
Application of SWATH Mass Spectrometry and Machine Learning in the Diagnosis of Inflammatory Bowel Disease Based on the Stool Proteome

 , , and
, , and
Abstract
1. Introduction
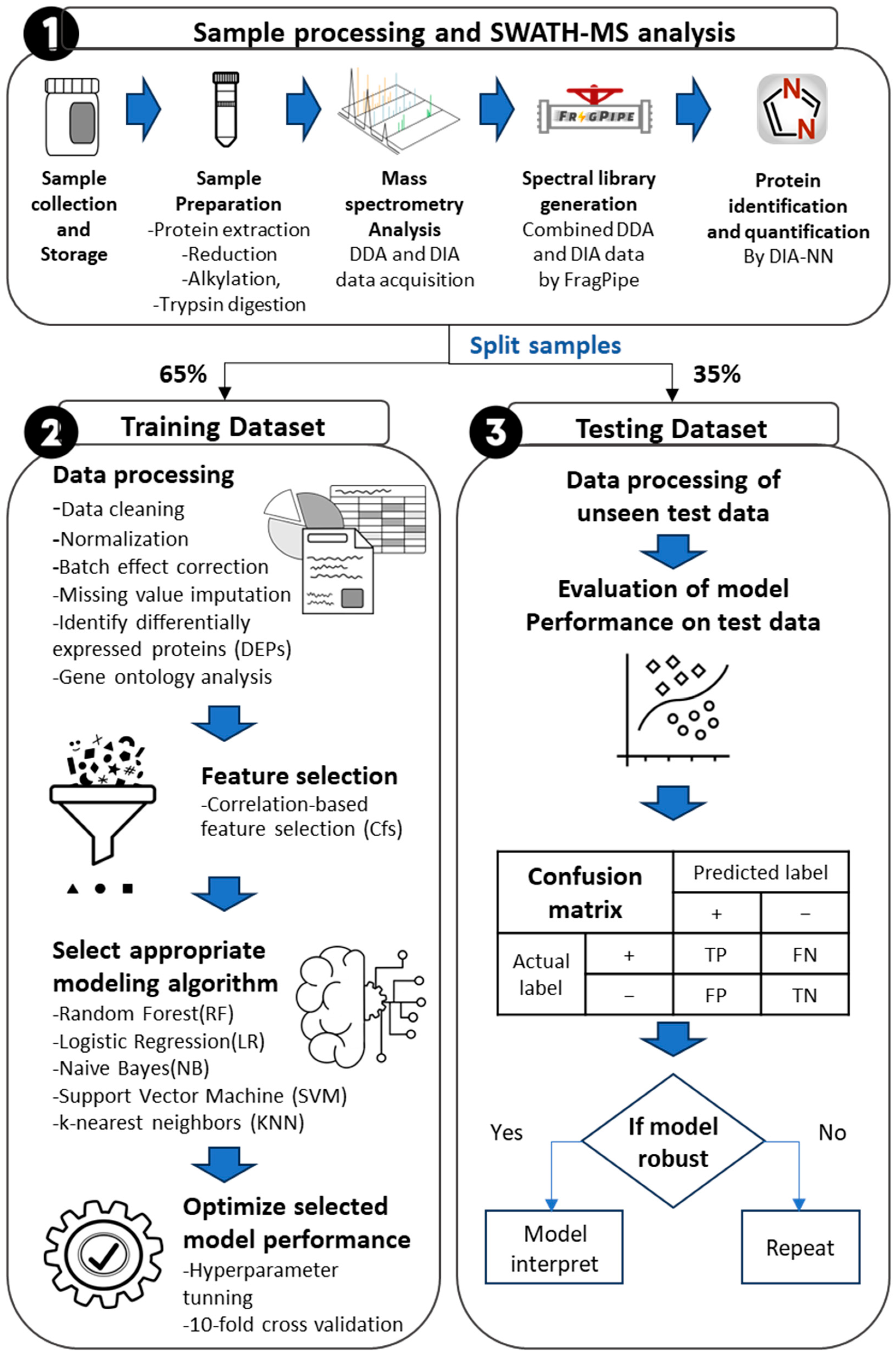
2. Materials and Methods
2.1. Sample Collection and Research Ethics
2.2. Sample Preparation
2.3. SWATH-MS Data Acquisition
2.4. Spectral Library Generation
2.5. Label Free Quantification Analysis
2.6. Statistical and Modeling Analysis
3. Results
3.1. Patient Demographics
3.2. MS Analysis and Generating the Spectral Library
3.3. Data Preprocessing
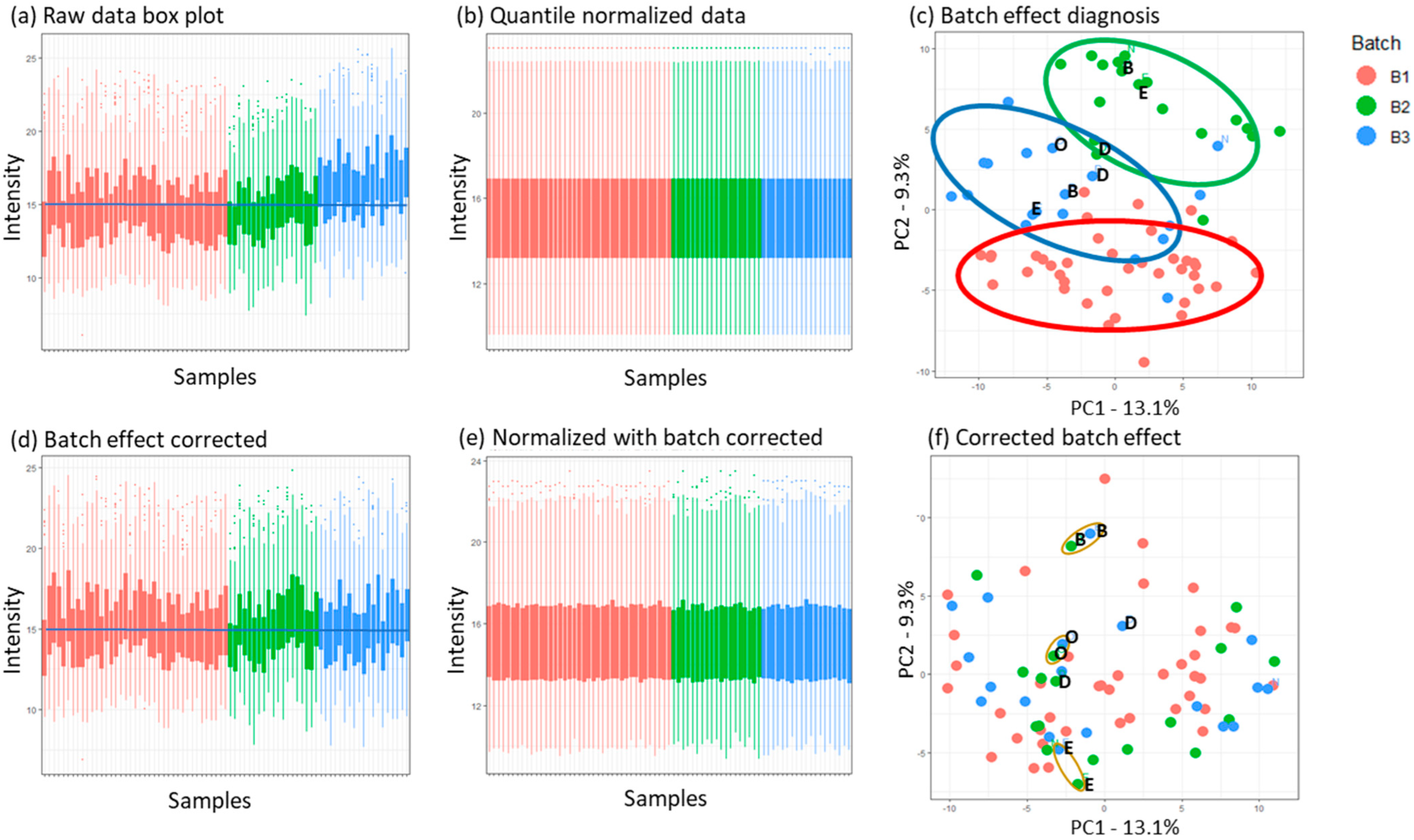
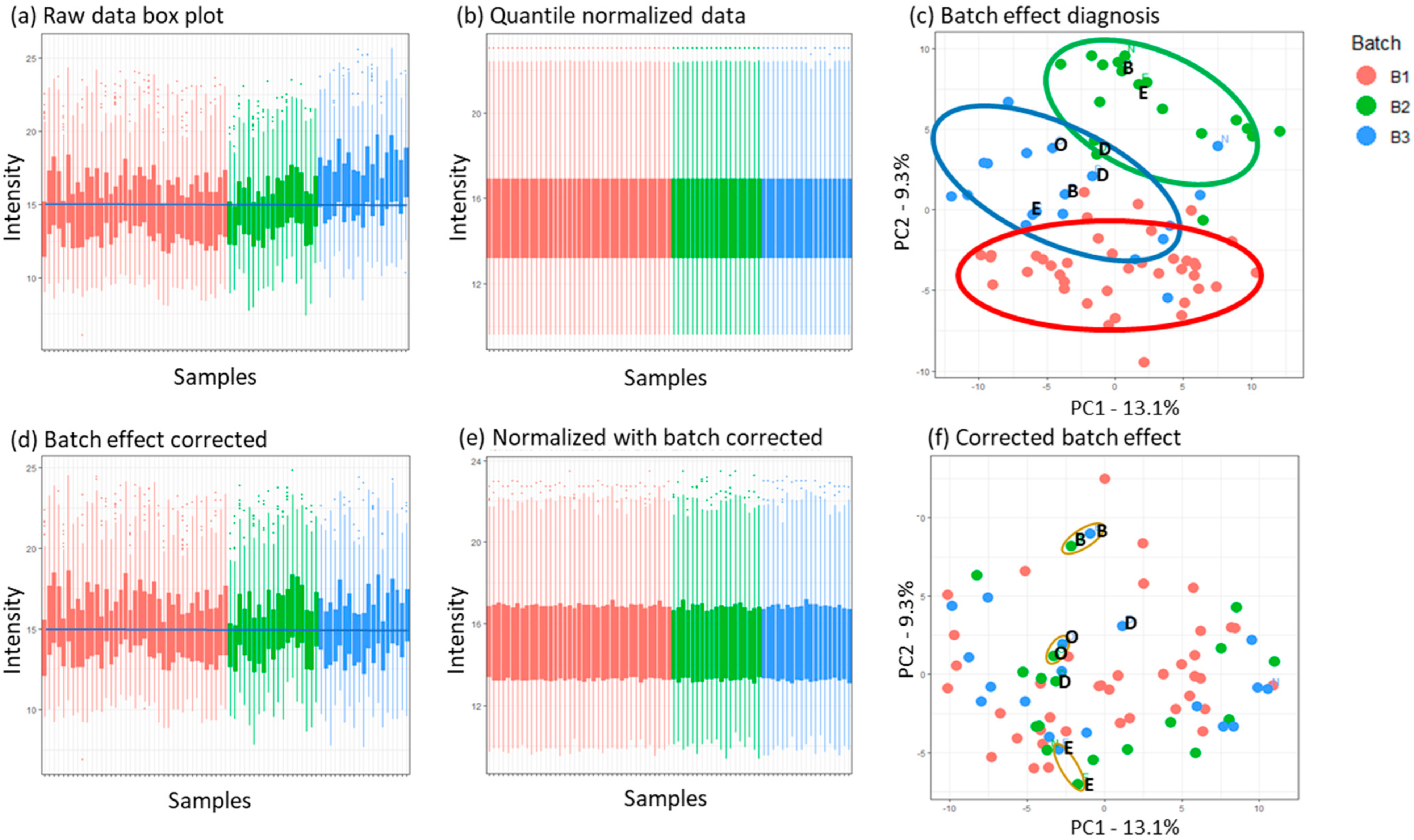
3.3.1. Normalization and Batch Effect Correction
3.3.2. Missing Value Imputation
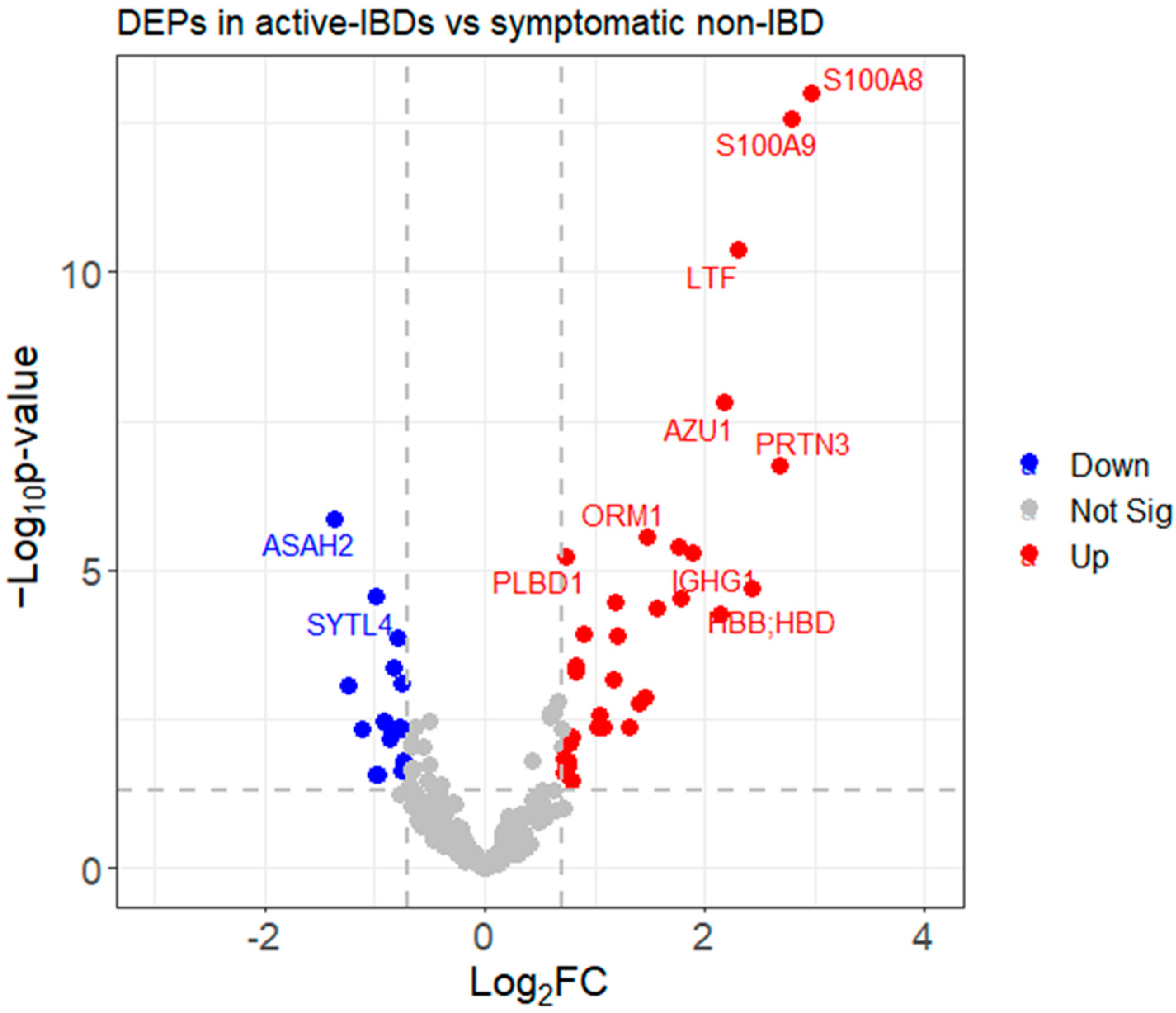
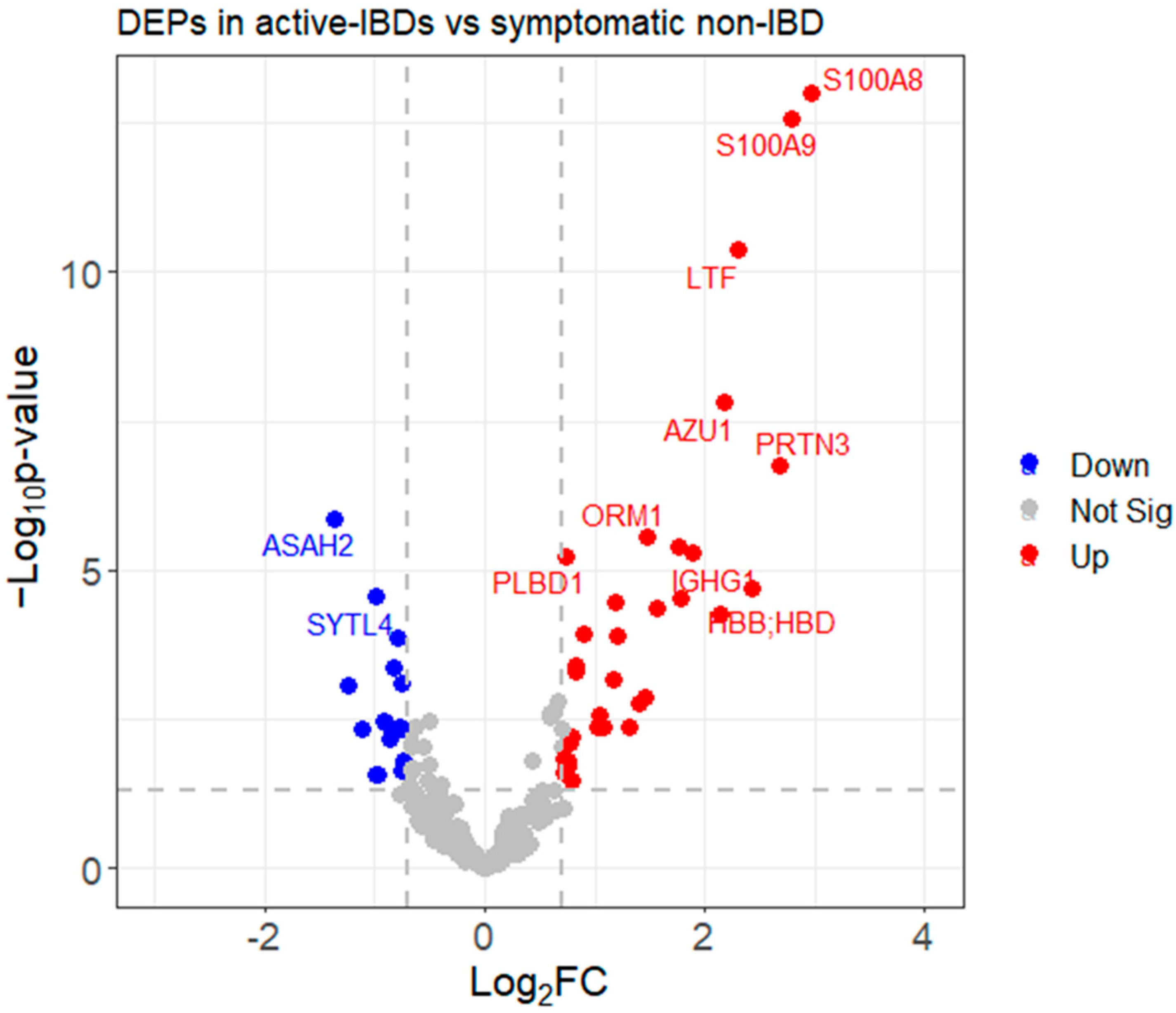
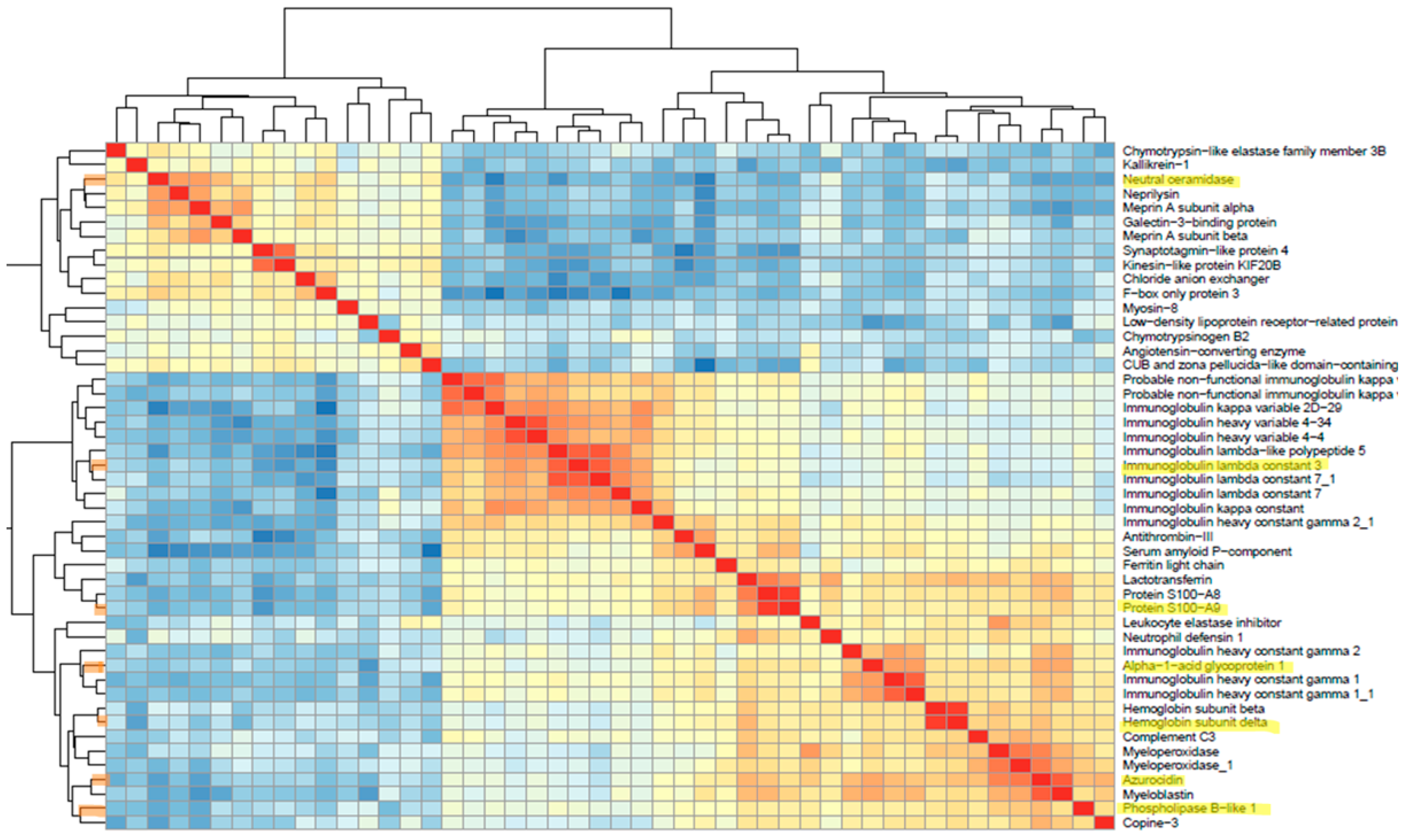
3.3.3. Identifying Differentially Expressed Proteins (DEPs)
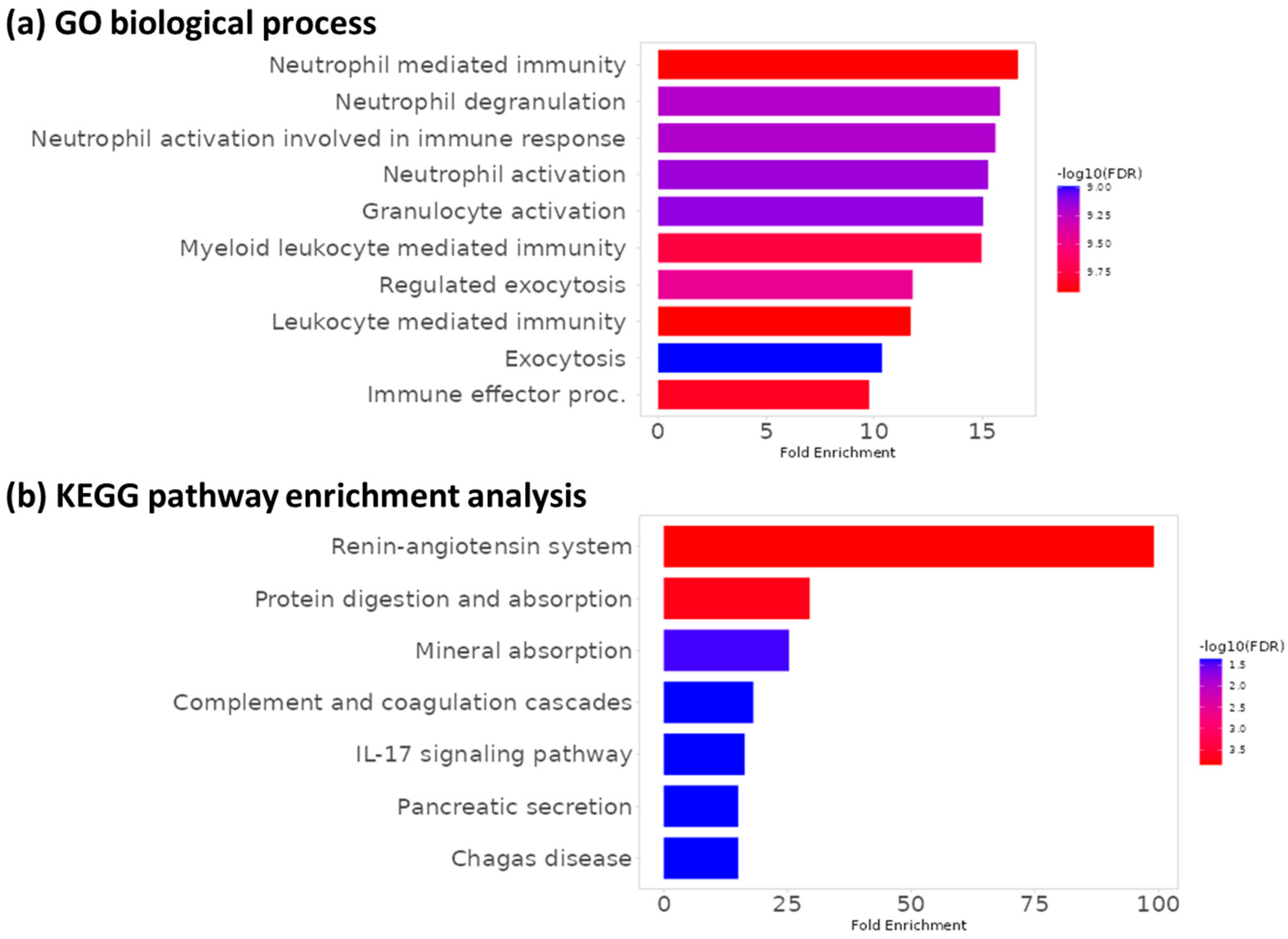
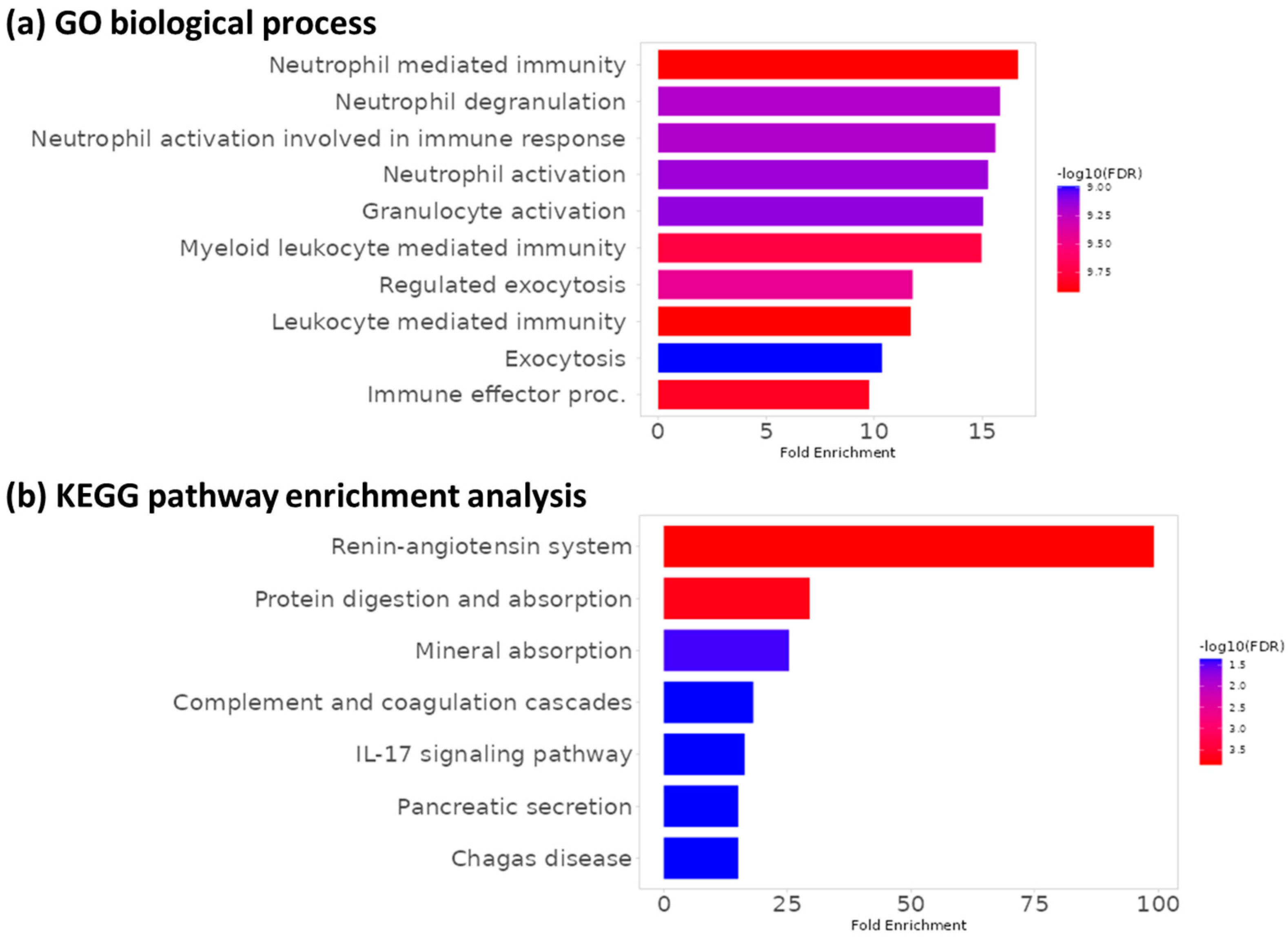
3.4. Functional Enrichment Analysis
