
Mannich Base Derived from Lawsone Inhibits PKM2 and Induces Neoplastic Cell Death

 , , ,
, , ,  ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Biological Assays
2.1.1. Cells and Reagents
2.1.2. Cell Viability Assay (Cytotoxicity)
2.1.3. Cell Migration Assay
2.1.4. Statistical Analysis
2.2. PKM2 Activity
2.2.1. Kinetic PKM2 Activity In Vitro
2.2.2. Quantification of ATP Production from PKM2 Reaction
2.2.3. Quantification of Intracellular ATP Production
2.3. In Silico Studies
2.3.1. Construction of the PKM2-MB-6a Complex
2.3.2. Molecular Docking
2.3.3. Molecular Dynamics Simulation
2.3.4. Interaction Energy Assessment
3. Results and Discussion
3.1. Compound MB-6a Cytotoxicity Is Related to the Interference in PKM2 Glycolytic Activity
3.1.1. Compound MB-6a Is More Active and Selective in OSCC Compared to Other Types of Cancer
3.1.2. Substance MB-6a Demonstrated Inhibitory Potential Against PKM2 Through an Enzymatic Assay
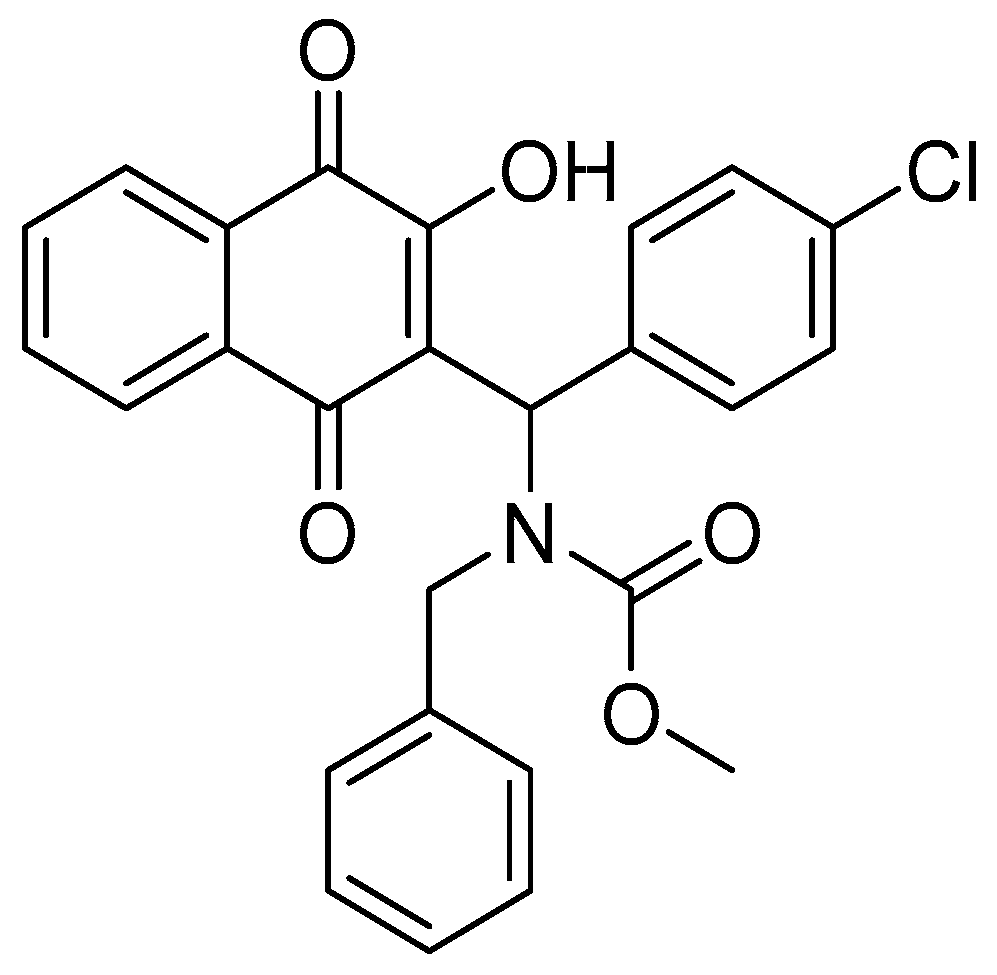
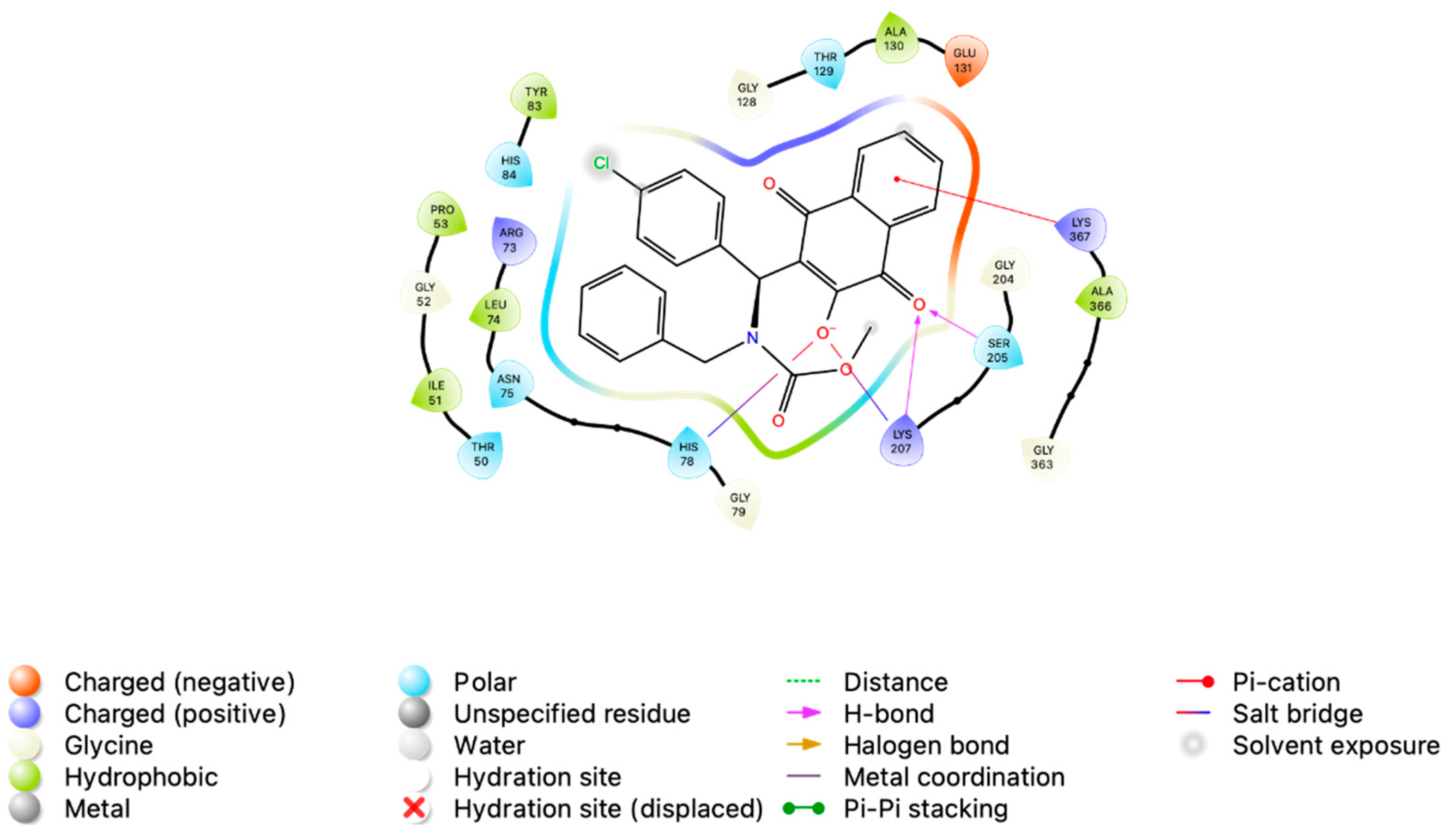
3.2. In Silico Analysis of MB-6a-PKM2 Interaction Indicates That PKM2 Is Potentially Targeted by MB-6a
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Deo, S.V.S.; Sharma, J.; Kumar, S. GLOBOCAN 2020 Report on Global Cancer Burden: Challenges and Opportunities for Surgical Oncologists. Ann. Surg. Oncol. 2022, 29, 6497–6500. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Wang, J.; Jiang, L.; James Kang, Y. Cancer and Stem Cells. Exp. Biol. Med. 2021, 246, 1791–1801. [Google Scholar] [CrossRef] [PubMed]
- Hawash, M. Advances in Cancer Therapy: A Comprehensive Review of CDK and EGFR Inhibitors. Cells 2024, 13, 1656. [Google Scholar] [CrossRef] [PubMed]
- Dayton, T.L.; Jacks, T.; Vander Heiden, M.G. PKM2, Cancer Metabolism, and the Road Ahead. EMBO Rep. 2016, 17, 1721–1730. [Google Scholar] [CrossRef]
- Wang, Z.-H.; Peng, W.-B.; Zhang, P.; Yang, X.-P.; Zhou, Q. Lactate in the Tumour Microenvironment: From Immune Modulation to Therapy. EBioMedicine 2021, 73, 103627. [Google Scholar] [CrossRef] [PubMed]
- Zahra, K.; Dey, T.; Ashish; Mishra, S.P.; Pandey, U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front. Oncol. 2020, 10, 159. [Google Scholar] [CrossRef]
