
Asoprisnil as a Novel Ligand Interacting with Stress-Associated Glucocorticoid Receptor




Abstract

1. Introduction
2. Materials and Methods
2.1. Ligand Selection
2.2. Ligand Properties Screening
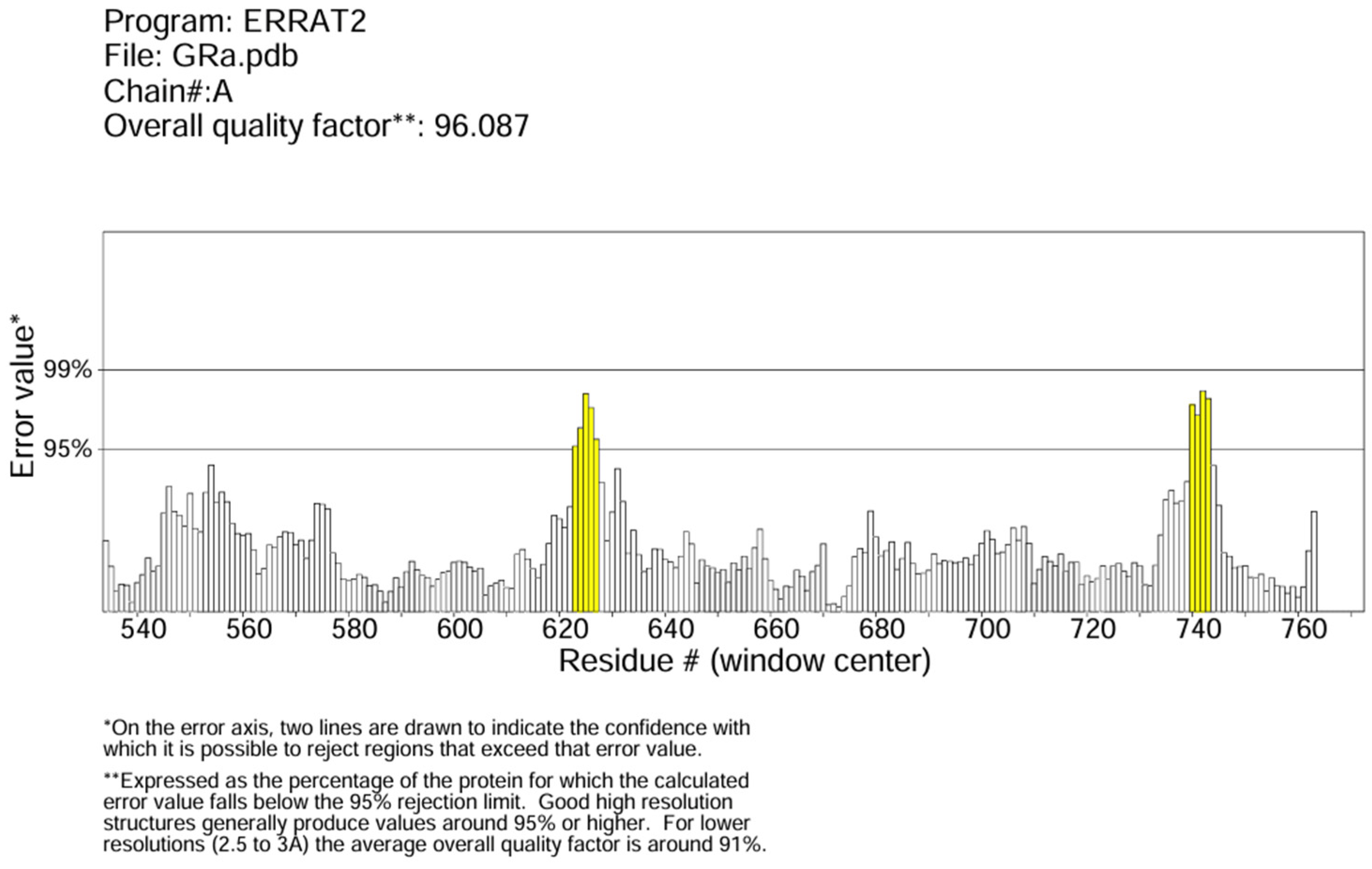
2.3. Protein Preparation and Quality Check
2.4. Molecular Docking
2.5. Molecular Docking Analysis
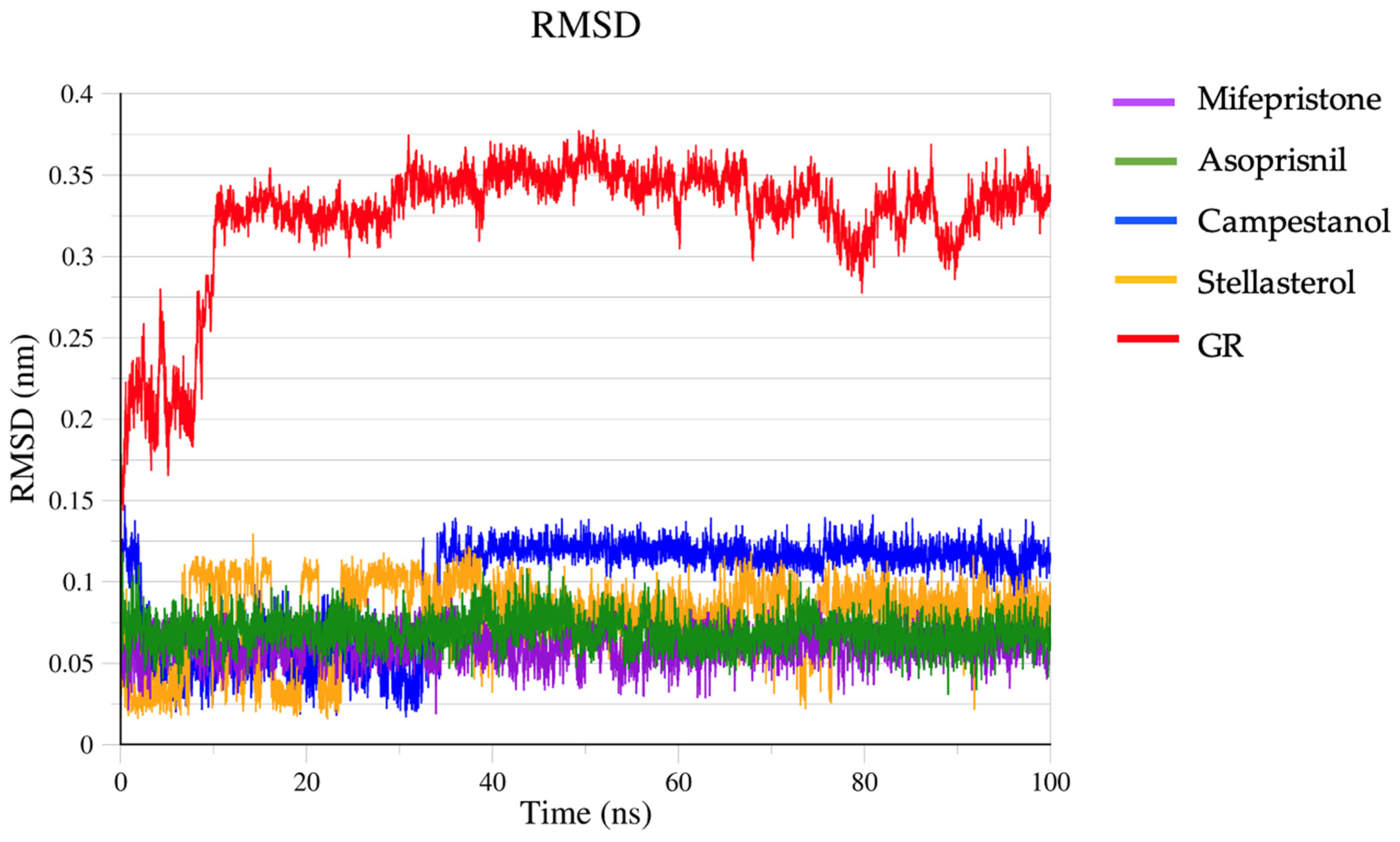
2.6. Molecular Dynamic Simulation
3. Results and Discussion
3.1. Pharmacokinetic and Toxicological Analysis of the Ligands
3.2. Protein Preparation and Quality Check Result
3.3. AutoDock Result Analysis
3.4. Molecular Interaction Analysis
3.5. Molecular Mechanics Poisson–Boltzmann Surface Area (MMPBSA) Calculations
3.6. Molecular Dynamic Simulation Result
4. Conclusions and Future Perspective
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adinoff, B.; Iranmanesh, A.; Veldhuis, J.; Fisher, L. Disturbances of the Stress Response: The Role of the HPA Axis during Alcohol Withdrawal and Abstinence. Alcohol. Health Res. World 1998, 22, 67–72. [Google Scholar]
- Thau, L.; Gandhi, J.; Sharma, S. Physiology, Cortisol. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Krahel, A.; Paszynska, E.; Slopien, A.; Gawriolek, M.; Otulakowska-Skrzynska, J.; Rzatowski, S.; Hernik, A.; Hanć, T.; Bryl, E.; Szczesniewska, P.; et al. Stress/Immune Biomarkers in Saliva among Children with ADHD Status. IJERPH 2021, 18, 769. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, D.; Weissenkampen, J.D.; Wasserman, E.; Krishnamurthy, V.B.; Millett, C.E.; Conway, S.; Saunders, E.F.H. Dysregulated Diurnal Cortisol Pattern and Heightened Night-Time Cortisol in Individuals with Bipolar Disorder. Neuropsychobiology 2022, 81, 51–59. [Google Scholar] [CrossRef]
- Belvederi Murri, M.; Prestia, D.; Mondelli, V.; Pariante, C.; Patti, S.; Olivieri, B.; Arzani, C.; Masotti, M.; Respino, M.; Antonioli, M.; et al. The HPA Axis in Bipolar Disorder: Systematic Review and Meta-Analysis. Psychoneuroendocrinology 2016, 63, 327–342. [Google Scholar] [CrossRef]