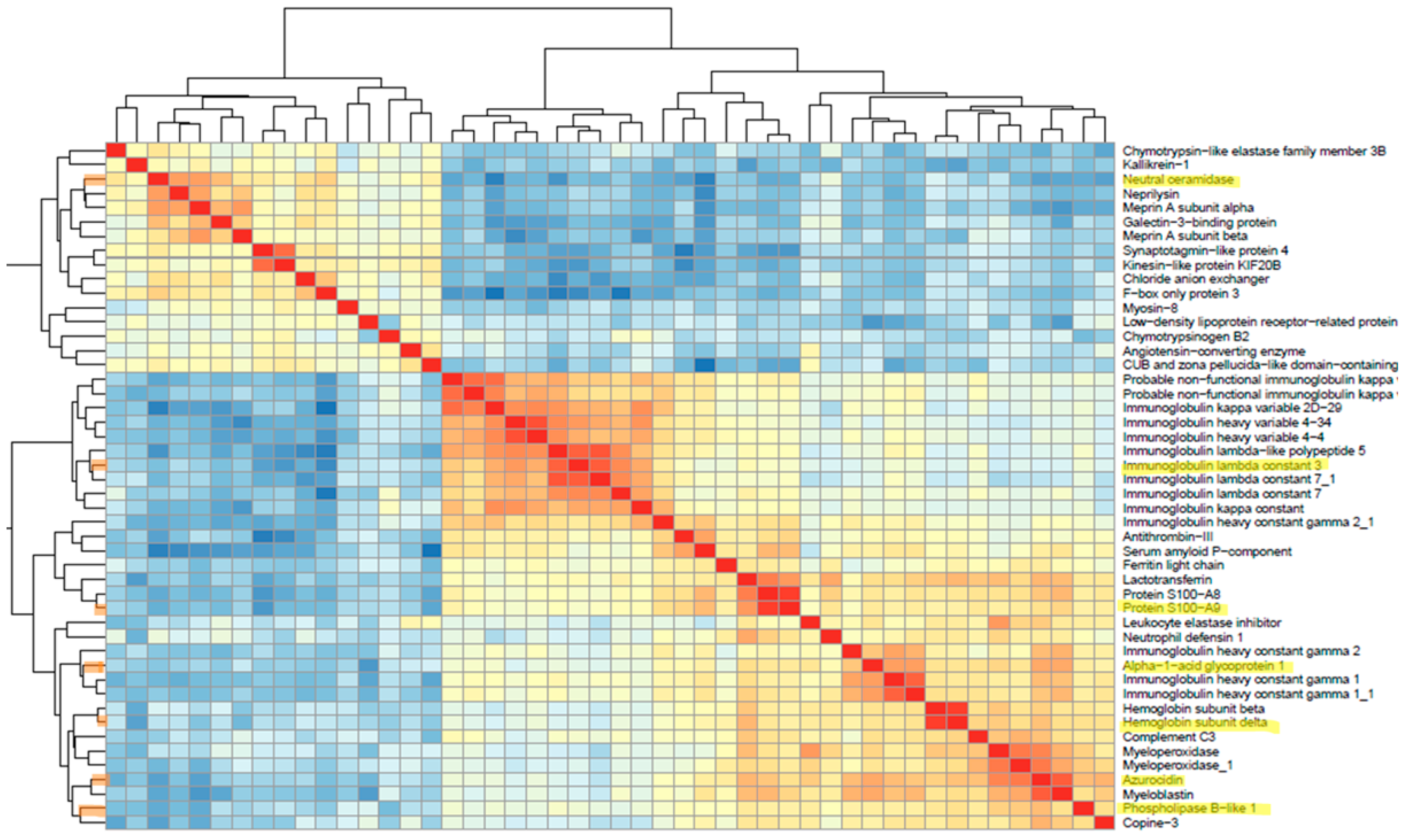
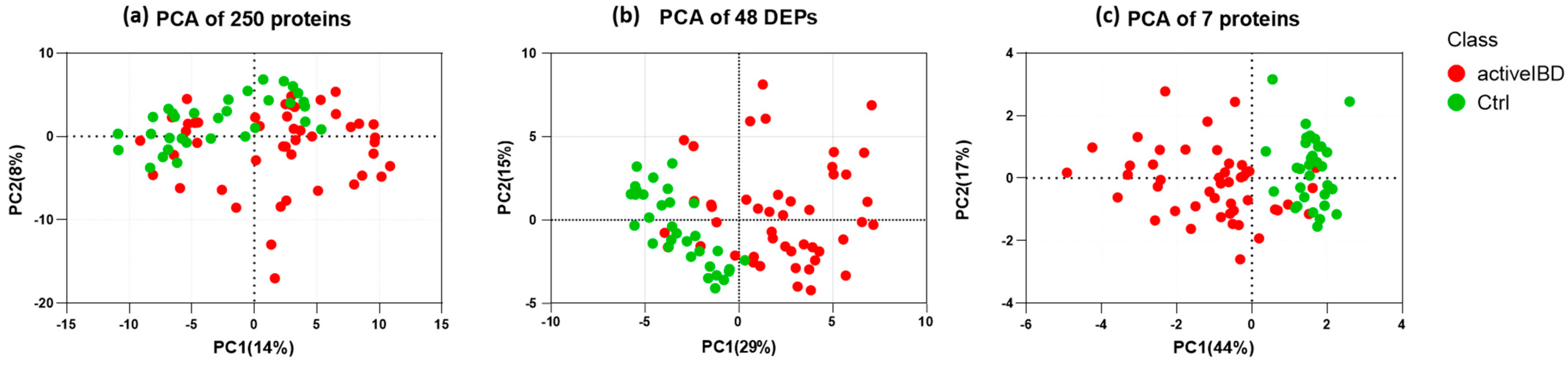
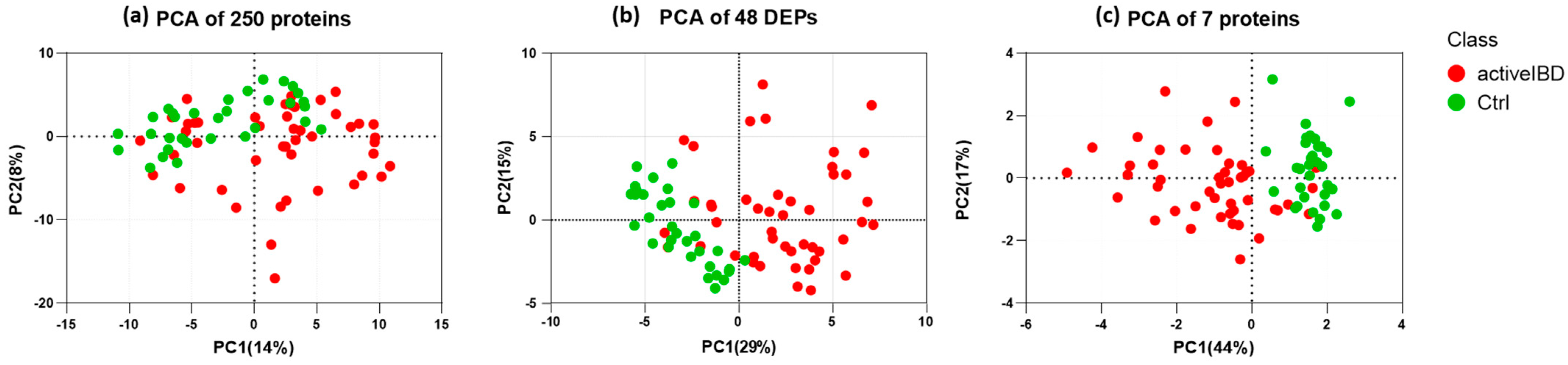
3.5. Feature Selection
3.6. Selecting the Appropriate Machine Learning Algorithm
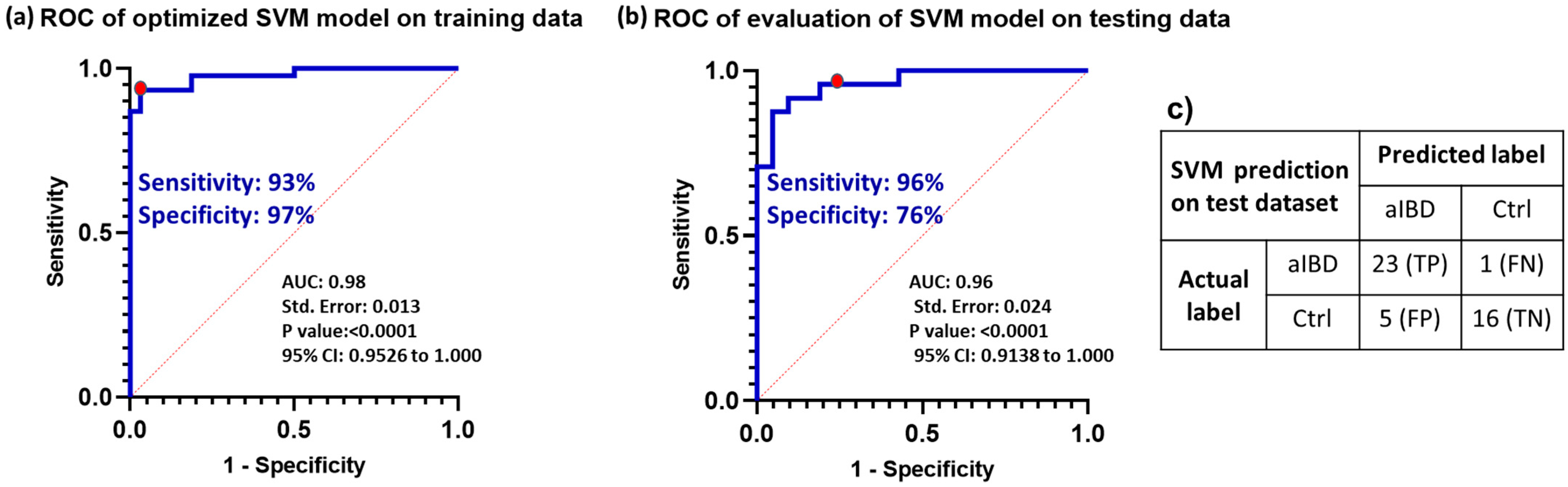
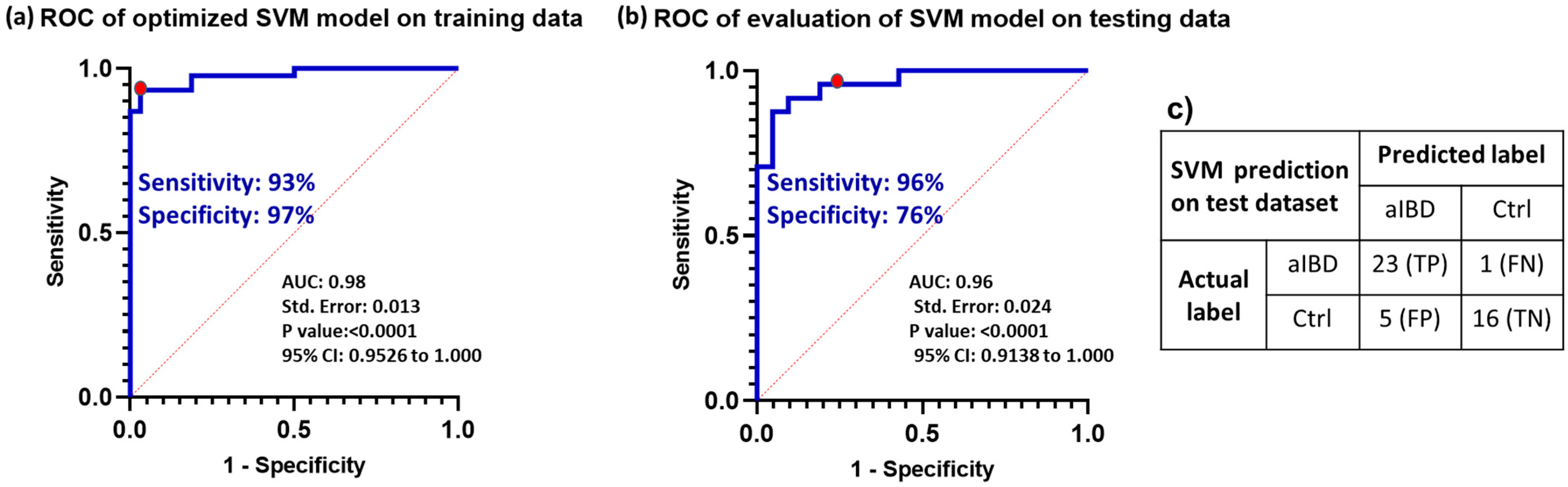
3.7. Optimizing the Selected Model Performance
3.8. Model Validation with Prospective Data
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baumgart, D.C.; Carding, S.R. Inflammatory bowel disease: Cause and immunobiology. Lancet 2007, 369, 1627–1640. [Google Scholar] [CrossRef]
- Pithadia, A.B.; Jain, S. Treatment of inflammatory bowel disease (IBD). Pharmacol. Rep. 2011, 63, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Langshaw, A.; Rosen, J.; Pensabene, L.; Borrelli, O.; Salvatore, S.; Thapar, N.; Concolino, D.; Saps, M. Overlap between functional abdominal pain disorders and organic diseases in children. Rev. Gastroenterol. México 2018, 83, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.A.; Maple, J.T.; Ben-Menachem, T.; Cash, B.D.; Decker, G.A.; Early, D.S.; Evans, J.A.; Fanelli, R.D.; Fukami, N.; Hwang, J.H. Complications of colonoscopy. Gastrointest. Endosc. 