
Generation of Transcript Length Variants and Reprogramming of mRNA Splicing During Atherosclerosis Progression in ApoE-Deficient Mice



Abstract
1. Background
2. Materials and Methods
2.1. Mice, Tissue and RNA Extraction
2.2. Microarray Expression Analysis
2.3. Sequence Identifiers, Definitions and Nomenclature
2.4. Data Analysis
2.5. Structural Analysis
3. Results
3.1. 3′UTR Lengthening in ATS Progression Is Partially Reversed by the αCD40 siRNA Treatment
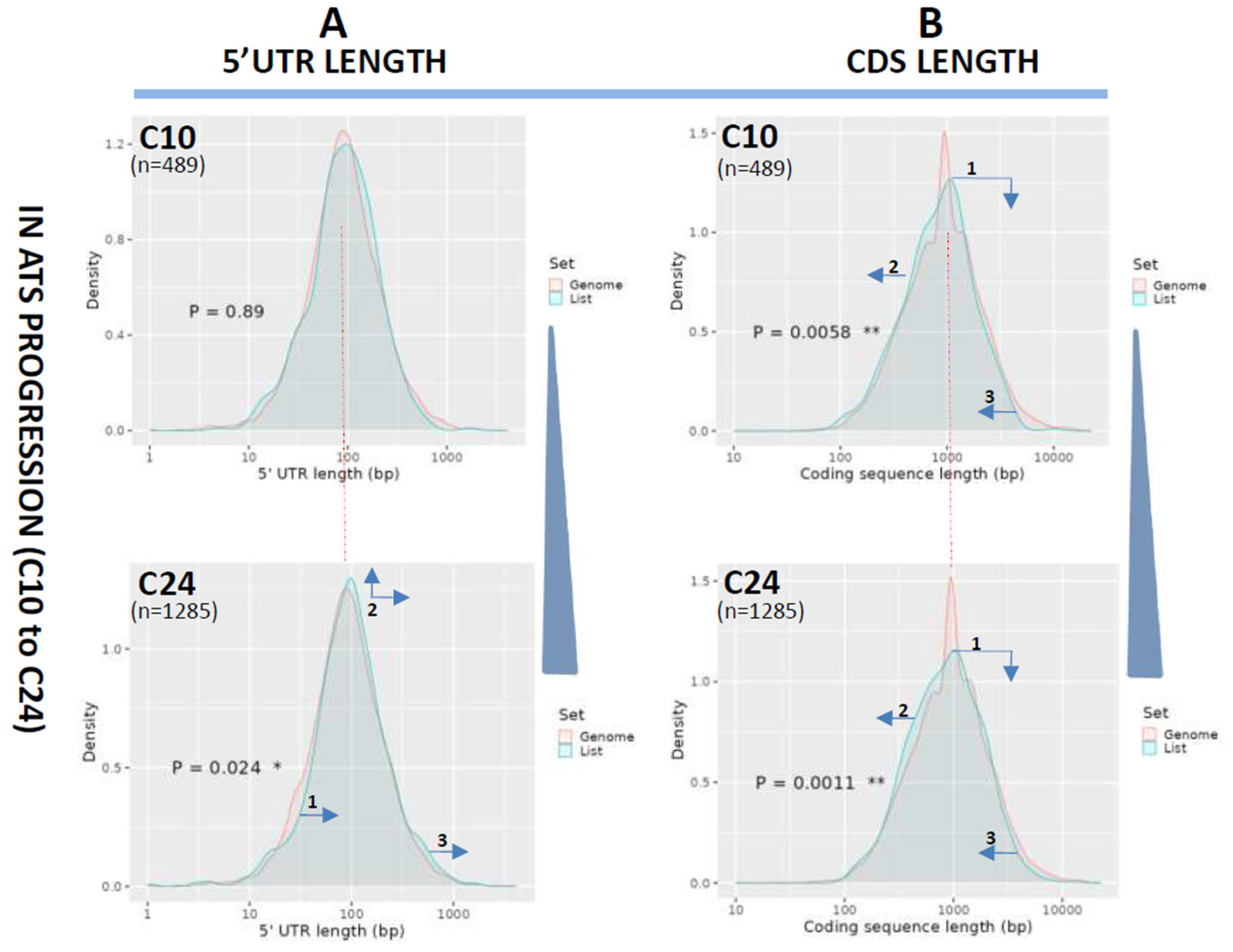
3.2. Length Changes in 5′UTRs and Coding Regions (CDS)
3.3. Variations in the Number of Exons per Coding Gene During ATS Progression and Treatment-Induced Regression
3.4. Differential Expression of Transcript Isoforms During ATS Progression or Treatment-Induced Regression
3.5. Dysregulation of 3′UTR Splicing Variants in ATS Progression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 5′SS | 5′ splice site |
| 3′SS | 3′ splice site |
| APA | Alternative polyadenylation |
| ATS | Atherosclerosis |
| C10/24 | Mice treated with the scrambled control siRNA at weeks 10/24 |
| CDS | Coding sequence |
| ceRNA | Competing endogenous RNA |
| CPSF | Cleavage and polyadenylation specificity factor |
| CstF64 | Cleavage stimulation factor 64 kDa |
| DEG | Differentially expressed gene |
| PA | Polyadenylation |
| PABPN | Poly(A)-Binding Protein Nuclear 1 |
| piRNA OR | Piwi-associated RNA Odorant receptor |
| RBP | RNA-binding protein |
| RNPS1 | RNA-Binding Protein with Serine-Rich Domain 1 |
| SRE | Splicing regulatory element |
| SoS | Suppressor of splicing |
| U1-RNPs | U1-ribonucleoprotein |
| T10/24 | Mice treated with αCD40 siRNA at weeks 10/24 |
References
- Deogharia, M.; Gurha, P. The “guiding” principles of noncoding RNA function. Wiley Interdiscip. Rev. RNA 2022, 13, e1704. [Google Scholar] [CrossRef]
- Cai, Y.; Yu, X.; Hu, S.; Yu, J. A brief review on the mechanisms of miRNA regulation. Genom. Proteom. Bioinform. 2009, 7, 147–154. [Google Scholar] [CrossRef]
- Navarro, E.; Mallen, A.; Hueso, M. Dynamic Variations of 3′UTR Length Reprogram the mRNA Regulatory Landscape. Biomedicines 2021, 9, 1560. [Google Scholar] [CrossRef]
- Mayr, C. 3′ UTRs Regulate Protein Functions by Providing a Nurturing Niche during Protein Synthesis. Cold Spring Harb. Symp. Quant. Biol. 2019, 84, 95–104. [Google Scholar] [CrossRef]
- Mayr, C. Regulation by 3′-Untranslated Regions. Annu. Rev. Genet. 2017, 51, 171–194. [Google Scholar] [CrossRef]
- Romo, L.; Findlay, S.D.; Burge, C.B. Regulatory features aid interpretation of 3′UTR variants. Am. J. Hum. Genet. 2024, 111, 350–363. [Google Scholar] [CrossRef]
- Chew, L.J.; Murphy, D.; Carter, D.A. Alternatively polyadenylated vasoactive intestinal peptide mRNAs are differentially regulated at the level of stability. Mol. Endocrinol. 1994, 8, 603–613. [Google Scholar] [CrossRef]