- Manenschijn, L.; Spijker, A.T.; Koper, J.W.; Jetten, A.M.; Giltay, E.J.; Haffmans, J.; Hoencamp, E.; Van Rossum, E.F.C. Long-Term Cortisol in Bipolar Disorder: Associations with Age of Onset and Psychiatric Co-Morbidity. Psychoneuroendocrinology 2012, 37, 1960–1968. [Google Scholar] [CrossRef]
- Faresjö, Å.; Theodorsson, E.; Chatziarzenis, M.; Sapouna, V.; Claesson, H.-P.; Koppner, J.; Faresjö, T. Higher Perceived Stress but Lower Cortisol Levels Found among Young Greek Adults Living in a Stressful Social Environment in Comparison with Swedish Young Adults. PLoS ONE 2013, 8, e73828. [Google Scholar] [CrossRef] [PubMed]
- Lyu, N.; Zhao, Q.; Fu, B.; Li, J.; Wang, H.; Yang, F.; Liu, S.; Huang, J.; Zhang, X.; Zhang, L.; et al. Hormonal and Inflammatory Signatures of Different Mood Episodes in Bipolar Disorder: A Large-Scale Clinical Study. BMC Psychiatry 2023, 23, 449. [Google Scholar] [CrossRef] [PubMed]
- Adrenocorticotropic Hormone (ACTH): What It Is & Function. Available online: https://my.clevelandclinic.org/health/articles/23151-adrenocorticotropic-hormone-acth (accessed on 8 April 2024).
- Dziurkowska, E.; Wesolowski, M. Cortisol as a Biomarker of Mental Disorder Severity. J. Clin. Med. 2021, 10, 5204. [Google Scholar] [CrossRef]
- Walker, E.F.; Trotman, H.D.; Pearce, B.D.; Addington, J.; Cadenhead, K.S.; Cornblatt, B.A.; Heinssen, R.; Mathalon, D.H.; Perkins, D.O.; Seidman, L.J.; et al. Cortisol Levels and Risk for Psychosis: Initial Findings from the North American Prodrome Longitudinal Study. Biol. Psychiatry 2013, 74, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Pivonello, R.; Simeoli, C.; De Martino, M.C.; Cozzolino, A.; De Leo, M.; Iacuaniello, D.; Pivonello, C.; Negri, M.; Pellecchia, M.T.; Iasevoli, F.; et al. Neuropsychiatric Disorders in Cushing’s Syndrome. Front. Neurosci. 2015, 9, 129. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.P.; McKlveen, J.M.; Ghosal, S.; Kopp, B.; Wulsin, A.; Makinson, R.; Scheimann, J.; Myers, B. Regulation of the Hypothalamic-Pituitary-Adrenocortical Stress Response. Compr. Physiol. 2016, 6, 603–621. [Google Scholar]