2011, 74, 745–752. [Google Scholar] [CrossRef]
- Noiseux, I.; Veilleux, S.; Bitton, A.; Kohen, R.; Vachon, L.; White Guay, B.; Rioux, J.D. Inflammatory bowel disease patient perceptions of diagnostic and monitoring tests and procedures. BMC Gastroenterol. 2019, 19, 30. [Google Scholar] [CrossRef] [PubMed]
- Lopez, R.N.; Leach, S.T.; Lemberg, D.A.; Duvoisin, G.; Gearry, R.B.; Day, A.S. Fecal biomarkers in inflammatory bowel disease. J. Gastroenterol. Hepatol. 2017, 32, 577–582. [Google Scholar] [CrossRef]
- Laserna-Mendieta, E.J.; Lucendo, A.J. Faecal calprotectin in inflammatory bowel diseases: A review focused on meta-analyses and routine usage limitations. Clin. Chem. Lab. Med. (CCLM) 2019, 57, 1295–1307. [Google Scholar] [CrossRef] [PubMed]
- Rokkas, T.; Portincasa, P.; Koutroubakis, I.E. Fecal calprotectin in assessing inflammatory bowel disease endoscopic activity: A diagnostic accuracy meta-analysis. J. Gastrointest. Liver Dis. 2018, 27, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.V.; Piersma, S.R.; Oudgenoeg, G.; Jimenez, C.R. Label-free mass spectrometry-based proteomics for biomarker discovery and validation. Expert Rev. Mol. Diagn. 2012, 12, 343–359. [Google Scholar] [CrossRef]
- Sajic, T.; Liu, Y.; Aebersold, R. Using data-independent, high-resolution mass spectrometry in protein biomarker research: Perspectives and clinical applications. PROTEOMICS–Clin. Appl. 2015, 9, 307–321. [Google Scholar] [CrossRef]
- Ludwig, C.; Gillet, L.; Rosenberger, G.; Amon, S.; Collins, B.C.; Aebersold, R. Data-independent acquisition-based SWATH-MS for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2018, 14, e8126. [Google Scholar] [CrossRef] [PubMed]
- Anjo, S.I.; Santa, C.; Manadas, B. SWATH-MS as a tool for biomarker discovery: From basic research to clinical applications. Proteomics 2017, 17, 1600278. [Google Scholar] [CrossRef]