- Legendre, M.; Ritchie, W.; Lopez, F.; Gautheret, D. Differential repression of alternative transcripts: A screen for miRNA targets. PLoS Comput. Biol. 2006, 2, e43. [Google Scholar] [CrossRef]
- Singh, S.; Sinha, T.; Panda, A.C. Regulation of microRNA by circular RNA. Wiley Interdiscip. Rev. RNA 2023, 15, e1820. [Google Scholar] [CrossRef]
- He, Y.; Chen, Q.; Zhang, J.; Yu, J.; Xia, M.; Wang, X. Pervasive 3′-UTR Isoform Switches During Mouse Oocyte Maturation. Front. Mol. Biosci. 2021, 8, 727614. [Google Scholar] [CrossRef]
- Boreikaite, V.; Passmore, L.A. 3′-End Processing of Eukaryotic mRNA: Machinery, Regulation, and Impact on Gene Expression. Annu. Rev. Biochem. 2023, 92, 199–225. [Google Scholar] [CrossRef]
- Derti, A.; Garrett-Engele, P.; Macisaac, K.D.; Stevens, R.C.; Sriram, S.; Chen, R.; Rohl, C.A.; Johnson, J.M.; Babak, T. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012, 22, 1173–1183. [Google Scholar] [CrossRef]
- North, M.; Sargent, C.; O’Brien, J.; Taylor, K.; Wolfe, J.; Affara, N.A.; Ferguson-Smith, M.A. Comparison of ZFY and ZFX gene structure and analysis of alternative 3′ untranslated regions of ZFY. Nucleic Acids Res. 1991, 19, 2579–2586. [Google Scholar] [CrossRef]
- Code, R.J.; Olmsted, J.B. Mouse microtubule-associated protein 4 (MAP4) transcript diversity generated by alternative polyadenylation. Gene 1992, 122, 367–370. [Google Scholar] [CrossRef]
- Li, Y.; Yuan, Y. Alternative RNA splicing and gastric cancer. Mutat. Res. 2017, 773, 263–273. [Google Scholar] [CrossRef]
- Ogorodnikov, A.; Kargapolova, Y.; Danckwardt, S. Processing and transcriptome expansion at the mRNA 3′ end in health and disease: Finding the right end. Pflug. Arch. 2016, 468, 993–1012. [Google Scholar] [CrossRef]
- Zeng, J.; Song, K.; Wang, J.; Wen, H.; Zhou, J.; Ni, T.; Lu, H.; Yu, Y. Characterization and optimization of 5 untranslated region containing poly-adenine tracts in Kluyveromyces marxianus using machine-learning model. Microb. Cell Fact. 2024, 23, 7. [Google Scholar] [CrossRef]
- Ryczek, N.; Lys, A.; Makalowska, I. The Functional Meaning of 5′UTR in Protein-Coding Genes. Int. J. Mol. Sci. 2023, 24, 2976. [Google Scholar] [CrossRef]
- Sarkar, A.; Maji, R.K.; Saha, S.; Ghosh, Z. piRNAQuest: Searching the piRNAome for silencers. BMC Genom. 2014, 15, 555. [Google Scholar] [CrossRef]
- Hess, M.A.; Duncan, R.F. Sequence and structure determinants of Drosophila Hsp70 mRNA translation: 5′UTR secondary structure specifically inhibits heat shock protein mRNA translation. Nucleic Acids Res. 1996, 24, 2441–2449. [Google Scholar] [CrossRef]
