

The Relevance and Implications of Monoclonal Antibody Therapies on Traumatic Brain Injury Pathologies

Abstract

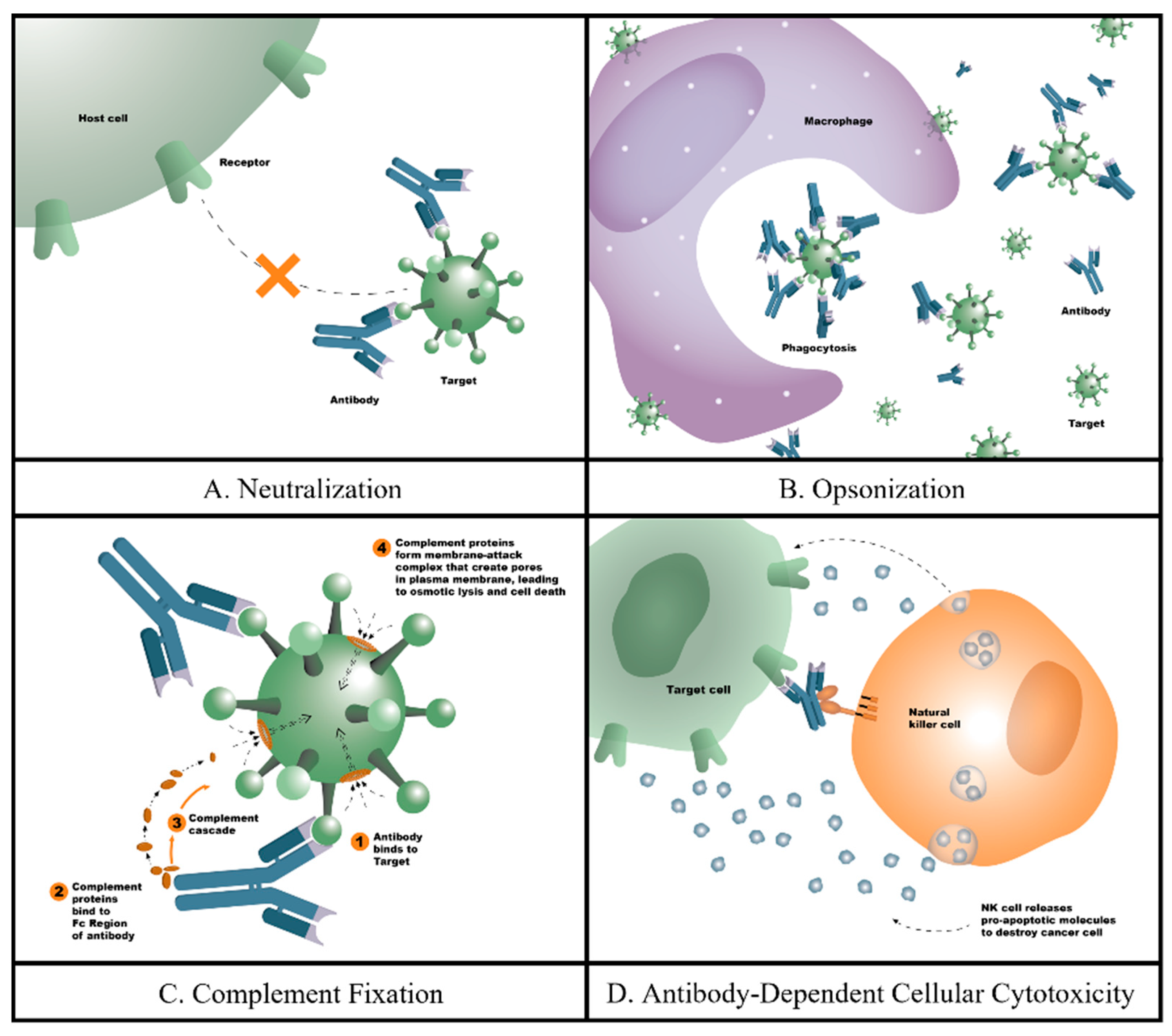
1. Introduction
2. mAbs and Inflammation
2.1. Pathophysiology Relevance
2.2. mAb and TBI Inflammatory Processes: FDA-Approved mAbs and Pre-Approval Testing Phases
2.3. Pre-Approval mAbs
2.3.1. High Mobility Group Box 1 (HMGB1)
2.3.2. Cluster of Differentiation 11/18 (CD11/CD18)
2.4. FDA-Approved mAbs
2.4.1. Interleukin 6 and Interleukin 6 Receptor (IL-6, IL-6R)
2.4.2. Interleukin-1β (IL-1β)
3. mAbs and Neurodegeneration
3.1. Pathophysiology Relevance
3.2. Pre-Approval mAbs
3.3. FDA-Approved mAbs
4. mAbs and Coagulopathy
4.1. Pathophysiology Relevance
4.2. FDA-Approved mAbs
5. mAbs and Vascular Function
5.1. Pathophysiology Relevance
5.2. FDA-Approved mAbs
6. mAbs and Excitotoxicity
Pre-Approval mAbs
7. mAbs and Oxidative Stress
Pre-Approval mAbs
8. mAbs and Safety
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018, 130, 1080–1097. [Google Scholar] [CrossRef] [PubMed]
- Swanson, T.M.; Isaacson, B.M.; Cyborski, C.M.; French, L.M.; Tsao, J.W.; Pasquina, P.F. Traumatic Brain Injury Incidence, Clinical Overview, and Policies in the US Military Health System Since 2000. Public Health Rep. 2017, 132, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Georges, A.; Joe, M.D. Traumatic Brain Injury. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Ng, S.Y.; Lee, A.Y.W. Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets. Front. Cell. Neurosci. 2019, 13, 528. [Google Scholar] [CrossRef]
- Narayan, R.K.; Michel, M.E.; Ansell, B.; Baethmann, A.; Biegon, A.; Bracken, M.B.; Bullock, M.R.; Choi, S.C.; Clifton, G.L.; Contant, C.F.; et al. Clinical trials in head injury. J. Neurotrauma 2002, 19, 503–557. [Google Scholar] [CrossRef]
- Loane, D.J.; Faden, A.I. Neuroprotection for traumatic brain injury: Translational challenges and emerging therapeutic strategies. Trends Pharmacol. Sci. 2010, 31, 596–604. [Google Scholar] [CrossRef]
- Maas, A.I.; Marmarou, A.; Murray, G.D.; Steyerberg, E.W. Clinical trials in traumatic brain injury: Current problems and future solutions. Acta Neurochir. Suppl. 2004, 89, 113–118. [Google Scholar] [CrossRef]
- Nichol, A.; French, C.; Little, L.; Haddad, S.; Presneill, J.; Arabi, Y.; Bailey, M.; Cooper, D.J.; Duranteau, J.; Huet, O.; et al. Erythropoietin in traumatic brain injury (EPO-TBI): A double-blind randomised controlled trial. Lancet 2015, 386, 2499–2506. [Google Scholar] [CrossRef]