- He, X.; Du, S.; Lei, T.; Li, X.; Liu, Y.; Wang, H.; Tong, R.; Wang, Y. PKM2 in Carcinogenesis and Oncotherapy. Oncotarget 2017, 8, 110656–110670. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Peng, G.; Liu, X.; Zhang, Y.; Han, H.; Liu, Z.-R. Pyruvate Kinase M2 Coordinates Metabolism Switch between Glycolysis and Glutaminolysis in Cancer Cells. iScience 2020, 23, 101684. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Zheng, Q.; Ni, W.; Wei, Z.; Yu, S.; Jia, Q.; Wang, M.; Wang, A.; Chen, W.; Lu, Y. Breaking Glucose Transporter 1/Pyruvate Kinase M2 Glycolytic Loop Is Required for Cantharidin Inhibition of Metastasis in Highly Metastatic Breast Cancer. Front. Pharmacol. 2019, 10, 590. [Google Scholar] [CrossRef]
- Liang, J.; Cao, R.; Wang, X.; Zhang, Y.; Wang, P.; Gao, H.; Li, C.; Yang, F.; Zeng, R.; Wei, P.; et al. Mitochondrial PKM2 Regulates Oxidative Stress-Induced Apoptosis by Stabilizing Bcl2. Cell Res. 2017, 27, 329–351. [Google Scholar] [CrossRef] [PubMed]
- Chhipa, A.S.; Patel, S. Targeting Pyruvate Kinase Muscle Isoform 2 (PKM2) in Cancer: What Do We Know so Far? Life Sci. 2021, 280, 119694. [Google Scholar] [CrossRef]
- Bolton, J.L.; Dunlap, T. Formation and Biological Targets of Quinones: Cytotoxic versus Cytoprotective Effects. Chem. Res. Toxicol. 2017, 30, 13–37. [Google Scholar] [CrossRef]
- de Souza, L.C.d.S.V.; Alcântara, L.M.; de Macêdo-Sales, P.A.; Reis, N.F.; de Oliveira, D.S.; Machado, R.L.D.; Geraldo, R.B.; dos Santos, A.L.S.; Ferreira, V.F.; Gonzaga, D.T.G.; et al. Synthetic Derivatives against Wild-Type and Non-Wild-Type Sporothrix Brasiliensis: In Vitro and In Silico Analyses. Pharmaceuticals 2022, 15, 55. [Google Scholar] [CrossRef]
- Pacheco, P.; Gonzaga, D.; Cirne-Santos, C.; Barros, C.; Gomes, M.; Gomes, R.; Gonçalves, M.; Ferreira, V.; Rabelo, V.; Abreu, P.; et al. Synthesis and Anti-Chikungunya Virus (CHIKV) Activity of Novel 1,4-Naphthoquinone Sulfonamide and Sulfonate Ester Derivatives. J. Braz. Chem. Soc. 2022, 33, 556–569. [Google Scholar] [CrossRef]
- Pacheco, P.A.F.; de Menezes Ribeiro, T.; dos Santos Galvão, R.M.; dos Santos, E.G.; Faria, A.F.M.; von Ranke, N.L.; Bello, M.L.; Rodrigues, C.R.; Ferreira, V.F.; Souza, A.L.A.; et al. Synthesis of New N,S-Acetal Analogs Derived from Juglone with Cytotoxic Activity against Trypanossoma Cruzi. J. Bioenerg. Biomembr. 2020, 52, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Cavalcanti Chipoline, I.; Carolina Carvalho da Fonseca, A.; Ribeiro Machado da Costa, G.; Pereira de Souza, M.; Won-Held Rabelo, V.; de Queiroz, L.N.; Luiz Ferraz de Souza, T.; Cardozo Paes de Almeida, E.; Alvarez Abreu, P.; Pontes, B.; et al. Molecular Mechanism of Action of New 1,4-Naphthoquinones Tethered to 1,2,3-1H-Triazoles with Cytotoxic and Selective Effect against Oral Squamous Cell Carcinoma. Bioorg. Chem. 2020, 101, 103984. [Google Scholar] [CrossRef]
