J.A.; Ko, J.S.; Gu, C.; Cai, G.; Ouyang, W.; Sen, G.; et al. A novel IL-17 signaling pathway controlling keratinocyte proliferation and tumorigenesis via the TRAF4-ERK5 axis. J. Exp. Med. 2015, 212, 1571–1587. [Google Scholar] [CrossRef] [PubMed]
- Lachman, L.B.; Hacker, M.P.; Handschumacher, R.E. Partial purification of human lymphocyte-activating factor (LAF) by ultrafiltration and electrophoretic techniques. J. Immunol. 1977, 119, 2019–2023. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef]
- Pyrillou, K.; Burzynski, L.C.; Clarke, M.C.H. Alternative Pathways of IL-1 Activation, and Its Role in Health and Disease. Front. Immunol. 2020, 11, 613170. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, Q. Uncoupled pyroptosis and IL-1β secretion downstream of inflammasome signaling. Front. Immunol. 2023, 14, 1128358. [Google Scholar] [CrossRef]
- Gabay, C.; Lamacchia, C.; Palmer, G. IL-1 pathways in inflammation and human diseases. Nat. Rev. Rheumatol. 2010, 6, 232–241. [Google Scholar] [CrossRef]
- Weber, A.; Wasiliew, P.; Kracht, M. Interleukin-1 (IL-1) Pathway. Sci. Signal. 2010, 3, cm1. [Google Scholar] [CrossRef]
- Brikos, C.; Wait, R.; Begum, S.; O’Neill, L.A.; Saklatvala, J. Mass spectrometric analysis of the endogenous type I interleukin-1 (IL-1) receptor signaling complex formed after IL-1 binding identifies IL-1RAcP, MyD88, and IRAK-4 as the stable components. Mol. Cell Proteom. 2007, 6, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, K.; Gohda, J.; Kanayama, A.; Miyamoto, Y.; Sakurai, H.; Yamamoto, M.; Akira, S.; Hayashi, H.; Su, B.; Inoue, J. Two mechanistically and temporally distinct NF-kappaB activation pathways in IL-1 signaling. Sci. Signal. 2009, 2, ra66. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Peng, B.; Li, Z.; Sclabas, G.M.; Fujioka, S.; Niu, J.; Schmidt-Supprian, M.; Evans, D.B.; Abbruzzese, J.L.; Chiao, P.J. Mechanisms of proinflammatory cytokine-induced biphasic NF-kappaB activation. Mol. Cell 2003, 12, 1287–1300. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Yang, J.; Lin, Y.; Walker, C.; Cheng, J.; Liu, Z.G.; Su, B. Differential regulation of interleukin 1 receptor and Toll-like receptor signaling by MEKK3. Nat. Immunol. 2004, 5, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.X.; Yu, S.; Gran, B.; Li, J.; Siglienti, I.; Chen, X.; Calida, D.; Ventura, E.; Kamoun, M.; Rostami, A. Role of IL-12 receptor beta 1 in regulation of T cell response by APC in experimental autoimmune encephalomyelitis. J. Immunol. 2003, 171, 4485–4492. [Google Scholar] [CrossRef] [PubMed]
- Sutton, C.E.; Lalor, S.J.; Sweeney, C.M.; Brereton, C.F.; Lavelle, E.C.; Mills, K.H. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 2009, 31, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Riol-Blanco, L.; Lazarevic, V.; Awasthi, A.; Mitsdoerffer, M.; Wilson, B.S.; Croxford, A.; Waisman, A.; Kuchroo, V.K.; Glimcher, L.H.; Oukka, M. IL-23 receptor regulates unconventional IL-17-producing T cells that control bacterial infections. J. Immunol. 2010, 184, 1710–1720. [Google Scholar] [CrossRef]
- Kastelein, R.A.; Hunter, C.A.; Cua, D.J. Discovery and Biology of IL-23 and IL-27: Related but Functionally Distinct Regulators of Inflammation. Annu. Rev. Immunol. 2007, 25, 221–242. [Google Scholar] [CrossRef] [PubMed]
- Parham, C.; Chirica, M.; Timans, J.; Vaisberg, E.; Travis, M.; Cheung, J.; Pflanz, S.; Zhang, R.; Singh, K.P.; Vega, F.; et al. A Receptor for the Heterodimeric Cytokine IL-23 Is Composed of IL-12Rβ1 and a Novel Cytokine Receptor Subunit, IL-23R1. J. Immunol. 2002, 168, 5699–5708. [Google Scholar] [CrossRef]