- Peng, W.; Xing, Z.; Yang, J.; Wang, Y.; Wang, W.; Huang, W. The efficacy of erythropoietin in treating experimental traumatic brain injury: A systematic review of controlled trials in animal models. J. Neurosurg. 2014, 121, 653–664. [Google Scholar] [CrossRef]
- Leist, M.; Ghezzi, P.; Grasso, G.; Bianchi, R.; Villa, P.; Fratelli, M.; Savino, C.; Bianchi, M.; Nielsen, J.; Gerwien, J.; et al. Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science 2004, 305, 239–242. [Google Scholar] [CrossRef]
- Galgano, M.; Toshkezi, G.; Qiu, X.; Russell, T.; Chin, L.; Zhao, L.R. Traumatic Brain Injury: Current Treatment Strategies and Future Endeavors. Cell Transplant. 2017, 26, 1118–1130. [Google Scholar] [CrossRef]
- Peterson, A.B.; Zhou, H.; Thomas, K.E. Disparities in traumatic brain injury-related deaths-United States, 2020. J. Saf. Res. 2022, 83, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Greve, M.W.; Zink, B.J. Pathophysiology of traumatic brain injury. Mt. Sinai J. Med. 2009, 76, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Garattini, L.; Padula, A. Precision medicine and monoclonal antibodies: Breach of promise? Croat. Med. J. 2019, 60, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.L.; Gilliland, G.L. Engineering antibody therapeutics. Curr. Opin. Struct. Biol. 2016, 38, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Kato, C.; Kato, A. Therapeutic antibodies: Their mechanisms of action and the pathological findings they induce in toxicity studies. J. Toxicol. Pathol. 2015, 28, 133–139. [Google Scholar] [CrossRef]
- Buss, N.A.; Henderson, S.J.; McFarlane, M.; Shenton, J.M.; de Haan, L. Monoclonal antibody therapeutics: History and future. Curr. Opin. Pharmacol. 2012, 12, 615–622. [Google Scholar] [CrossRef]
- Cavallo, F.; Calogero, R.A.; Forni, G. Are oncoantigens suitable targets for anti-tumour therapy? Nat. Rev. Cancer 2007, 7, 707–713. [Google Scholar] [CrossRef]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Posner, J.; Barrington, P.; Brier, T.; Datta-Mannan, A. Monoclonal Antibodies: Past, Present and Future. In Concepts and Principles of Pharmacology; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 260, pp. 81–141. [Google Scholar] [CrossRef]
- Morrison, S.L.; Johnson, M.J.; Herzenberg, L.A.; Oi, V.T. Chimeric human antibody molecules: Mouse antigen-binding domains with human constant region domains. Proc. Natl. Acad. Sci. USA 1984, 81, 6851–6855. [Google Scholar] [CrossRef]