- Borges, A.A.; de Souza, M.P.; da Fonseca, A.C.C.; Wermelinger, G.F.; Ribeiro, R.C.B.; Amaral, A.A.P.; de Carvalho, C.J.C.; Abreu, L.S.; de Queiroz, L.N.; de Almeida, E.C.P.; et al. Chemoselective Synthesis of Mannich Adducts from 1,4-Naphthoquinones and Profile as Autophagic Inducers in Oral Squamous Cell Carcinoma. Molecules 2022, 28, 309. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, A.C.C.d.; de Queiroz, L.N.; Sales Felisberto, J.; Jessé Ramos, Y.; Mesquita Marques, A.; Wermelinger, G.F.; Pontes, B.; de Lima Moreira, D.; Robbs, B.K. Cytotoxic Effect of Pure Compounds from Piper Rivinoides Kunth against Oral Squamous Cell Carcinoma. Nat. Prod. Res. 2021, 35, 6163–6167. [Google Scholar] [CrossRef]
- da Cruz Galhardo Camargo, G.A.; de Oliveira Barbosa, L.M.; Stumbo, M.B.; Thurler Júnior, J.C.; da Costa, G.R.M.; Domingos-Vieira, A.D.C.; Pascoal, V.D.B.; Robbs, B.K.; Lopes, R.T.; de Araujo, O.M.O.; et al. Effects of Infrared Light Laser Therapy on in Vivo and in Vitro Periodontitis Models. J. Periodontol. 2022, 93, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Borges, A.D.A.; Ouverney, G.; Arruda, A.T.S.; Ribeiro, A.V.; Ribeiro, R.C.B.; Souza, A.S.; da Fonseca, A.C.C.; Nicolau de Queiroz, L.N.; de Almeida, E.C.P.; Pontes, B.; et al. Determination of Inhibitory Effect of PKM2 Enzyme and Antitumoral Activity of Novel Coumarin-Naphthoquinone Hybrids. Curr. Med. Chem. 2025, 32, 359–379. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Cherinka, B.; Andrews, B.H.; Sánchez-Gallego, J.; Brownstein, J.; Argudo-Fernández, M.; Blanton, M.; Bundy, K.; Jones, A.; Masters, K.; Law, D.R.; et al. Marvin: A Tool Kit for Streamlined Access and Visualization of the SDSS-IV MaNGA Data Set. Astron. J. 2019, 158, 74. [Google Scholar] [CrossRef]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of PKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS Biomolecular Solvation Software Suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; MacKerell, A.D. Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Steinbach, P.J.; Brooks, B.R. New Spherical-cutoff Methods for Long-range Forces in Macromolecular Simulation. J. Comput. Chem. 1994, 15, 667–683. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover Chains: The Canonical Ensemble via Continuous Dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Posch, H.A.; Hoover, W.G.; Vesely, F.J. Canonical Dynamics of the Nosé Oscillator: Stability, Order, and Chaos. Phys. Rev. A 1986, 33, 4253–4265. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. Gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; Van Gunsteren, W.F.; Mark, A.E. Peptide Folding: When Simulation Meets Experiment. Angew. Chem.-Int. Ed. 1999, 38, 236–240. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson−Boltzmann Surface Area Method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate Atomic Surfaces from Linear Combinations of Pairwise Overlaps (LCPO). J. Comput. Chem. 1999, 20, 217–230. [Google Scholar] [CrossRef]
- Tan, C.; Tan, Y.-H.; Luo, R. Implicit Nonpolar Solvent Models. J. Phys. Chem. B 2007, 111, 12263–12274. [Google Scholar] [CrossRef] [PubMed]
- Ho, G.Y.; Woodward, N.; Coward, J.I.G. Cisplatin versus Carboplatin: Comparative Review of Therapeutic Management in Solid Malignancies. Crit. Rev. Oncol. Hematol. 2016, 102, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Pokhriyal, R.; Hariprasad, R.; Kumar, L.; Hariprasad, G. Chemotherapy Resistance in Advanced Ovarian Cancer Patients. Biomark Cancer 2019, 11, 1179299X19860815. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Luo, Y.; Piao, X.; Shen, G.; Meng, L.; Zhang, Y.; Wang, J.; Li, J.; Wang, H.; Xu, W.; et al. Novel 1,4-naphthoquinone Derivatives Induce Reactive Oxygen Species-mediated Apoptosis in Liver Cancer Cells. Mol. Med. Rep. 2019, 19, 1654–1664. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mapa, M.S.T.; Sprando, R.L. Liver Toxicity of Anthraquinones: A Combined in Vitro Cytotoxicity and in Silico Reverse Dosimetry Evaluation. Food Chem. Toxicol. 2020, 140, 111313. [Google Scholar] [CrossRef]
- Wang, S.-H.; Lo, C.-Y.; Gwo, Z.-H.; Lin, H.-J.; Chen, L.-G.; Kuo, C.-D.; Wu, J.-Y. Synthesis and Biological Evaluation of Lipophilic 1,4-Naphthoquinone Derivatives against Human Cancer Cell Lines. Molecules 2015, 20, 11994–12015. [Google Scholar] [CrossRef] [PubMed]
- Zorzanelli, B.C.; Ouverney, G.; Pauli, F.P.; da Fonseca, A.C.C.; de Almeida, E.C.P.; de Carvalho, D.G.; Possik, P.A.; Rabelo, V.W.-H.; Abreu, P.A.; Pontes, B.; et al. Pro-Apoptotic Antitumoral Effect of Novel Acridine-Core Naphthoquinone Compounds against Oral Squamous Cell Carcinoma. Molecules 2022, 27, 5148. [Google Scholar] [CrossRef]
- Wu, L.; Liu, Y.; Li, Y. Synthesis of Spirooxindole-O-Naphthoquinone-Tetrazolo [1,5-a]Pyrimidine Hybrids as Potential Anticancer Agents. Molecules 2018, 23, 2330. [Google Scholar] [CrossRef]