- Zhao, G.; Wang, Q.; Zhang, Y.; Gu, R.; Liu, M.; Li, Q.; Zhang, J.; Yuan, H.; Feng, T.; Ou, D.; et al. DDX17 induces epithelial-mesenchymal transition and metastasis through the miR-149-3p/CYBRD1 pathway in colorectal cancer. Cell Death Dis. 2023, 14, 1. [Google Scholar] [CrossRef]
- Arbiol-Roca, A.; Padro-Miquel, A.; Hueso, M.; Navarro, E.; Alia-Ramos, P.; Gonzalez-Alvarez, M.T.; Rama, I.; Torras, J.; Grinyo, J.M.; Cruzado, J.M.; et al. Association of ANRIL gene polymorphisms with major adverse cardiovascular events in hemodialysis patients. Clin. Chim. Acta 2017, 466, 61–67. [Google Scholar] [CrossRef]
- Hueso, M.; De Ramon, L.; Navarro, E.; Ripoll, E.; Cruzado, J.M.; Grinyo, J.M.; Torras, J. Silencing of CD40 in vivo reduces progression of experimental atherogenesis through an NF-kappaB/miR-125b axis and reveals new potential mediators in the pathogenesis of atherosclerosis. Atherosclerosis 2016, 255, 80–89. [Google Scholar] [CrossRef]
- Hueso, M.; Cruzado, J.M.; Torras, J.; Navarro, E. An Exonic Switch Regulates Differential Accession of microRNAs to the Cd34 Transcript in Atherosclerosis Progression. Genes 2019, 10, 70. [Google Scholar] [CrossRef]
- Hueso, M.; Grinan, R.; Mallen, A.; Navarro, E.; Purqueras, E.; Goma, M.; Sbraga, F.; Blasco-Lucas, A.; Revilla, G.; Santos, D.; et al. MiR-125b downregulates macrophage scavenger receptor type B1 and reverse cholesterol transport. Biomed. Pharmacother. 2022, 146, 112596. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Mitschka, S.; Mayr, C. Context-specific regulation and function of mRNA alternative polyadenylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 779–796. [Google Scholar] [CrossRef]
- Huang, Y. The novel regulatory role of lncRNA-miRNA-mRNA axis in cardiovascular diseases. J. Cell. Mol. Med. 2018, 22, 5768–5775. [Google Scholar] [CrossRef]
- Hong, D.; Jeong, S. 3′UTR Diversity: Expanding Repertoire of RNA Alterations in Human mRNAs. Mol. Cells 2023, 46, 48–56. [Google Scholar] [CrossRef]
- Kandhari, N.; Kraupner-Taylor, C.A.; Harrison, P.F.; Powell, D.R.; Beilharz, T.H. The detection and bioinformatic analysis of alternative 3′UTR isoforms as potential cancer biomarkers. Int. J. Mol. Sci. 2021, 22, 5322. [Google Scholar] [CrossRef]
- Huang, D.; Wang, X.; Huang, Z.; Liu, Y.; Liu, X.; Gin, T.; Wong, S.H.; Yu, J.; Zhang, L.; Chan, M.T.V.; et al. 3′untranslated regions of tumor suppressor genes evolved specific features to favor cancer resistance. Oncogene 2022, 41, 3278–3288. [Google Scholar] [CrossRef]
- Yang, H.D.; Nam, S.W. Pathogenic diversity of RNA variants and RNA variation-associated factors in cancer development. Exp. Mol. Med. 2020, 52, 582–593. [Google Scholar] [CrossRef]
- Akman, H.B.; Oyken, M.; Tuncer, T.; Can, T.; Erson-Bensan, A.E. 3′UTR shortening and EGF signaling: Implications for breast cancer. Hum. Mol. Genet. 2015, 24, 6910–6920. [Google Scholar] [CrossRef]