- Sidoli, S.; Lin, S.; Xiong, L.; Bhanu, N.V.; Karch, K.R.; Johansen, E.; Hunter, C.; Mollah, S.; Garcia, B.A. Sequential Window Acquisition of all Theoretical Mass Spectra (SWATH) Analysis for Characterization and Quantification of Histone Post-translational Modifications*[S]. Mol. Cell. Proteom. 2015, 14, 2420–2428. [Google Scholar] [CrossRef]
- Fabian, O.; Bajer, L.; Drastich, P.; Harant, K.; Sticova, E.; Daskova, N.; Modos, I.; Tichanek, F.; Cahova, M. A Current State of Proteomics in Adult and Pediatric Inflammatory Bowel Diseases: A Systematic Search and Review. Int. J. Mol. Sci. 2023, 24, 9386. [Google Scholar] [CrossRef]
- Basso, D.; Padoan, A.; D’Incà, R.; Arrigoni, G.; Scapellato, M.L.; Contran, N.; Franchin, C.; Lorenzon, G.; Mescoli, C.; Moz, S. Peptidomic and proteomic analysis of stool for diagnosing IBD and deciphering disease pathogenesis. Clin. Chem. Lab. Med. (CCLM) 2020, 58, 968–979. [Google Scholar] [CrossRef]
- Vitali, R.; Palone, F.; Armuzzi, A.; Fulci, V.; Negroni, A.; Carissimi, C.; Cucchiara, S.; Stronati, L. Proteomic analysis identifies three reliable biomarkers of intestinal inflammation in the stools of patients with Inflammatory Bowel Disease. J. Crohn’s Colitis 2023, 17, 92–102. [Google Scholar] [CrossRef]
- Gagné, D.; Shajari, E.; Thibault, M.-P.; Noël, J.-F.; Boisvert, F.-M.; Babakissa, C.; Levy, E.; Gagnon, H.; Brunet, M.A.; Grynspan, D. Proteomics Profiling of Stool Samples from Preterm Neonates with SWATH/DIA Mass Spectrometry for Predicting Necrotizing Enterocolitis. Int. J. Mol. Sci. 2022, 23, 11601. [Google Scholar] [CrossRef]
- Adusumilli, R.; Mallick, P. Data conversion with ProteoWizard msConvert. Proteom. Methods Protoc. 2017, 1550, 339–368. [Google Scholar]
- Kong, A.T.; Leprevost, F.V.; Avtonomov, D.M.; Mellacheruvu, D.; Nesvizhskii, A.I. MSFragger: Ultrafast and comprehensive peptide identification in mass spectrometry–based proteomics. Nat. Methods 2017, 14, 513–520. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Hastie, T.; Tibshirani, R.; Narasimhan, B.; Chu, G. Impute: Imputation for Microarray Data, R Package Version 1.76.0 2023. Available online: https://bioconductor.org/packages/impute (accessed on 1 April 2023).