- Jana, M.; Pahan, K. Induction of lymphotoxin-α by interleukin-12 p40 homodimer, the so-called biologically inactive molecule, but not IL-12 p70. Immunology 2009, 127, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Chizzolini, C.; Chicheportiche, R.; Alvarez, M.; de Rham, C.; Roux-Lombard, P.; Ferrari-Lacraz, S.; Dayer, J.M. Prostaglandin E2 synergistically with interleukin-23 favors human Th17 expansion. Blood 2008, 112, 3696–3703. [Google Scholar] [CrossRef]
- Kalim, K.W.; Groettrup, M. Prostaglandin E2 inhibits IL-23 and IL-12 production by human monocytes through down-regulation of their common p40 subunit. Mol. Immunol. 2013, 53, 274–282. [Google Scholar] [CrossRef]
- Martora, F.; Megna, M.; Battista, T.; Potestio, L.; Annunziata, M.C.; Marasca, C.; Villani, A.; Fabbrocini, G. Adalimumab, Ustekinumab, and Secukinumab in the Management of Hidradenitis Suppurativa: A Review of the Real-Life Experience. Clin. Cosmet. Investig. Dermatol. 2023, 16, 135–148. [Google Scholar] [CrossRef]
- Reddy, S.P.; Lin, E.J.; Shah, V.V.; Wu, J.J. Chapter 10—Adalimumab. In Therapy for Severe Psoriasis; Wu, J.J., Feldman, S.R., Lebwohl, M.G., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 111–126. [Google Scholar]
- Kobayashi, S.; Kashiwagi, T.; Kimura, J. Real-world effectiveness and safety of adalimumab for treatment of ankylosing spondylitis in Japan. Mod. Rheumatol. 2019, 29, 1007–1012. [Google Scholar] [CrossRef]
- Efficacy of high dose infliximab in hidradenitis suppurativa. J. Am. Acad. Dermatol. 2019, 81, AB54. [CrossRef]
- Ghias, M.H.; Johnston, A.D.; Kutner, A.J.; Micheletti, R.G.; Hosgood, H.D.; Cohen, S.R. High-dose, high-frequency infliximab: A novel treatment paradigm for hidradenitis suppurativa. J. Am. Acad. Dermatol. 2020, 82, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.T.; Flood, K.S.; Porter, M.L.; Kimball, A.B. TNF-α inhibitors in the treatment of hidradenitis suppurativa. Ther. Adv. Chronic Dis. 2019, 10, 2040622319851640. [Google Scholar] [CrossRef] [PubMed]
- Giamarellos-Bourboulis, E.J.; Pelekanou, E.; Antonopoulou, A.; Petropoulou, H.; Baziaka, F.; Karagianni, V.; Stavrianeas, N.; Giamarellou, H. An open-label phase II study of the safety and efficacy of etanercept for the therapy of hidradenitis suppurativa. Br. J. Dermatol. 2008, 158, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Cusack, C.; Buckley, C. Etanercept: Effective in the management of hidradenitis suppurativa. Br. J. Dermatol. 2006, 154, 726–729. [Google Scholar] [CrossRef] [PubMed]
- Sotiriou, E.; Apalla, Z.; Ioannidos, D. Etanercept for the treatment of hidradenitis suppurativa. Acta Derm. Venereol. 2009, 89, 82–83. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.A.; Dommasch, E.; Treat, J.; Sciacca-Kirby, J.; Chachkin, S.; Williams, J.; Shin, D.B.; Leyden, J.J.; Vittorio, C.; Gelfand, J.M. A prospective clinical trial of open-label etanercept for the treatment of hidradenitis suppurativa. J. Am. Acad. Dermatol. 2009, 60, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Shadid, A.; Alobaida, S.; Binamer, Y. Certolizumab to treat hidradenitis suppurativa. Dermatol. Rep. 2023, 15, 9566. [Google Scholar] [CrossRef]
- Tursi, A. Concomitant hidradenitis suppurativa and pyostomatitis vegetans in silent ulcerative colitis successfully treated with golimumab. Dig. Liver Dis. 2016, 48, 1511–1512. [Google Scholar] [CrossRef]
- van der Zee, H.H.; Prens, E.P. Failure of anti-interleukin-1 therapy in severe hidradenitis suppurativa: A case report. Dermatology 2013, 226, 97–100. [Google Scholar] [CrossRef]