- Jones, P.T.; Dear, P.H.; Foote, J.; Neuberger, M.S.; Winter, G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 1986, 321, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. MAbs 2015, 7, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2019. MAbs 2019, 11, 219–238. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Watier, H.; Janice, M.R. Evolution of antibody therapeutics. In Protein Therapeutics; Vaughan, T., Osbourn, J., Jallal, B., Eds.; Wiley: Hoboken, NJ, USA, 2017. [Google Scholar]
- Wu, C.H.; Liu, I.J.; Lu, R.M.; Wu, H.C. Advancement and applications of peptide phage display technology in biomedical science. J. Biomed. Sci. 2016, 23, 8. [Google Scholar] [CrossRef]
- Grilo, A.L.; Mantalaris, A. The Increasingly Human and Profitable Monoclonal Antibody Market. Trends Biotechnol. 2019, 37, 9–16. [Google Scholar] [CrossRef]
- Okuma, Y.; Wake, H.; Teshigawara, K.; Takahashi, Y.; Hishikawa, T.; Yasuhara, T.; Mori, S.; Takahashi, H.K.; Date, I.; Nishibori, M. Anti-High Mobility Group Box 1 Antibody Therapy May Prevent Cognitive Dysfunction After Traumatic Brain Injury. World Neurosurg. 2019, 122, e864–e871. [Google Scholar] [CrossRef]
- Wang, D.; Liu, K.; Wake, H.; Teshigawara, K.; Mori, S.; Nishibori, M. Anti-high mobility group box-1 (HMGB1) antibody inhibits hemorrhage-induced brain injury and improved neurological deficits in rats. Sci. Rep. 2017, 7, 46243. [Google Scholar] [CrossRef]
- Bao, F.; Chen, Y.; Dekaban, G.A.; Weaver, L.C. An anti-CD11d integrin antibody reduces cyclooxygenase-2 expression and protein and DNA oxidation after spinal cord injury in rats. J. Neurochem. 2004, 90, 1194–1204. [Google Scholar] [CrossRef]
- Bao, F.; Chen, Y.; Dekaban, G.A.; Weaver, L.C. Early anti-inflammatory treatment reduces lipid peroxidation and protein nitration after spinal cord injury in rats. J. Neurochem. 2004, 88, 1335–1344. [Google Scholar] [CrossRef]
- Bao, F.; Dekaban, G.A.; Weaver, L.C. Anti-CD11d antibody treatment reduces free radical formation and cell death in the injured spinal cord of rats. J. Neurochem. 2005, 94, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Saville, L.R.; Pospisil, C.H.; Mawhinney, L.A.; Bao, F.; Simedrea, F.C.; Peters, A.A.; O’Connell, P.J.; Weaver, L.C.; Dekaban, G.A. A monoclonal antibody to CD11d reduces the inflammatory infiltrate into the injured spinal cord: A potential neuroprotective treatment. J. Neuroimmunol. 2004, 156, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Gris, D.; Marsh, D.R.; Oatway, M.A.; Chen, Y.; Hamilton, E.F.; Dekaban, G.A.; Weaver, L.C. Transient blockade of the CD11d/CD18 integrin reduces secondary damage after spinal cord injury, improving sensory, autonomic, and motor function. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 4043–4051. [Google Scholar] [CrossRef] [PubMed]