- Gjerstad, J.K.; Lightman, S.L.; Spiga, F. Role of Glucocorticoid Negative Feedback in the Regulation of HPA Axis Pulsatility. Stress 2018, 21, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Mikulska, J.; Juszczyk, G.; Gawrońska-Grzywacz, M.; Herbet, M. HPA Axis in the Pathomechanism of Depression and Schizophrenia: New Therapeutic Strategies Based on Its Participation. Brain Sci. 2021, 11, 1298. [Google Scholar] [CrossRef]
- Goncharova, N.; Chigarova, O.; Rudenko, N.; Oganyan, T. Glucocorticoid Negative Feedback in Regulation of the Hypothalamic-Pituitary-Adrenal Axis in Rhesus Monkeys With Various Types of Adaptive Behavior: Individual and Age-Related Differences. Front. Endocrinol. 2019, 10, 24. [Google Scholar] [CrossRef]
- Menke, A. The HPA Axis as Target for Depression. Curr. Neuropharmacol. 2024, 22, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Sanders, A.; Fernandez, C.J.; Gama, R. Chapter 3—Adrenal Cortical Hormones and Blood Pressure Regulation. In Endocrine Hypertension; Pappachan, J.M., Fernandez, C.J., Eds.; Academic Press: Cambridge, MA, USA, 2023; pp. 35–52. ISBN 978-0-323-96120-2. [Google Scholar]
- Oakley, R.H.; Cidlowski, J.A. The Biology of the Glucocorticoid Receptor: New Signaling Mechanisms in Health and Disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Nicolaides, N.C.; Chrousos, G.; Kino, T. Glucocorticoid Receptor. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Glucocorticoid Receptor Alpha—An Overview | ScienceDirect Topics. Available online: https://www.sciencedirect.com/topics/medicine-and-dentistry/glucocorticoid-receptor-alpha (accessed on 30 October 2024).