- Chen, C.H.; Lin, H.Y.; Pan, C.L.; Chen, F.C. The genomic features that affect the lengths of 5′ untranslated regions in multicellular eukaryotes. BMC Bioinform. 2011, 12 (Suppl. S9), S3. [Google Scholar] [CrossRef]
- Girardot, M.; Martin, J.; Guibert, S.; Leveziel, H.; Julien, R.; Oulmouden, A. Widespread expression of the bovine Agouti gene results from at least three alternative promoters. Pigment. Cell Res. 2005, 18, 34–41. [Google Scholar] [CrossRef]
- Kajdasz, A.; Niewiadomska, D.; Sekrecki, M.; Sobczak, K. Distribution of alternative untranslated regions within the mRNA of the CELF1 splicing factor affects its expression. Sci. Rep. 2022, 12, 190. [Google Scholar] [CrossRef]
- Singh, J.; Raina, A.; Sangwan, N.; Chauhan, A.; Avti, P.K. Structural, molecular hybridization and network based identification of miR-373-3p and miR-520e-3p as regulators of NR4A2 human gene involved in neurodegeneration. Nucleosides Nucleotides Nucleic Acids 2022, 41, 419–443. [Google Scholar] [CrossRef]
- Niyazova, R.; Berillo, O.; Atambayeva, S.; Pyrkova, A.; Alybayeva, A.; Ivashchenko, A. miR-1322 Binding Sites in Paralogous and Orthologous Genes. Biomed. Res. Int. 2015, 2015, 962637. [Google Scholar] [CrossRef]
- Gu, W.; Xu, Y.; Xie, X.; Wang, T.; Ko, J.H.; Zhou, T. The role of RNA structure at 5′ untranslated region in microRNA-mediated gene regulation. RNA 2014, 20, 1369–1375. [Google Scholar] [CrossRef]
- Riolo, G.; Cantara, S.; Ricci, C. What’s Wrong in a Jump? Prediction and Validation of Splice Site Variants. Methods Protoc. 2021, 4, 62. [Google Scholar] [CrossRef]
- Doulazmi, M.; Cros, C.; Dusart, I.; Trembleau, A.; Dubacq, C. Alternative polyadenylation produces multiple 3′ untranslated regions of odorant receptor mRNAs in mouse olfactory sensory neurons. BMC Genom. 2019, 20, 577. [Google Scholar] [CrossRef] [PubMed]
- Ule, J.; Blencowe, B.J. Alternative Splicing Regulatory Networks: Functions, Mechanisms, and Evolution. Mol. Cell 2019, 76, 329–345. [Google Scholar] [CrossRef]
- Malard, F.; Mackereth, C.D.; Campagne, S. Principles and correction of 5′-splice site selection. RNA Biol. 2022, 19, 943–960. [Google Scholar] [CrossRef]
- Li, B.; Wachtel, C.; Miriami, E.; Yahalom, G.; Friedlander, G.; Sharon, G.; Sperling, R.; Sperling, J. Stop codons affect 5′ splice site selection by surveillance of splicing. Proc. Natl. Acad. Sci. USA 2002, 16, 5277–5282. [Google Scholar] [CrossRef]
- Brillen, A.L.; Schöneweis, K.; Walotka, L.; Hartmann, L.; Müller, L.; Ptok, J.; Kaisers, W.; Poschmann, G.; Stühler, K.; Buratti, E.; et al. Succession of splicing regulatory elements determines cryptic 5′ss functionality. Nucleic Acids Res. 2017, 45, 4202–4216. [Google Scholar] [CrossRef]