- Wallner, L.; Dai, J.; Escara-Wilke, J.; Zhang, J.; Yao, Z.; Lu, Y.; Trikha, M.; Nemeth, J.A.; Zaki, M.H.; Keller, E.T. Inhibition of interleukin-6 with CNTO328, an anti-interleukin-6 monoclonal antibody, inhibits conversion of androgen-dependent prostate cancer to an androgen-independent phenotype in orchiectomized mice. Cancer Res. 2006, 66, 3087–3095. [Google Scholar] [CrossRef]
- van Rhee, F.; Fayad, L.; Voorhees, P.; Furman, R.; Lonial, S.; Borghaei, H.; Sokol, L.; Crawford, J.; Cornfeld, M.; Qi, M.; et al. Siltuximab, a novel anti-interleukin-6 monoclonal antibody, for Castleman’s disease. J. Clin. Oncol. 2010, 28, 3701–3708. [Google Scholar] [CrossRef]
- Smith, D.E.; Hanna, R.; Della, F.; Moore, H.; Chen, H.; Farese, A.M.; MacVittie, T.J.; Virca, G.D.; Sims, J.E. The soluble form of IL-1 receptor accessory protein enhances the ability of soluble type II IL-1 receptor to inhibit IL-1 action. Immunity 2003, 18, 87–96. [Google Scholar] [CrossRef]
- Kondo, A.; Shahpasand, K.; Mannix, R.; Qiu, J.; Moncaster, J.; Chen, C.-H.; Yao, Y.; Lin, Y.-M.; Driver, J.A.; Sun, Y.; et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 2015, 523, 431–436. [Google Scholar] [CrossRef]
- Bravo, C.P.; Krukowski, K.; Barker, S.; Wang, C.; Li, Y.Q.; Fan, L.; Vázquez-Rosa, E.; Shin, M.K.; Wong, M.Y.; McCullough, L.D.; et al. Anti-acetylated-tau immunotherapy is neuroprotective in tauopathy and brain injury. Mol. Neurodegener. 2024, 19, 51. [Google Scholar] [CrossRef]
- Aducanumab Approved to Treat Alzheimer’s Disease; Alzforum: Cambridge, MA, USA, 2021.
- Sims, J.R.; Zimmer, J.A.; Evans, C.D.; Lu, M.; Ardayfio, P.; Sparks, J.; Wessels, A.M.; Shcherbinin, S.; Wang, H.; Monkul Nery, E.S.; et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA 2023, 330, 512–527. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Sabbagh, M.; Cohen, S. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 1631–1632. [Google Scholar] [CrossRef]
- Prins, N.D.; Scheltens, P. Treating Alzheimer’s disease with monoclonal antibodies: Current status and outlook for the future. Alzheimers Res. Ther. 2013, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.K.; Kuan, Y.C.; Lin, H.W.; Hu, C.J. Clinical trials of new drugs for Alzheimer disease: A 2020–2023 update. J. Biomed. Sci. 2023, 30, 83. [Google Scholar] [CrossRef] [PubMed]
- Flavell, L. Treatments that Target Amyloid-Beta 2020. Available online: https://alzheimersnewstoday.com/aducanumab/ (accessed on 15 December 2020).