- Lockett, J.; Inder, W.J.; Clifton, V.L. The Glucocorticoid Receptor: Isoforms, Functions, and Contribution to Glucocorticoid Sensitivity. Endocr. Rev. 2024, 45, 593–624. [Google Scholar] [CrossRef] [PubMed]
- Briassoulis, G.; Damjanovic, S.; Xekouki, P.; Lefebvre, H.; Stratakis, C.A. The Glucocorticoid Receptor and Its Expression in the Anterior Pituitary and the Adrenal Cortex: A Source of Variation in Hypothalamic-Pituitary-Adrenal Axis Function; Implications for Pituitary and Adrenal Tumors. Endocr. Pract. 2011, 17, 941–948. [Google Scholar] [CrossRef]
- Mifepristone. Available online: https://go.drugbank.com/drugs/DB00834 (accessed on 12 April 2024).
- Blasey, C.M.; Block, T.S.; Belanoff, J.K.; Roe, R.L. Efficacy and Safety of Mifepristone for the Treatment of Psychotic Depression. J. Clin. Psychopharmacol. 2011, 31, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Delivanis, D.A.; Sharma, A.; Hamidi, O.; Shah, M.; Bancos, I. Advances in the Diagnosis and Medical Management of Cushing’s Syndrome. In Advances in Treatment and Management in Surgical Endocrinology; Elsevier: Amsterdam, The Netherlands, 2020; pp. 151–174. ISBN 978-0-323-66195-9. [Google Scholar]
- Reyes-Contreras, M.; Glauser, G.; Rennison, D.J.; Taborsky, B. Early-Life Manipulation of Cortisol and Its Receptor Alters Stress Axis Programming and Social Competence. Phil. Trans. R. Soc. B 2019, 374, 20180119. [Google Scholar] [CrossRef]
- Garner, B.; Phillips, L.J.; Bendall, S.; Hetrick, S.E. Antiglucocorticoid and Related Treatments for Psychosis. Cochrane Database Syst. Rev. 2016, CD006995. [Google Scholar] [CrossRef]
- Karena, Z.V.; Shah, H.; Vaghela, H.; Chauhan, K.; Desai, P.K.; Chitalwala, A.R. Clinical Utility of Mifepristone: Apprising the Expanding Horizons. Cureus 2022, 14, e28318. [Google Scholar] [CrossRef]
- Leichombam, R.; Bawiskar, D.; Leichombam, R.; Bawiskar, D. Exploring the Safety and Efficacy of Medical Termination of Pregnancy: A Comprehensive Review. Cureus 2023, 15, e46444. [Google Scholar] [CrossRef] [PubMed]
- Shih, G. Mifepristone Is Under Scrutiny in the Courts, But It Has Been Used Safely and Effectively Around the World for Decades. Available online: http://theconversation.com/mifepristone-is-under-scrutiny-in-the-courts-but-it-has-been-used-safely-and-effectively-around-the-world-for-decades-204163 (accessed on 30 October 2024).