- Wieczorek, S.; Combes, F.; Lazar, C.; Giai Gianetto, Q.; Gatto, L.; Dorffer, A.; Hesse, A.-M.; Coute, Y.; Ferro, M.; Bruley, C. DAPAR & ProStaR: Software to perform statistical analyses in quantitative discovery proteomics. Bioinformatics 2017, 33, 135–136. [Google Scholar]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA data mining software: An update. ACM SIGKDD Explor. Newsl. 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Kuhn, M. A Short Introduction to the caret Package. R Found Stat. Comput. 2015, 1, 1–10. [Google Scholar]
- Deane-Mayer, Z.A.; Knowles, J.E.; Deane-Mayer, M.Z.A. Package ‘caretEnsemble’. 2016. Available online: https://mirrors.nic.cz/R/web/packages/caretEnsemble/caretEnsemble.pdf (accessed on 1 May 2023).
- Kursa, M.B.; Rudnicki, W.R. Feature selection with the Boruta package. J. Stat. Softw. 2010, 36, 1–13. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef]
- Bai, M.; Deng, J.; Dai, C.; Pfeuffer, J.; Sachsenberg, T.; Perez-Riverol, Y. LFQ-Based Peptide and Protein Intensity Differential Expression Analysis. J. Proteome Res. 2023, 22, 2114–2123. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Hou, J.; Tanner, J.J.; Cheng, J. Bioinformatics methods for mass spectrometry-based proteomics data analysis. Int. J. Mol. Sci. 2020, 21, 2873. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.-H.; Wu, P.-S.; Wong, T.-H.; Lin, I.-Y.; Lin, J.; Cox, J.; Yu, S.-H. Benchmarking differential expression, imputation and quantification methods for proteomics data. Brief. Bioinform. 2022, 23, bbac138. [Google Scholar] [CrossRef] [PubMed]
- Spratt, H.M.; Ju, H. Statistical Approaches to Candidate Biomarker Panel Selection. Adv. Exp. Med. Biol. 2016, 919, 463–492. [Google Scholar] [CrossRef] [PubMed]
- Dubois, E.; Galindo, A.N.; Dayon, L.; Cominetti, O. Comparison of normalization methods in clinical research applications of mass spectrometry-based proteomics. In Proceedings of the 2020 IEEE Conference on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), Vina del Mar, Chile, 27–29 October 2020; IEEE Publisher: Vina del Mar, Chile, 2020; pp. 1–10. [Google Scholar] [CrossRef]
- Callister, S.J.; Barry, R.C.; Adkins, J.N.; Johnson, E.T.; Qian, W.-j.; Webb-Robertson, B.-J.M.; Smith, R.D.; Lipton, M.S. Normalization approaches for removing systematic biases associated with mass spectrometry and label-free proteomics. J. Proteome Res. 2006, 5, 277–286. [Google Scholar] [CrossRef]
- Zhao, Y.; Wong, L.; Goh, W.W.B. How to do quantile normalization correctly for gene expression data analyses. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Čuklina, J.; Lee, C.H.; Williams, E.G.; Sajic, T.; Collins, B.C.; Rodríguez Martínez, M.; Sharma, V.S.; Wendt, F.; Goetze, S.; Keele, G.R. Diagnostics and correction of batch effects in large-scale proteomic studies: A tutorial. Mol. Syst. Biol. 2021, 17, e10240. [Google Scholar] [CrossRef]
- Kong, W.; Hui, H.W.H.; Peng, H.; Goh, W.W.B. Dealing with missing values in proteomics data. Proteomics 2022, 22, 2200092. [Google Scholar] [CrossRef]
- Wei, R.; Wang, J.; Su, M.; Jia, E.; Chen, S.; Chen, T.; Ni, Y. Missing value imputation approach for mass spectrometry-based metabolomics data. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Hasan, M.K.; Alam, M.A.; Roy, S.; Dutta, A.; Jawad, M.T.; Das, S. Missing value imputation affects the performance of machine learning: A review and analysis of the literature (2010–2021). Inform. Med. Unlocked 2021, 27, 100799. [Google Scholar] [CrossRef]
- Stekhoven, D.J.; Bühlmann, P. MissForest—Non-parametric missing value imputation for mixed-type data. Bioinformatics 2012, 28, 112–118. [Google Scholar] [CrossRef]
- Troyanskaya, O.; Cantor, M.; Sherlock, G.; Brown, P.; Hastie, T.; Tibshirani, R.; Botstein, D.; Altman, R.B. Missing value estimation methods for DNA microarrays. Bioinformatics 2001, 17, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, W.; Hu, L.; Cheng, J.; Yang, H.; Liu, Y. NAguideR: Performing and prioritizing missing value imputations for consistent bottom-up proteomic analyses. Nucleic Acids Res. 2020, 48, e83. [Google Scholar] [CrossRef]
- West, R.M. Best practice in statistics: Use the Welch t-test when testing the difference between two groups. Ann. Clin. Biochem. 2021, 58, 267–269. [Google Scholar] [CrossRef]
- Wieczorek, S.; Combes, F.; Borges, H.; Burger, T. Protein-level statistical analysis of quantitative label-free proteomics data with ProStaR. Proteom. Biomark. Discov. Methods Protoc. 2019, 1959, 225–246. [Google Scholar]
- Giai Gianetto, Q.; Combes, F.; Ramus, C.; Bruley, C.; Couté, Y.; Burger, T. Calibration plot for proteomics: A graphical tool to visually check the assumptions underlying FDR control in quantitative experiments. Proteomics 2016, 16, 29–32. [Google Scholar] [CrossRef]
- Lo, S.W.; Segal, J.P.; Lubel, J.S.; Garg, M. What do we know about the renin angiotensin system and inflammatory bowel disease? Expert Opin. Ther. Targets 2022, 26, 897–909. [Google Scholar] [CrossRef]
- Peuhkuri, K.; Vapaatalo, H.; Korpela, R. Even low-grade inflammation impacts on small intestinal function. World J. Gastroenterol. WJG 2010, 16, 1057. [Google Scholar] [CrossRef] [PubMed]
- Geremia, A.; Jewell, D.P. The IL-23/IL-17 pathway in inflammatory bowel disease. Expert Rev. Gastroenterol. Hepatol. 2012, 6, 223–237. [Google Scholar] [CrossRef]
- Liu, H.; Motoda, H. Computational Methods of Feature Selection; CRC Press: Boca Raton, FL, USA, 2007; 440p. [Google Scholar] [CrossRef]
- Hall, M.A. Correlation-Based Feature Selection for Machine Learning. Ph.D. Thesis, The University of Waikato, Hamilton, New Zaeland, 1999. Available online: https://hdl.handle.net/10289/15043 (accessed on 12 June 2023).