- Zella, S.M.A.; Metzdorf, J.; Ciftci, E.; Ostendorf, F.; Muhlack, S.; Gold, R.; Tonges, L. Emerging Immunotherapies for Parkinson Disease. Neurol. Ther. 2019, 8, 29–44. [Google Scholar] [CrossRef] [PubMed]
- MEDI1341. In Alzforum; Alzforum: Cambridge, MA, USA, 2020.
- AstraZeneca. Multiple Ascending Dose Study of MEDI1341 in Patients with Parkinson’s Disease; United States National Library of Medicine at the National Institutes of Health: Bethesda, MD, USA, 2020.
- AbbVie. A Study to Evaluate the Safety and Tolerability of ABBV-0805 in Patients with Parkinson’s Disease; United States National Library of Medicine at the National Institutes of Health: Bethesda, MD, USA, 2020.
- Abciximab. Available online: https://www.drugs.com (accessed on 20 April 2010).
- FDA Approves First Therapy for the Treatment of Adult Patients with a Rare Blood Clotting Disorder; U.S. Food and Drug Administration (FDA): Silver Spring, MD, USA, 2019.
- Adakveo; Novartis Pharmaceuticals Corporation: East Hanover, NJ, USA, 2019.
- Drug Approval Package: Avastin (Bevacizum) NDA #125085. U.S. Food and Drug Administration (FDA): Silver Spring, MD, USA, 8 March 2005. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2004/STN-125085_Avastin.cfm (accessed on 8 April 2021).
- Drug Approval Package: Beovu (brolucizumab-dbll); U.S. Food and Drug Administration (FDA): Silver Spring, MD, USA, 2019.
- FDA Professional Drug Information on Cyramza. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/125477s002lbl.pdf (accessed on 20 November 2022).
- Pitt, D.; Werner, P.; Raine, C.S. Glutamate excitotoxicity in a model of multiple sclerosis. Nat. Med. 2000, 6, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Romanic, A.M.; White, R.F.; Arleth, A.J.; Ohlstein, E.H.; Barone, F.C. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: Inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke 1998, 29, 1020–1030. [Google Scholar] [CrossRef]
- Goffin, L.; Fagagnini, S.; Vicari, A.; Mamie, C.; Melhem, H.; Weder, B.; Lutz, C.; Lang, S.; Scharl, M.; Rogler, G.; et al. Anti-MMP-9 Antibody: A Promising Therapeutic Strategy for Treatment of Inflammatory Bowel Disease Complications with Fibrosis. Inflamm. Bowel Dis. 2016, 22, 2041–2057. [Google Scholar] [CrossRef]
- Shah, M.A.; Bodoky, G.; Starodub, A.; Cunningham, D.; Yip, D.; Wainberg, Z.A.; Bendell, J.; Thai, D.; He, J.; Bhargava, P.; et al. Phase III Study to Evaluate Efficacy and Safety of Andecaliximab with mFOLFOX6 as First-Line Treatment in Patients with Advanced Gastric or GEJ Adenocarcinoma (GAMMA-1). J. Clin. Oncol. 2021, 39, 990–1000. [Google Scholar] [CrossRef]
- Kalra, S.; Malik, R.; Singh, G.; Bhatia, S.; Al-Harrasi, A.; Mohan, S.; Albratty, M.; Albarrati, A.; Tambuwala, M.M. Pathogenesis and management of traumatic brain injury (TBI): Role of neuroinflammation and anti-inflammatory drugs. Inflammopharmacology 2022, 30, 1153–1166. [Google Scholar] [CrossRef]
- Morganti-Kossmann, M.C.; Satgunaseelan, L.; Bye, N.; Kossmann, T. Modulation of immune response by head injury. Injury 2007, 38, 1392–1400. [Google Scholar] [CrossRef]
- Corps, K.N.; Roth, T.L.; McGavern, D.B. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 2015, 72, 355–362. [Google Scholar] [CrossRef]