- Bakour, N.; Moriarty, F.; Moore, G.; Robson, T.; Annett, S.L. Prognostic Significance of Glucocorticoid Receptor Expression in Cancer: A Systematic Review and Meta-Analysis. Cancers 2021, 13, 1649. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.S.; Rahman, M.O.; Alqahtani, A.S.; Sultana, N.; Almarfadi, O.M.; Ali, M.A.; Lee, J. Anticancer Potential of Phytochemicals from Oroxylum indicum Targeting Lactate Dehydrogenase A through Bioinformatic Approach. Toxicol. Rep. 2023, 10, 56–75. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Banerjee, P.; Kemmler, E.; Dunkel, M.; Preissner, R. ProTox 3.0: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res. 2024, 52, W513–W520. [Google Scholar] [CrossRef] [PubMed]
- Borba, J.V.B.; Alves, V.M.; Braga, R.C.; Korn, D.R.; Overdahl, K.; Silva, A.C.; Hall, S.U.S.; Overdahl, E.; Kleinstreuer, N.; Strickland, J.; et al. STopTox: An in Silico Alternative to Animal Testing for Acute Systemic and Topical Toxicity. Environ. Health Perspect. 2022, 130, 027012. [Google Scholar] [CrossRef]
- Jakubec, D.; Skoda, P.; Krivak, R.; Novotny, M.; Hoksza, D. PrankWeb 3: Accelerated Ligand-Binding Site Predictions for Experimental and Modelled Protein Structures. Nucleic Acids Res. 2022, 50, W593–W597. [Google Scholar] [CrossRef] [PubMed]
- Jendele, L.; Krivak, R.; Skoda, P.; Novotny, M.; Hoksza, D. PrankWeb: A Web Server for Ligand Binding Site Prediction and Visualization. Nucleic Acids Res. 2019, 47, W345–W349. [Google Scholar] [CrossRef]
- Krivák, R.; Hoksza, D. P2Rank: Machine Learning Based Tool for Rapid and Accurate Prediction of Ligand Binding Sites from Protein Structure. J. Cheminform 2018, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of Protein Structures: Patterns of Nonbonded Atomic Interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Youkharibache, P.; Marchler-Bauer, A.; Lanczycki, C.; Zhang, D.; Lu, S.; Madej, T.; Marchler, G.H.; Cheng, T.; Chong, L.C.; et al. iCn3D: From Web-Based 3D Viewer to Structural Analysis Tool in Batch Mode. Front. Mol. Biosci. 2022, 9, 831740. [Google Scholar] [CrossRef]
- Wang, J.; Youkharibache, P.; Zhang, D.; Lanczycki, C.J.; Geer, R.C.; Madej, T.; Phan, L.; Ward, M.; Lu, S.; Marchler, G.H.; et al. iCn3D, a Web-Based 3D Viewer for Sharing 1D/2D/3D Representations of Biomolecular Structures. Bioinformatics 2020, 36, 131–135. [Google Scholar] [CrossRef]
- Zentrum Für Bioinformatik: Universität Hamburg—Proteins Plus Server. Available online: https://proteins.plus/ (accessed on 28 January 2024).
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Fromm, M.F. Importance of P-Glycoprotein at Blood–Tissue Barriers. Trends Pharmacol. Sci. 2004, 25, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Fromm, M.F. P-Glycoprotein: A Defense Mechanism Limiting Oral Bioavailability and CNS Accumulation of Drugs. Int. J. Clin. Pharmacol. Ther. 2000, 38, 69–74. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings1. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Davis, R.J. Cell Signaling and Stress Responses. Cold Spring Harb. Perspect. Biol. 2016, 8, a006072. [Google Scholar] [CrossRef] [PubMed]
- Jennings, P. Stress Response Pathways, Toxicity Pathways and Adverse Outcome Pathways. Arch. Toxicol. 2013, 87, 13–14. [Google Scholar] [CrossRef]
- Polasek, T.M.; Lin, F.P.Y.; Miners, J.O.; Doogue, M.P. Perpetrators of Pharmacokinetic Drug–Drug Interactions Arising from Altered Cytochrome P450 Activity: A Criteria-Based Assessment. Br. J. Clin. Pharmacol. 2011, 71, 727–736. [Google Scholar] [CrossRef]