- Ning, X.; Qi, H.; Li, R.; Jin, Y.; McNutt, M.A.; Yin, Y. Synthesis and Antitumor Activity of Novel 2, 3-Didithiocarbamate Substituted Naphthoquinones as Inhibitors of Pyruvate Kinase M2 Isoform. J. Enzym. Inhib. Med. Chem. 2018, 33, 126–129. [Google Scholar] [CrossRef]
- Chable-Bessia, C.; Boullé, C.; Neyret, A.; Swain, J.; Hénaut, M.; Merida, P.; Gros, N.; Makinson, A.; Lyonnais, S.; Chesnais, C.; et al. Low Selectivity Indices of Ivermectin and Macrocyclic Lactones on SARS-CoV-2 Replication In Vitro. COVID 2022, 2, 60–75. [Google Scholar] [CrossRef]
- Paul, P.; Chakraborty, P.; Chatterjee, A.; Sarker, R.K.; Dastidar, D.G.; Kundu, T.; Sarkar, N.; Das, A.; Tribedi, P. 1,4-Naphthoquinone Accumulates Reactive Oxygen Species in Staphylococcus Aureus: A Promising Approach towards Effective Management of Biofilm Threat. Arch. Microbiol. 2021, 203, 1183–1193. [Google Scholar] [CrossRef]
- Angulo-Elizari, E.; Henriquez-Figuereo, A.; Morán-Serradilla, C.; Plano, D.; Sanmartín, C. Unlocking the Potential of 1,4-Naphthoquinones: A Comprehensive Review of Their Anticancer Properties. Eur. J. Med. Chem. 2024, 268, 116249. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Wang, D.; Tang, Y. PKM2 Promotes Cell Metastasis and Inhibits Autophagy via the JAK/STAT3 Pathway in Hepatocellular Carcinoma. Mol. Cell. Biochem. 2021, 476, 2001–2010. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.-R.; Gopula, B.; Chu, P.-C.; Hu, J.-L.; Feng, C.-H. A PKM2 Inhibitor Induces Apoptosis and Autophagy through JAK2 in Human Oral Squamous Cell Carcinoma Cells. Chem. Biol. Interact. 2023, 380, 110538. [Google Scholar] [CrossRef] [PubMed]
- El-Far, A.H.; Al Jaouni, S.K.; Li, X.; Fu, J. Cancer Metabolism Control by Natural Products: Pyruvate Kinase M2 Targeting Therapeutics. Phytother. Res. 2022, 36, 3181–3201. [Google Scholar] [CrossRef]
- Zhou, Y.; Huang, Z.; Su, J.; Li, J.; Zhao, S.; Wu, L.; Zhang, J.; He, Y.; Zhang, G.; Tao, J.; et al. Benserazide Is a Novel Inhibitor Targeting PKM2 for Melanoma Treatment. Int. J. Cancer 2020, 147, 139–151. [Google Scholar] [CrossRef]
- Chen, Y.-L. Mechanisms of Pyruvate Kinase M2 Isoform Inhibits Cell Motility in Hepatocellular Carcinoma Cells. World J. Gastroenterol. 2015, 21, 9093. [Google Scholar] [CrossRef]
- Chen, M.; Liu, H.; Li, Z.; Ming, A.L.; Chen, H. Mechanism of PKM2 Affecting Cancer Immunity and Metabolism in Tumor Microenvironment. J. Cancer 2021, 12, 3566–3574. [Google Scholar] [CrossRef]