- Boehm, V.; Britto-Borges, T.; Steckelberg, A.; Singh, K.K.; Gerbracht, J.V.; Gueney, E.; Blazquez, L.; Altmüller, J.; Dieterich, C.; Gehring, N.H. Exon Junction Complexes Suppress Spurious Splice Sites to Safeguard Transcriptome Integrity. Mol. Cell 2018, 72, 482–495.e7. [Google Scholar] [CrossRef]
- Mabin, J.W.; Lewis, P.W.; Brow, D.A.; Dvinge, H. Human spliceosomal snRNA sequence variants generate variant spliceosomes. RNA 2021, 27, 1186–1203. [Google Scholar] [CrossRef]
- Dvinge, H.; Guenthoer, J.; Porter, P.L.; Bradley, R.K. RNA components of the spliceosome regulate tissue- and cancer-specific alternative splicing. Genome Res. 2019, 29, 1591–1604. [Google Scholar] [CrossRef]
- Arafat, M.; Sperling, R. A Quality Control Mechanism of Splice Site Selection Abrogated under Stress and in Cancer. Cancers 2022, 14, 1750. [Google Scholar] [CrossRef]
- Movassat, M.; Crabb, T.L.; Busch, A.; Yao, C.; Reynolds, D.J.; Shi, Y.; Hertel, K.J. Coupling between alternative polyadenylation and alternative splicing is limited to terminal introns. RNA Biol. 2016, 13, 646–655. [Google Scholar] [CrossRef]
- Cano-Besquet, S.; Park, M.; Berkley, N.; Wong, M.; Ashiqueali, S.; Noureddine, S.; Gesing, A.; Schneider, A.; Mason, J.; Masternak, M.M.; et al. Gene and transcript expression patterns, coupled with isoform switching and long non-coding RNA dynamics in adipose tissue, underlie the longevity of Ames dwarf mice. Geroscience 2024. [Google Scholar] [CrossRef] [PubMed]
- Dolgalev, G.; Poverennaya, E. Quantitative Analysis of Isoform Switching in Cancer. Int. J. Mol. Sci. 2023, 24, 10065. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.B.; Mukherjee, S.; Detroja, R.; Frenkel-Morgenstern, M. The landscape of differential splicing and transcript alternations in severe COVID-19 infection. FEBS J. 2023, 290, 3128–3144. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jin, T.; Hu, L.; Ding, J. Alternative splicing regulation and its therapeutic potential in bladder cancer. Front. Oncol. 2024, 14, 1402350. [Google Scholar] [CrossRef]
- Raguraman, P.; Balachandran, A.A.; Chen, S.; Diermeier, S.D.; Veedu, R.N. Antisense Oligonucleotide-Mediated Splice Switching: Potential Therapeutic Approach for Cancer Mitigation. Cancers 2021, 13, 5555. [Google Scholar] [CrossRef]
- Du, J.X.; Zhu, G.Q.; Cai, J.L.; Wang, B.; Luo, Y.H.; Chen, C.; Cai, C.Z.; Zhang, S.J.; Zhou, J.; Fan, J.; et al. Splicing factors: Insights into their regulatory network in alternative splicing in cancer. Cancer Lett. 2021, 501, 83–104. [Google Scholar] [CrossRef]
- Salton, M.; Misteli, T. Small Molecule Modulators of Pre-mRNA Splicing in Cancer Therapy. Trends Mol. Med. 2016, 22, 28–37. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genesymbol | Isoforms in C10 (Seqname) | New Isoforms in C24 (Seqname) | Length Variations (C24 vs. C10) in: | Mechanisms of Length Variation | |||