- Kapetas, A.J.; Abuhelwa, A.Y.; Sorich, M.J.; McKinnon, R.A.; Rodrigues, A.D.; Rowland, A.; Hopkins, A.M. Evidence-Based Guidelines for Drug Interaction Studies: Model-Informed Time Course of Intestinal and Hepatic CYP3A4 Inhibition by Clarithromycin. AAPS J. 2021, 23, 104. [Google Scholar] [CrossRef] [PubMed]
- Autry, B.M.; Wadhwa, R. Mifepristone. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Dresser, G.K.; Spence, J.D.; Bailey, D.G. Pharmacokinetic-Pharmacodynamic Consequences and Clinical Relevance of Cytochrome P450 3A4 Inhibition. Clin. Pharmacokinet. 2000, 38, 41–57. [Google Scholar] [CrossRef]
- Wang, X.; Dowty, M.E.; Wouters, A.; Tatulych, S.; Connell, C.A.; Le, V.H.; Tripathy, S.; O’Gorman, M.T.; Winton, J.A.; Yin, N.; et al. Assessment of the Effects of Inhibition or Induction of CYP2C19 and CYP2C9 Enzymes, or Inhibition of OAT3, on the Pharmacokinetics of Abrocitinib and Its Metabolites in Healthy Individuals. Eur. J. Drug Metab. Pharmacokinet. 2022, 47, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Cicali, E.J.; Smith, D.M.; Duong, B.Q.; Kovar, L.G.; Cavallari, L.H.; Johnson, J.A. A Scoping Review of the Evidence Behind Cytochrome P450 2D6 Isoenzyme Inhibitor Classifications. Clin. Pharmacol. Ther. 2020, 108, 116–125. [Google Scholar] [CrossRef]
- Drug Metabolism—The Importance of Cytochrome P450 3A4. Available online: https://www.medsafe.govt.nz/profs/puarticles/march2014drugmetabolismcytochromep4503a4.htm (accessed on 30 October 2024).
- Kumar, V.; Wahlstrom, J.L.; Rock, D.A.; Warren, C.J.; Gorman, L.A.; Tracy, T.S. CYP2C9 Inhibition: Impact of Probe Selection and Pharmacogenetics on in Vitro Inhibition Profiles. Drug Metab. Dispos. 2006, 34, 1966–1975. [Google Scholar] [CrossRef]
- Gallagher, P.; Young, A.H. Mifepristone (RU-486) Treatment for Depression and Psychosis: A Review of the Therapeutic Implications. Neuropsychiatr. Dis. Treat. 2006, 2, 33–42. [Google Scholar]
- Young, A.H.; Gallagher, P.; Watson, S.; Del-Estal, D.; Owen, B.M.; Nicol Ferrier, I. Improvements in Neurocognitive Function and Mood Following Adjunctive Treatment with Mifepristone (RU-486) in Bipolar Disorder. Neuropsychopharmacol. 2004, 29, 1538–1545. [Google Scholar] [CrossRef] [PubMed]
- Nayana, J.; Shankaranarayana Rao, B.S.; Srikumar, B.N. Mifepristone’s Effects on Depression- and Anxiety-like Behavior in Rodents. Steroids 2022, 184, 109058. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.A.; Diamond, M.P.; Williams, A.R.W.; Carr, B.R.; Myers, E.R.; Feldman, R.A.; Elger, W.; Mattia-Goldberg, C.; Schwefel, B.M.; Chwalisz, K. Safety and Efficacy of the Selective Progesterone Receptor Modulator Asoprisnil for Heavy Menstrual Bleeding with Uterine Fibroids: Pooled Analysis of Two 12-Month, Placebo-Controlled, Randomized Trials. Hum. Reprod. 2019, 34, 623–634. [Google Scholar] [CrossRef]
- DeManno, D.; Elger, W.; Garg, R.; Lee, R.; Schneider, B.; Hess-Stumpp, H.; Schubert, G.; Chwalisz, K. Asoprisnil (J867): A Selective Progesterone Receptor Modulator for Gynecological Therapy. Steroids 2003, 68, 1019–1032. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.; Gholam, M.; Zullo, L.; Kerksiek, A.; Castelao, E.; Von Gunten, A.; Preisig, M.; Lütjohann, D.; Popp, J. Plant Sterols and Cholesterol Metabolism Are Associated with Five-Year Cognitive Decline in the Elderly Population. iScience 2023, 26, 106740. [Google Scholar] [CrossRef] [PubMed]
- Vanmierlo, T.; Bogie, J.F.J.; Mailleux, J.; Vanmol, J.; Lütjohann, D.; Mulder, M.; Hendriks, J.J.A. Plant Sterols: Friend or Foe in CNS Disorders? Prog. Lipid Res. 2015, 58, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Rog, J.; Wingralek, Z.; Nowak, K.; Grudzień, M.; Grunwald, A.; Banaszek, A.; Karakula-Juchnowicz, H. The Potential Role of the Ketogenic Diet in Serious Mental Illness: Current Evidence, Safety, and Practical Advice. J. Clin. Med. 2024, 13, 2819. [Google Scholar] [CrossRef]