- Greener, J.G.; Kandathil, S.M.; Moffat, L.; Jones, D.T. A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 2022, 23, 40–55. [Google Scholar] [CrossRef]
- Ying, X. An Overview of Overfitting and Its Solutions. Proc. J. Phys. Conf. Ser. 2019, 1168, 022022. [Google Scholar] [CrossRef]
- Bischl, B.; Binder, M.; Lang, M.; Pielok, T.; Richter, J.; Coors, S.; Thomas, J.; Ullmann, T.; Becker, M.; Boulesteix, A.L. Hyperparameter optimization: Foundations, algorithms, best practices, and open challenges. Wiley Interdiscip. Rev. Data Min. Knowl. Discov. 2023, 13, e1484. [Google Scholar] [CrossRef]
- Mantovani, R.G.; Rossi, A.L.; Vanschoren, J.; Bischl, B.; De Carvalho, A.C. Effectiveness of random search in SVM hyper-parameter tuning. In Proceedings of the 2015 International Joint Conference on Neural Networks (IJCNN), Killarney, Ireland, 12–17 July 2015; IEEE Publisher: Killarney, Ireland; pp. 1–8. [Google Scholar] [CrossRef]
- Mooiweer, E.; Fidder, H.H.; Siersema, P.D.; Laheij, R.J.; Oldenburg, B. Fecal hemoglobin and calprotectin are equally effective in identifying patients with inflammatory bowel disease with active endoscopic inflammation. Inflamm. Bowel Dis. 2014, 20, 307–314. [Google Scholar] [CrossRef]
- Schröder, O.; Naumann, M.; Shastri, Y.; Povse, N.; Stein, J. Prospective evaluation of faecal neutrophil-derived proteins in identifying intestinal inflammation: Combination of parameters does not improve diagnostic accuracy of calprotectin. Aliment. Pharmacol. Ther. 2007, 26, 1035–1042. [Google Scholar] [CrossRef]
- dos Santos Ramos, A.; Viana, G.C.S.; de Macedo Brigido, M.; Almeida, J.F. Neutrophil extracellular traps in inflammatory bowel diseases: Implications in pathogenesis and therapeutic targets. Pharmacol. Res. 2021, 171, 105779. [Google Scholar] [CrossRef]
- Matsumori, A.; Shimada, T.; Shimada, M.; Drayson, M.T. Immunoglobulin free light chains: An inflammatory biomarker of diabetes. Inflamm. Res. 2020, 69, 715–718. [Google Scholar] [CrossRef]
- Napodano, C.; Pocino, K.; Rigante, D.; Stefanile, A.; Gulli, F.; Marino, M.; Basile, V.; Rapaccini, G.L.; Basile, U. Free light chains and autoimmunity. Autoimmun. Rev. 2019, 18, 484–492. [Google Scholar] [CrossRef]
- Xu, S.; Zhao, L.; Larsson, A.; Venge, P. The identification of a phospholipase B precursor in human neutrophils. FEBS J. 2009, 276, 175–186. [Google Scholar] [CrossRef]
- Fournier, T.; Medjoubi-N, N.; Porquet, D. Alpha-1-acid glycoprotein. Biochim. Et Biophys. Acta (BBA)-Protein Struct. Mol. Enzymol. 2000, 1482, 157–171. [Google Scholar] [CrossRef]
- Watanabe, T.; Aoyagi, K.; Nimura, S.; Eguchi, K.; Tomioka, Y.; Sakisaka, S. New fecal biomarker, α1-acid glycoprotein, for evaluation of inflammatory bowel disease: Comparison with calprotectin and lactoferrin. Fukuoka Univ. Med. J. 2013, 40, 155–162. [Google Scholar]
- Bock, J.; Liebisch, G.; Schweimer, J.; Schmitz, G.; Rogler, G. Exogenous sphingomyelinase causes impaired intestinal epithelial barrier function. World J. Gastroenterol. WJG 2007, 13, 5217. [Google Scholar] [CrossRef]
- Parveen, F.; Bender, D.; Law, S.-H.; Mishra, V.K.; Chen, C.-C.; Ke, L.-Y. Role of ceramidases in sphingolipid metabolism and human diseases. Cells 2019, 8, 1573. [Google Scholar] [CrossRef]
- Snider, A.J.; Wu, B.X.; Jenkins, R.W.; Sticca, J.A.; Kawamori, T.; Hannun, Y.A.; Obeid, L.M. Loss of neutral ceramidase increases inflammation in a mouse model of inflammatory bowel disease. Prostaglandins Other Lipid Mediat. 2012, 99, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Karamizadeh, S.; Abdullah, S.M.; Halimi, M.; Shayan, J.; javad Rajabi, M. Advantage and drawback of support vector machine functionality. In Proceedings of the 2014 International Conference on Computer, Communications, and Control Technology (I4CT), Langkawi, Malaysia, 2–4 September 2014; IEEE Publisher: Langkawi, Malaysia; pp. 63–65. [Google Scholar] [CrossRef]
- Burbidge, R.; Buxton, B. An introduction to support vector machines for data mining. Keynote Pap. Young OR12 2001, 3–15. Available online: https://api.semanticscholar.org/CorpusID:8133449 (accessed on 15 June 2023).
- Singh, A.; Thakur, N.; Sharma, A. A review of supervised machine learning algorithms. In Proceedings of the 2016 3rd International Conference on Computing for Sustainable Global Development (INDIACom), New Delhi, India, 16–18 March 2016; IEEE Publisher: New Delhi, India; pp. 1310–1315. [Google Scholar]
- Hsu, C.-W.; Chang, C.-C.; Lin, C.-J. A Practical Guide to Support Vector Classification; National Taiwan University: Taipei, Taiwan, 2003; pp. 1396–1400. Available online: https://www.csie.ntu.edu.tw/~cjlin/papers/guide/guide.pdf (accessed on 2 July 2023).
- Huang, S.; Cai, N.; Pacheco, P.P.; Narrandes, S.; Wang, Y.; Xu, W. Applications of support vector machine (SVM) learning in cancer genomics. Cancer Genom. Proteom. 2018, 15, 41–51. [Google Scholar]
- Vapnik, V.N. Pattern recognition using generalized portrait method. Autom. Remote Control 1963, 24, 774–780. [Google Scholar]
- Goel, A.; Srivastava, S.K. Role of kernel parameters in performance evaluation of SVM. In Proceedings of the 2016 Second International Conference on Computational Intelligence & Communication Technology (CICT), Ghaziabad, India, 12–13 February 2016; IEEE Publisher: Ghaziabad, India; pp. 166–169. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Gene | Fold Change | p-Value | Attribute Weight |
|---|---|---|---|---|
| Protein S100-A9 | S100A9 | 6.9 | 0.0000 | −3.9813 |
| Azurocidin | AZU1 | 4.5 | 0.0000 | −2.7925 |
| Immunoglobulin lambda constant 3 | IGLC3 | 2.0 | 0.0044 | −2.4284 |
| Hemoglobin subunit delta | HBB | 5.4 | 0.0000 | −2.2529 |
| Phospholipase B-like 1 | PLBD1 | 1.7 | 0.0000 | −1.6708 |
| Alpha-1-acid glycoprotein 1 | ORM1 | 2.8 | 0.0000 | −0.9056 |
| Neutral ceramidase | ASAH2 | −2.6 | 0.0000 | 1.1675 |
| Classifier | Accuracy | Precision | Recall | F-Score | AU-ROC | AU-PRC |
|---|---|---|---|---|---|---|
| SVM | 95% | 0.97 | 0.93 | 0.96 | 0.95 | 0.96 |
| NB | 90% | 0.94 | 0.90 | 0.92 | 0.93 | 0.94 |
| LR | 88% | 0.89 | 0.91 | 0.90 | 0.92 | 0.92 |
| KNN | 88% | 0.91 | 0.89 | 0.90 | 0.90 | 0.89 |
| RF | 87% | 0.89 | 0.89 | 0.89 | 0.94 | 0.93 |
| Measure | Evaluation Focus |
|---|---|
| Accuracy |
|
| Precision |
|
| Recall (Sensitivity) |
|
| F-score |
|
| ROC Area |
|
| PRC Area |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shajari, E.; Gagné, D.; Malick, M.; Roy, P.; Noël, J.-F.; Gagnon, H.; Brunet, M.A.; Delisle, M.; Boisvert, F.-M.; Beaulieu, J.-F. Application of SWATH Mass Spectrometry and Machine Learning in the Diagnosis of Inflammatory Bowel Disease Based on the Stool Proteome. Biomedicines 2024, 12, 333. https://doi.org/10.3390/biomedicines12020333
Shajari E, Gagné D, Malick M, Roy P, Noël J-F, Gagnon H, Brunet MA, Delisle M, Boisvert F-M, Beaulieu J-F. Application of SWATH Mass Spectrometry and Machine Learning in the Diagnosis of Inflammatory Bowel Disease Based on the Stool Proteome. Biomedicines. 2024; 12(2):333. https://doi.org/10.3390/biomedicines12020333
Chicago/Turabian StyleShajari, Elmira, David Gagné, Mandy Malick, Patricia Roy, Jean-François Noël, Hugo Gagnon, Marie A. Brunet, Maxime Delisle, François-Michel Boisvert, and Jean-François Beaulieu. 2024. "Application of SWATH Mass Spectrometry and Machine Learning in the Diagnosis of Inflammatory Bowel Disease Based on the Stool Proteome" Biomedicines 12, no. 2: 333. https://doi.org/10.3390/biomedicines12020333
APA StyleShajari, E., Gagné, D., Malick, M., Roy, P., Noël, J.-F., Gagnon, H., Brunet, M. A., Delisle, M., Boisvert, F.-M., & Beaulieu, J.-F. (2024). Application of SWATH Mass Spectrometry and Machine Learning in the Diagnosis of Inflammatory Bowel Disease Based on the Stool Proteome. Biomedicines, 12(2), 333. https://doi.org/10.3390/biomedicines12020333