|---|---|---|---|---|---|---|---|
| FL | 5′UTR | CDS | 3′UTR | ||||
| Dusp22 | NM_001037955 | NM_134068 | S | = | S | S | Cryptic splicing at the end of the CDS-3′UTR generates a shorter NM_134068 |
| Ly6e | NM_001164037 NM_0011640381 NM_0011640401 | NM_001164037 | L | L | = | = | Three alternative 3′Splice aceptor sites generate three different 5′UTRs |
| Mta3 | NM_001171053 | NM_001171052 | L | = | L | L | An alternative 5′ splice donor site generates a new CDS and 3′UTR |
| Rab11fip5 | NM_177466 | NM_001003955 | L | = | L | = | Inclusion of an alternative exon in the CDS of NM_001003955 |
| Rbmx | NM_001166623 | NM_011252 | S2 | S2 | = | = | Shorter 5′UTR in NM_011252 by activation of an alternative 3′ splice acceptor site at an exon–exon junction at the 5′UTR |
| Skor | NM_001163757 | NM_001163758 | S | L | S | = | Shorter CDS in NM_001163758 than in NM_001163757 through use of two alternative start codons |
| Srsf5 | NM_001079694 | NM_009159 | S | S | = | = | 4 nts missing in the 5′UTR of NM_009159 |
| Genesymbol | Isoforms in T10 (Seqname) | New Isoforms in T24 (Seqname) | Length Variations (T24 vs. T10) in: | Mechanisms of Length Variation | |||
|---|---|---|---|---|---|---|---|
| FL | 5′UTR | CDS | 3′UTR | ||||
| Ilf3 | NM_001042708 | NM_001042707 | S | = | L | S | Alternative exons in CDS and 3′UTR. Alternative splicing (METEs) |
| Pwwp2a | NM_027557 | NM_001164231 | L | = | S | L | An alternative 5′ splice donor site generates a new CDS and 3′UTR |
| Rbmx | NM_001166623 | NM_011252 | S1 | S1 | = | = | Shorter 5′UTR in NM_011252 by activation of an alternative 3′ splice acceptor site at an exon–exon junction at the 5′UTR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hueso, M.; Mallén, A.; Navarro, E. Generation of Transcript Length Variants and Reprogramming of mRNA Splicing During Atherosclerosis Progression in ApoE-Deficient Mice. Biomedicines 2024, 12, 2703. https://doi.org/10.3390/biomedicines12122703
Hueso M, Mallén A, Navarro E. Generation of Transcript Length Variants and Reprogramming of mRNA Splicing During Atherosclerosis Progression in ApoE-Deficient Mice. Biomedicines. 2024; 12(12):2703. https://doi.org/10.3390/biomedicines12122703
Chicago/Turabian StyleHueso, Miguel, Adrián Mallén, and Estanis Navarro. 2024. "Generation of Transcript Length Variants and Reprogramming of mRNA Splicing During Atherosclerosis Progression in ApoE-Deficient Mice" Biomedicines 12, no. 12: 2703. https://doi.org/10.3390/biomedicines12122703
APA StyleHueso, M., Mallén, A., & Navarro, E. (2024). Generation of Transcript Length Variants and Reprogramming of mRNA Splicing During Atherosclerosis Progression in ApoE-Deficient Mice. Biomedicines, 12(12), 2703. https://doi.org/10.3390/biomedicines12122703