- Lull, M.E.; Block, M.L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.G.; Chen, S.D. The changing phenotype of microglia from homeostasis to disease. Transl. Neurodegener. 2012, 1, 9. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.V.; McGavern, D.B. Inflammatory neuroprotection following traumatic brain injury. Science 2016, 353, 783–785. [Google Scholar] [CrossRef] [PubMed]
- Ziebell, J.M.; Morganti-Kossmann, M.C. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 2010, 7, 22–30. [Google Scholar] [CrossRef]
- Lozano, D.; Gonzales-Portillo, G.S.; Acosta, S.; de la Pena, I.; Tajiri, N.; Kaneko, Y.; Borlongan, C.V. Neuroinflammatory responses to traumatic brain injury: Etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr. Dis. Treat. 2015, 11, 97–106. [Google Scholar] [CrossRef]
- Faden, A.I.; Wu, J.; Stoica, B.A.; Loane, D.J. Progressive inflammation-mediated neurodegeneration after traumatic brain or spinal cord injury. Br. J. Pharmacol. 2016, 173, 681–691. [Google Scholar] [CrossRef]
- Braun, M.; Vaibhav, K.; Saad, N.M.; Fatima, S.; Vender, J.R.; Baban, B.; Hoda, M.N.; Dhandapani, K.M. White matter damage after traumatic brain injury: A role for damage associated molecular patterns. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2614–2626. [Google Scholar] [CrossRef]
- Needham, E.J.; Helmy, A.; Zanier, E.R.; Jones, J.L.; Coles, A.J.; Menon, D.K. The immunological response to traumatic brain injury. J. Neuroimmunol. 2019, 332, 112–125. [Google Scholar] [CrossRef]
- Helmy, A.; De Simoni, M.G.; Guilfoyle, M.R.; Carpenter, K.L.; Hutchinson, P.J. Cytokines and innate inflammation in the pathogenesis of human traumatic brain injury. Prog. Neurobiol. 2011, 95, 352–372. [Google Scholar] [CrossRef]
- Chodobski, A.; Zink, B.J.; Szmydynger-Chodobska, J. Blood-brain barrier pathophysiology in traumatic brain injury. Transl. Stroke Res. 2011, 2, 492–516. [Google Scholar] [CrossRef]
- Corrigan, F.; Mander, K.A.; Leonard, A.V.; Vink, R. Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. J. Neuroinflamm. 2016, 13, 264. [Google Scholar] [CrossRef] [PubMed]
- Burda, J.E.; Bernstein, A.M.; Sofroniew, M.V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 2016, 275 Pt 3, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Michinaga, S.; Koyama, Y. Pathophysiological Responses and Roles of Astrocytes in Traumatic Brain Injury. Int. J. Mol. Sci. 2021, 22, 6418. [Google Scholar] [CrossRef] [PubMed]
- Okuma, Y.; Liu, K.; Wake, H.; Zhang, J.; Maruo, T.; Date, I.; Yoshino, T.; Ohtsuka, A.; Otani, N.; Tomura, S.; et al. Anti-high mobility group box-1 antibody therapy for traumatic brain injury. Ann. Neurol. 2012, 72, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Motoki, K.; Tagawa, K.; Chen, X.; Hama, H.; Nakajima, K.; Homma, H.; Tamura, T.; Watanabe, H.; Katsuno, M.; et al. HMGB1, a pathogenic molecule that induces neurite degeneration via TLR4-MARCKS, is a potential therapeutic target for Alzheimer’s disease. Sci. Rep. 2016, 6, 31895. [Google Scholar] [CrossRef]
- Sasaki, T.; Liu, K.; Agari, T.; Yasuhara, T.; Morimoto, J.; Okazaki, M.; Takeuchi, H.; Toyoshima, A.; Sasada, S.; Shinko, A.; et al. Anti-high mobility group box 1 antibody exerts neuroprotection in a rat model of Parkinson’s disease. Exp. Neurol. 2016, 275 Pt 1, 220–231. [Google Scholar] [CrossRef]