- Rahman, M.A.; Dash, R.; Sohag, A.A.M.; Alam, M.; Rhim, H.; Ha, H.; Moon, I.S.; Uddin, M.J.; Hannan, M.A. Prospects of Marine Sterols against Pathobiology of Alzheimer’s Disease: Pharmacological Insights and Technological Advances. Mar. Drugs 2021, 19, 167. [Google Scholar] [CrossRef]
- Pantsar, T.; Poso, A. Binding Affinity via Docking: Fact and Fiction. Molecules 2018, 23, 1899. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.K.; Verma, S. An In-Silico Evaluation of Dietary Components for Structural Inhibition of SARS-Cov-2 Main Protease. J. Biomol. Struct. Dyn. 2022, 40, 136–142. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Wakefield, A.E.; Kozakov, D.; Vajda, S. Mapping the Binding Sites of Challenging Drug Targets. Curr. Opin. Struct. Biol. 2022, 75, 102396. [Google Scholar] [CrossRef]
- Van Dijk, E.; Hoogeveen, A.; Abeln, S. The Hydrophobic Temperature Dependence of Amino Acids Directly Calculated from Protein Structures. PLOS Comput. Biol. 2015, 11, e1004277. [Google Scholar] [CrossRef] [PubMed]
- Gancia, E.; Montana, J.G.; Manallack, D.T. Theoretical Hydrogen Bonding Parameters for Drug Design. J. Mol. Graph. Model. 2001, 19, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Adeyemi, O.S.; Johnson, T.; Maduakolam-Aniobi, T.; Kato, K. Molecular Modelling and Experimental Validation Identified a New Therapeutic Inhibitor of Toxoplasmosis. Comput. Biol. Med. 2024, 183, 109236. [Google Scholar] [CrossRef] [PubMed]
- Rampogu, S.; Lee, G.; Park, J.S.; Lee, K.W.; Kim, M.O. Molecular Docking and Molecular Dynamics Simulations Discover Curcumin Analogue as a Plausible Dual Inhibitor for SARS-CoV-2. Int. J. Mol. Sci. 2022, 23, 1771. [Google Scholar] [CrossRef] [PubMed]
- Ejiohuo, O. A Perspective on the Synergistic Use of 3D Printing and Electrospinning to Improve Nanomaterials for Biomedical Applications. Nano Trends 2023, 4, 100025. [Google Scholar] [CrossRef]
- Giri, B.R.; Kwon, J.; Vo, A.Q.; Bhagurkar, A.M.; Bandari, S.; Kim, D.W. Hot-Melt Extruded Amorphous Solid Dispersion for Solubility, Stability, and Bioavailability Enhancement of Telmisartan. Pharmaceuticals 2021, 14, 73. [Google Scholar] [CrossRef]
- Chen, Y.; Mutukuri, T.T.; Wilson, N.E.; Zhou, Q. (Tony) Pharmaceutical Protein Solids: Drying Technology, Solid-State Characterization and Stability. Adv. Drug Deliv. Rev. 2021, 172, 211–233. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug Transport across the Blood–Brain Barrier. J. Cereb. Blood Flow. Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef]
- Kim, J.; Ahn, S.I.; Kim, Y. Nanotherapeutics Engineered to Cross the Blood-Brain Barrier for Advanced Drug Delivery to the Central Nervous System. J. Ind. Eng. Chem. 2019, 73, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Ejiohuo, O.; Folami, S.O.; Edi, D.; Isaac, J. Polyphenol Encapsulated Nanofibers in Wound Healing and Drug Delivery. Eur. J. Med. Chem. Rep. 2024, 12, 100184. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | |||||
|---|---|---|---|---|---|
| Pharmacokinetics | |||||
| Molecular Weight g/mol | Water Solubility LogS (class) | Druglikeness (Lipinski’s) | Lipophilicity (Log Po/w) | ||
| Mifepristone | 429.59 | Poorly soluble | 1 violation: MlogP > 4.15 | 4.60 | |
| Asoprisnil | 449.58 | Poorly soluble | 0 violation | 4.27 | |
| Campestanol | 402.70 | Moderately soluble | 1 violation: MlogP > 4.15 | 6.77 | |