- Liao, M.; Yao, D.; Wu, L.; Luo, C.; Wang, Z.; Zhang, J.; Liu, B. Targeting the Warburg Effect: A Revisited Perspective from Molecular Mechanisms to Traditional and Innovative Therapeutic Strategies in Cancer. Acta Pharm. Sin. B 2024, 14, 953–1008. [Google Scholar] [CrossRef]
- Su, Q.; Luo, S.; Tan, Q.; Deng, J.; Zhou, S.; Peng, M.; Tao, T.; Yang, X. The Role of Pyruvate Kinase M2 in Anticancer Therapeutic Treatments (Review). Oncol. Lett. 2019, 18, 5663–5672. [Google Scholar] [CrossRef] [PubMed]
- Shankar Babu, M.; Mahanta, S.; Lakhter, A.J.; Hato, T.; Paul, S.; Naidu, S.R. Lapachol Inhibits Glycolysis in Cancer Cells by Targeting Pyruvate Kinase M2. PLoS ONE 2018, 13, e0191419. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Christofk, H.R.; Schuman, E.; Subtelny, A.O.; Sharfi, H.; Harlow, E.E.; Xian, J.; Cantley, L.C. Identification of Small Molecule Inhibitors of Pyruvate Kinase M2. Biochem. Pharmacol. 2010, 79, 1118–1124. [Google Scholar] [CrossRef]
- Sarfraz, I.; Rasul, A.; Jabeen, F.; Sultana, T.; Adem, Ş. Identification of Natural Compounds as Inhibitors of Pyruvate Kinase M2 for Cancer Treatment. Molecules 2022, 27, 7113. [Google Scholar] [CrossRef]
- Newsholme, P.; Lima, M.M.R.; Procopio, J.; Pithon-Curi, T.C.; Doi, S.Q.; Bazotte, R.B.; Curi, R. Glutamine and Glutamate as Vital Metabolites. Braz. J. Med. Biol. Res. 2003, 36, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Chen, Y.; Wu, X.; Chen, J.; Zhou, Q.; Liu, B.; Zhang, L.; Yi, C. Interplay of Energy Metabolism and Autophagy. Autophagy 2024, 20, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yao, S.; Yang, H.; Liu, S.; Wang, Y. Autophagy: Regulator of Cell Death. Cell Death Dis. 2023, 14, 648. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Lin, Y.; Wang, D.; Pan, D.; Zhang, Y.; Jin, Y.; Zheng, C. Enhancing 5-Fluorouracil Efficacy through Suppression of PKM2 in Colorectal Cancer Cells. Cancer Chemother. Pharmacol. 2018, 82, 1081–1086. [Google Scholar] [CrossRef]
- Morgan, H.P.; O’Reilly, F.J.; Wear, M.A.; O’Neill, J.R.; Fothergill-Gilmore, L.A.; Hupp, T.; Walkinshaw, M.D. M2 Pyruvate Kinase Provides a Mechanism for Nutrient Sensing and Regulation of Cell Proliferation. Proc. Natl. Acad. Sci. USA 2013, 110, 5881–5886. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cancer Cells | Normal Cell | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SCC-9 | Hep-G2 | HT-29 | B16-F10 | HGF | ||||||||||
| IC50 (µM) | SD | SI | IC50 (µM) | SD | SI | IC50 (µM) | SD | SI | IC50 (µM) | SD | SI | IC50 (µM) | SD | |