- Kimball, A.B.; Jemec, G.B.E.; Alavi, A.; Reguiai, Z.; Gottlieb, A.B.; Bechara, F.G.; Paul, C.; Giamarellos Bourboulis, E.J.; Villani, A.P.; Schwinn, A.; et al. Secukinumab in moderate-to-severe hidradenitis suppurativa (SUNSHINE and SUNRISE): Week 16 and week 52 results of two identical, multicentre, randomised, placebo-controlled, double-blind phase 3 trials. Lancet 2023, 401, 747–761. [Google Scholar] [CrossRef]
- Chima, M.A.; Lebwohl, M.G. 28—Interleukin 17 Inhibitors. In Comprehensive Dermatologic Drug Therapy, 4th ed.; Wolverton, S.E., Ed.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 312–320.e2. [Google Scholar]
- Frew, J.W.; Navrazhina, K.; Grand, D.; Sullivan-Whalen, M.; Gilleaudeau, P.; Garcet, S.; Ungar, J.; Krueger, J.G. The effect of subcutaneous brodalumab on clinical disease activity in hidradenitis suppurativa: An open-label cohort study. J. Am. Acad. Dermatol. 2020, 83, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Merola, J.F.; Landewé, R.; McInnes, I.B.; Mease, P.J.; Ritchlin, C.T.; Tanaka, Y.; Asahina, A.; Behrens, F.; Gladman, D.D.; Gossec, L.; et al. Bimekizumab in patients with active psoriatic arthritis and previous inadequate response or intolerance to tumour necrosis factor-α inhibitors: A randomised, double-blind, placebo-controlled, phase 3 trial (BE COMPLETE). Lancet 2023, 401, 38–48. [Google Scholar] [CrossRef]
- Świerczewska, Z.; Lewandowski, M.; Surowiecka, A.; Barańska-Rybak, W. Immunomodulatory Drugs in the Treatment of Hidradenitis Suppurativa-Possibilities and Limitations. Int. J. Mol. Sci. 2022, 23, 9716. [Google Scholar] [CrossRef]
- Esme, P.; Botsali, A.; Akoglu, G.; Caliskan, E. An Anti-Interleukin-17A Monoclonal Antibody, Ixekizumab, in the Treatment of Resistant Hidradenitis Suppurativa: A Case Series. Skin. Appendage Disord. 2022, 8, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Ocker, L.; Abu Rached, N.; Seifert, C.; Scheel, C.; Bechara, F.G. Current Medical and Surgical Treatment of Hidradenitis Suppurativa—A Comprehensive Review. J. Clin. Med. 2022, 11, 7240. [Google Scholar] [CrossRef]
- Leslie, K.S.; Tripathi, S.V.; Nguyen, T.V.; Pauli, M.; Rosenblum, M.D. An open-label study of anakinra for the treatment of moderate to severe hidradenitis suppurativa. J. Am. Acad. Dermatol. 2014, 70, 243–251. [Google Scholar] [CrossRef]
- Tzanetakou, V.; Kanni, T.; Giatrakou, S.; Katoulis, A.; Papadavid, E.; Netea, M.G.; Dinarello, C.A.; van der Meer, J.W.M.; Rigopoulos, D.; Giamarellos-Bourboulis, E.J. Safety and Efficacy of Anakinra in Severe Hidradenitis Suppurativa: A Randomized Clinical Trial. JAMA Dermatol. 2016, 152, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Dhimolea, E. Canakinumab. MAbs 2010, 2, 3–13. [Google Scholar] [CrossRef]
- Gottlieb, A.; Natsis, N.E.; Kerdel, F.; Forman, S.; Gonzalez, E.; Jimenez, G.; Hernandez, L.; Kaffenberger, J.; Guido, G.; Lucas, K.; et al. A Phase II Open-Label Study of Bermekimab in Patients with Hidradenitis Suppurativa Shows Resolution of Inflammatory Lesions and Pain. J. Investig. Dermatol. 2020, 140, 1538–1545.E2. [Google Scholar] [CrossRef]
- Benson, J.M.; Peritt, D.; Scallon, B.J.; Heavner, G.A.; Shealy, D.J.; Giles-Komar, J.M.; Mascelli, M.A. Discovery and mechanism of ustekinumab: A human monoclonal antibody targeting interleukin-12 and interleukin-23 for treatment of immune-mediated disorders. MAbs 2011, 3, 535–545. [Google Scholar] [CrossRef]
- Montero-Vilchez, T.; Pozo-Román, T.; Sánchez-Velicia, L.; Vega-Gutiérrez, J.; Arias-Santiago, S.; Molina-Leyva, A. Ustekinumab in the treatment of patients with hidradenitis suppurativa: Multicenter case series and systematic review. J. Dermatol. Treat. 2022, 33, 348–353. [Google Scholar] [CrossRef]