- Haruma, J.; Teshigawara, K.; Hishikawa, T.; Wang, D.; Liu, K.; Wake, H.; Mori, S.; Takahashi, H.K.; Sugiu, K.; Date, I.; et al. Anti-high mobility group box-1 (HMGB1) antibody attenuates delayed cerebral vasospasm and brain injury after subarachnoid hemorrhage in rats. Sci. Rep. 2016, 6, 37755. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Shaikh, M.F.; Chakraborti, A.; Kumari, Y.; Aledo-Serrano, Á.; Aleksovska, K.; Alvim, M.K.M.; Othman, I. HMGB1: A Common Biomarker and Potential Target for TBI, Neuroinflammation, Epilepsy, and Cognitive Dysfunction. Front. Neurosci. 2018, 12, 628. [Google Scholar] [CrossRef]
- Gao, T.L.; Yuan, X.T.; Yang, D.; Dai, H.L.; Wang, W.J.; Peng, X.; Shao, H.J.; Jin, Z.F.; Fu, Z.J. Expression of HMGB1 and RAGE in rat and human brains after traumatic brain injury. J. Trauma. Acute Care Surg. 2012, 72, 643–649. [Google Scholar] [CrossRef]
- Bao, F.; Shultz, S.R.; Hepburn, J.D.; Omana, V.; Weaver, L.C.; Cain, D.P.; Brown, A. A CD11d monoclonal antibody treatment reduces tissue injury and improves neurological outcome after fluid percussion brain injury in rats. J. Neurotrauma 2012, 29, 2375–2392. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Muller-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998, 334 Pt 2, 297–314. [Google Scholar] [CrossRef]
- Narazaki, M.; Tanaka, T.; Kishimoto, T. The role and therapeutic targeting of IL-6 in rheumatoid arthritis. Expert Rev. Clin. Immunol. 2017, 13, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C.; Lamacchia, C.; Palmer, G. IL-1 pathways in inflammation and human diseases. Nat. Rev. Rheumatol. 2010, 6, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Dhimolea, E. Canakinumab. MAbs 2010, 2, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Nordstrom, A.; Nordstrom, P. Traumatic brain injury and the risk of dementia diagnosis: A nationwide cohort study. PLoS Med. 2018, 15, e1002496. [Google Scholar] [CrossRef]
- Jafari, S.; Etminan, M.; Aminzadeh, F.; Samii, A. Head injury and risk of Parkinson disease: A systematic review and meta-analysis. Mov. Disord. 2013, 28, 1222–1229. [Google Scholar] [CrossRef]
- Crane, P.K.; Gibbons, L.E.; Dams-O’Connor, K.; Trittschuh, E.; Leverenz, J.B.; Keene, C.D.; Sonnen, J.; Montine, T.J.; Bennett, D.A.; Leurgans, S.; et al. Association of Traumatic Brain Injury with Late-Life Neurodegenerative Conditions and Neuropathologic Findings. JAMA Neurol. 2016, 73, 1062–1069. [Google Scholar] [CrossRef]
- Gardner, R.C.; Byers, A.L.; Barnes, D.E.; Li, Y.; Boscardin, J.; Yaffe, K. Mild TBI and risk of Parkinson disease: A Chronic Effects of Neurotrauma Consortium Study. Neurology 2018, 90, e1771–e1779. [Google Scholar] [CrossRef]
- Graham, N.S.; Sharp, D.J. Understanding neurodegeneration after traumatic brain injury: From mechanisms to clinical trials in dementia. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1221–1233. [Google Scholar] [CrossRef]