| MB-6a | 56.74 | ±0.11 | 4.63 | 76.69 | ±0.51 | 3.4 | 129.0 | ±0.05 | 2.03 | 66.42 | ±0.03 | 3.9 | 262.9 | ±0.04 |
| Carboplatin | 265.3 | ±0.06 | 1.69 | 86.62 | ±0.02 | 3.03 | 174.1 | ±0.03 | 1.5 | 263.1 | ±0.02 | 0.99 | 448.8 | ±0.06 |
| Complex | Individual Score (kcal/mol) | Average Score (kcal/mol) | SD (kcal/mol) |
|---|---|---|---|
| APKM2-MB-6a-A | −7.4 | −7.16 | ±0.21 |
| −7.13 | |||
| −6.97 | |||
| BPKM2-MB-6a-B | −9.79 | −9.79 | ±0.005 |
| −9.79 | |||
| −9.8 | |||
| CPKM2-MB-6a-C | −7.62 | −6.94 | ±0.61 |
| −6.43 | |||
| −6.78 | |||
| DPKM2-MB-6a-D | −9.47 | −9.50 | ±0.07 |
| −9.46 | |||
| −9.59 |
| Complex | ΔVDWAALS (kcal/mol) | ΔEEL (kcal/mol) | ΔEPB (kcal/mol) | ΔENPOLAR (kcal/mol) | ΔGbind Total (kcal/mol) |
|---|---|---|---|---|---|
| APKM2-MB-6a-A | −28.34 ± 4.33 | −153.08 ± 30.42 | 158.55 ± 27.18 | −3.84 ± 0.51 | −26.71 ± 7.29 |
| BPKM2-MB-6a-B | −43.60 ± 5.52 | −157.56 ± 18.43 | 177.60 ± 15.85 | −5.31 ± 0.13 | −28.88 ± 8.58 |
| CPKM2-MB-6a-C | NA | NA | NA | NA | NA |
| DPKM2-MB-6a-D | −32.78 ± 5.08 | −133.88 ± 17.64 | 147.86 ± 16.48 | −3.56 ± 0.36 | −22.36 ± 5.20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubini-Dias, L.; Fernandes, T.V.A.; de Souza, M.P.; Hottz, D.; Arruda, A.T.; Borges, A.d.A.; Ouverney, G.; da Silva, F.d.C.; Forezi, L.d.S.M.; Limaverde-Sousa, G.; et al. Mannich Base Derived from Lawsone Inhibits PKM2 and Induces Neoplastic Cell Death. Biomedicines 2024, 12, 2916. https://doi.org/10.3390/biomedicines12122916
Rubini-Dias L, Fernandes TVA, de Souza MP, Hottz D, Arruda AT, Borges AdA, Ouverney G, da Silva FdC, Forezi LdSM, Limaverde-Sousa G, et al. Mannich Base Derived from Lawsone Inhibits PKM2 and Induces Neoplastic Cell Death. Biomedicines. 2024; 12(12):2916. https://doi.org/10.3390/biomedicines12122916
Chicago/Turabian StyleRubini-Dias, Lucas, Tácio V. A. Fernandes, Michele P. de Souza, Déborah Hottz, Afonso T. Arruda, Amanda de A. Borges, Gabriel Ouverney, Fernando de C. da Silva, Luana da S. M. Forezi, Gabriel Limaverde-Sousa, and et al. 2024. "Mannich Base Derived from Lawsone Inhibits PKM2 and Induces Neoplastic Cell Death" Biomedicines 12, no. 12: 2916. https://doi.org/10.3390/biomedicines12122916
APA StyleRubini-Dias, L., Fernandes, T. V. A., de Souza, M. P., Hottz, D., Arruda, A. T., Borges, A. d. A., Ouverney, G., da Silva, F. d. C., Forezi, L. d. S. M., Limaverde-Sousa, G., & Robbs, B. K. (2024). Mannich Base Derived from Lawsone Inhibits PKM2 and Induces Neoplastic Cell Death. Biomedicines, 12(12), 2916. https://doi.org/10.3390/biomedicines12122916