- Gargiulo, L.; Ibba, L.; Malagoli, P.; Angileri, R.G.; Bardazzi, F.; Bernardini, N.; Burlando, M.; Carrera, C.G.; Chiricozzi, A.; Dapavo, P.; et al. Real-life effectiveness and safety of guselkumab in patients with psoriasis who have an inadequate response to ustekinumab: A 104-week multicenter retrospective study—IL PSO (ITALIAN LANDSCAPE PSORIASIS). J. Eur. Acad. Dermatol. Venereol. 2023, 37, 1017–1027. [Google Scholar] [CrossRef]
- Dudink, K.; Bouwman, K.; Chen, Y.; DePrimo, S.E.; Munoz-Elias, E.J.; Aarts, P.; Schappin, R.; Florencia, E.F.; van Heeswijk, B.; Prens, L.M.; et al. Guselkumab for hidradenitis suppurativa: A phase II, open-label, mode-of-action study. Br. J. Dermatol. 2023, 188, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Baeten, D.; Østergaard, M.; Wei, J.C.-C.; Sieper, J.; Järvinen, P.; Tam, L.-S.; Salvarani, C.; Kim, T.-H.; Solinger, A.; Datsenko, Y.; et al. Risankizumab, an IL-23 inhibitor, for ankylosing spondylitis: Results of a randomised, double-blind, placebo-controlled, proof-of-concept, dose-finding phase 2 study. Ann. Rheum. Dis. 2018, 77, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Kimball, A.B.; Prens, E.P.; Passeron, T.; Maverakis, E.; Turchin, I.; Beeck, S.; Drogaris, L.; Geng, Z.; Zhan, T.; Messina, I.; et al. Efficacy and Safety of Risankizumab for the Treatment of Hidradenitis Suppurativa: A Phase 2, Randomized, Placebo-Controlled Trial. Dermatol. Ther. 2023, 13, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Alavi, A.; Hamzavi, I.; Brown, K.; Santos, L.L.; Zhu, Z.; Liu, H.; Howell, M.D.; Kirby, J.S. Janus kinase 1 inhibitor INCB054707 for patients with moderate-to-severe hidradenitis suppurativa: Results from two phase II studies. Br. J. Dermatol. 2022, 186, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.T.; Santillan, M.R.; Flood, K.S.; Charrow, A.; Porter, M.L.; Kimball, A.B. Tofacitinib shows benefit in conjunction with other therapies in recalcitrant hidradenitis suppurativa patients. JAAD Case Rep. 2020, 6, 99–102. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Cytokine | Receptor | Activated Pathway | Induced Reaction |
|---|---|---|---|
| TNF-α | TNFR1 TNFR2 | NFκB, MAPKS, Caspase8, MLKL NFκB, MAPKS, AKT | Cytotoxic and proinflammation Cell activation, migration, proliferation |
| IL-17 | IL-17R A, C, D | NFκB MEK5 | Inflammation Keratinocyte proliferation |
| IL-1β | IL-1R1 Co-receptor: IL-1RAcP | NFκB, JNK, p38 MAPK | Naïve T-cell and CD4+ memory T-cell expansion Keratinocyte proliferation |
| IL-12 | IL-12R (IL-12Rβ1 + IL-12Rβ2) | TYK2, STAT4 | Th1 proliferation and TFN-γ production |
| IL-23 | IL-12Rβ1 + IL-23Rα | JAK, RTK, STAT, ROR-γt | Th17 release IL-17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, Y.-L.; Yu, S. Hidradenitis Suppurativa: An Understanding of Genetic Factors and Treatment. Biomedicines 2024, 12, 338. https://doi.org/10.3390/biomedicines12020338
Chu Y-L, Yu S. Hidradenitis Suppurativa: An Understanding of Genetic Factors and Treatment. Biomedicines. 2024; 12(2):338. https://doi.org/10.3390/biomedicines12020338
Chicago/Turabian StyleChu, Yi-Lun, and Sebastian Yu. 2024. "Hidradenitis Suppurativa: An Understanding of Genetic Factors and Treatment" Biomedicines 12, no. 2: 338. https://doi.org/10.3390/biomedicines12020338
APA StyleChu, Y.-L., & Yu, S. (2024). Hidradenitis Suppurativa: An Understanding of Genetic Factors and Treatment. Biomedicines, 12(2), 338. https://doi.org/10.3390/biomedicines12020338