| Stellasterol | 398.66 | Moderately soluble | 1 violation: MlogP > 4.15 | 6.58 | |
| (B) | |||||
| Predicted Toxicity Profile | |||||
| Prediction (Probability) | |||||
| Classification | Target | Mifepristone | Asoprisnil | Campestanol | Stellasterol |
| Organ toxicity | Hepatotoxicity | Inactive (0.96) | Inactive (0.73) | Inactive (0.79) | Inactive (0.75) |
| Neurotoxicity | Active (0.78) | Inactive (0.85) | Inactive (0.56) | Active (0.57) | |
| Toxicity end points | Carcinogenicity | Inactive (0.53) | Inactive (0.53) | Inactive (0.77) | Inactive (0.60) |
| Immunotoxicity | Active (0.81) | Active (0.93) | Active (0.97) | Active (0.94) | |
| Mutagenicity | Inactive (0.90) | Inactive (0.57) | Inactive (0.87) | Inactive (0.96) | |
| Cytotoxicity | Inactive (0.80) | Inactive (0.75) | Inactive (0.88) | Inactive (0.96) | |
| Tox21-Stress response pathways | Nuclear factor (erythroid-derived 2)-like 2/antioxidant responsive element (nrf2/ARE) | Inactive (0.97) | Inactive (0.88) | Inactive (0.93) | Inactive (0.73) |
| Heat shock factor response element (HSE) | Inactive (0.97) | Inactive (0.88) | Inactive (0.93) | Inactive (0.73) | |
| Mitochondrial membrane potential (MMP) | Active (0.99) | Inactive (0.65) | Inactive (0.86) | Inactive (0.55) | |
| Phosphoprotein (tumor suppressor) p53 | Active (1.0) | Inactive (0.81) | Inactive (0.99) | Inactive (0.99) | |
| ATPase family AAA domain-containing protein 5 (ATAD5) | Inactive (0.93) | Inactive (0.88) | Inactive (1.0) | Inactive (0.99) | |
| Cytochrome | CYP1A2 | Inactive (0.99) | Inactive (0.88) | Inactive (0.90) | Inactive (0.98) |
| CYP2C19 | Active (1.0) | Inactive (0.65) | Inactive (0.73) | Inactive (0.76) | |
| CYP2C9 | Inactive (0.51) | Inactive (0.54) | Active (0.67) | Active (0.75) | |
| CYP2D6 | Active (0.78) | Inactive (0.71) | Inactive (0.93) | Inactive (0.91) | |
| CYP3A4 | Active (0.79) | Inactive (0.58) | Inactive (0.98) | Inactive (0.94) | |
| CYP2E1 | Inactive (1.0) | Inactive (0.98) | Inactive (1.0) | Inactive (0.99) | |
| Acute inhalation toxicity | Yes | No | No | No | |
| Acute oral toxicity | Yes | No | Yes | No | |
| Acute dermal toxicity | No | No | No | No | |
| Eye irritation and corrosion | No | No | No | No | |
| Skin sensitisation | No | No | No | No | |
| Skin irritation and corrosion | No | No | Yes | No | |
| Ligand | RMSD (Å) | Binding Affinity (kcal/mol) | Inhibition Constant (Ki) |
|---|---|---|---|
| Mifepristone | 11.96 | −12.39 | 821.48 pM |
| Asoprisnil | 9.37 | −11.68 | 2.76 nM |
| Campestanol | 10.15 | −8.90 | 300.27 nM |
| Stellasterol | 11.90 | −12.07 | 1.41 nM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ejiohuo, O.; Bajia, D.; Pawlak, J.; Szczepankiewicz, A. Asoprisnil as a Novel Ligand Interacting with Stress-Associated Glucocorticoid Receptor. Biomedicines 2024, 12, 2745. https://doi.org/10.3390/biomedicines12122745
Ejiohuo O, Bajia D, Pawlak J, Szczepankiewicz A. Asoprisnil as a Novel Ligand Interacting with Stress-Associated Glucocorticoid Receptor. Biomedicines. 2024; 12(12):2745. https://doi.org/10.3390/biomedicines12122745
Chicago/Turabian StyleEjiohuo, Ovinuchi, Donald Bajia, Joanna Pawlak, and Aleksandra Szczepankiewicz. 2024. "Asoprisnil as a Novel Ligand Interacting with Stress-Associated Glucocorticoid Receptor" Biomedicines 12, no. 12: 2745. https://doi.org/10.3390/biomedicines12122745
APA StyleEjiohuo, O., Bajia, D., Pawlak, J., & Szczepankiewicz, A. (2024). Asoprisnil as a Novel Ligand Interacting with Stress-Associated Glucocorticoid Receptor. Biomedicines, 12(12), 2745. https://doi.org/10.3390/biomedicines12122745