- Christman, C.W.; Grady, M.S.; Walker, S.A.; Holloway, K.L.; Povlishock, J.T. Ultrastructural studies of diffuse axonal injury in humans. J. Neurotrauma 1994, 11, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Giza, C.C.; Hovda, D.A. The new neurometabolic cascade of concussion. Neurosurgery 2014, 75 (Suppl. S4), S24–S33. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.E.; Stewart, J.E.; Begbie, F.D.; Trojanowski, J.Q.; Smith, D.H.; Stewart, W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 2013, 136, 28–42. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, S.T.; Asken, B.M. Injury cascades in TBI-related neurodegeneration. Brain Inj. 2017, 31, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Knauer, M.F.; Soreghan, B.; Burdick, D.; Kosmoski, J.; Glabe, C.G. Intracellular accumulation and resistance to degradation of the Alzheimer amyloid A4/beta protein. Proc. Natl. Acad. Sci. USA 1992, 89, 7437–7441. [Google Scholar] [CrossRef]
- Nicolas, G.; Wallon, D.; Goupil, C.; Richard, A.C.; Pottier, C.; Dorval, V.; Sarov-Riviere, M.; Riant, F.; Herve, D.; Amouyel, P.; et al. Mutation in the 3’untranslated region of APP as a genetic determinant of cerebral amyloid angiopathy. Eur. J. Hum. Genet. 2016, 24, 92–98. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 77sr71. [Google Scholar] [CrossRef]
- Trojanowski, J.Q.; Goedert, M.; Iwatsubo, T.; Lee, V.M. Fatal attractions: Abnormal protein aggregation and neuron death in Parkinson’s disease and Lewy body dementia. Cell Death Differ. 1998, 5, 832–837. [Google Scholar] [CrossRef]
- Kellie, J.F.; Higgs, R.E.; Ryder, J.W.; Major, A.; Beach, T.G.; Adler, C.H.; Merchant, K.; Knierman, M.D. Quantitative measurement of intact alpha-synuclein proteoforms from post-mortem control and Parkinson’s disease brain tissue by intact protein mass spectrometry. Sci. Rep. 2014, 4, 5797. [Google Scholar] [CrossRef]
- Yasuda, T.; Nakata, Y.; Mochizuki, H. alpha-Synuclein and neuronal cell death. Mol. Neurobiol. 2013, 47, 466–483. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, S.M.; Nash, M.J.; Sweeting, C.J.; Graham, D.I.; Roberts, G.W. Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci. Lett. 1993, 160, 139–144. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Daneshvar, D.H. The neuropathology of traumatic brain injury. In Handbook of Clinical Neurology; Elsevier B.V.: Amsterdam, The Netherlands, 2015; Volume 127, pp. 45–66. [Google Scholar] [CrossRef]
- Goldstein, L.E.; Fisher, A.M.; Tagge, C.A.; Zhang, X.L.; Velisek, L.; Sullivan, J.A.; Upreti, C.; Kracht, J.M.; Ericsson, M.; Wojnarowicz, M.W.; et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med. 2012, 4, 134ra160. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. 2012, 22, 142–149. [Google Scholar] [CrossRef]
- Tavakoli, Z.; Jahandar, H.; Shahpasand, K.; Zaeifi, D.; Mousavi, S.E. Targeting cis-p-tau and neuro-related gene expression in traumatic brain injury: Therapeutic insights from TC-DAPK6 treatment in mice. Mol. Biol. Rep. 2024, 51, 1010. [Google Scholar] [CrossRef]
- Mietelska-Porowska, A.; Wasik, U.; Goras, M.; Filipek, A.; Niewiadomska, G. Tau protein modifications and interactions: Their role in function and dysfunction. Int. J. Mol. Sci. 2014, 15, 4671–4713. [Google Scholar] [CrossRef]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef]
- Sotiropoulos, I.; Galas, M.C.; Silva, J.M.; Skoulakis, E.; Wegmann, S.; Maina, M.B.; Blum, D.; Sayas, C.L.; Mandelkow, E.M.; Mandelkow, E.; et al. Atypical, non-standard functions of the microtubule associated Tau protein. Acta Neuropathol. Commun. 2017, 5, 91. [Google Scholar] [CrossRef]
- Delic, V.; Beck, K.D.; Pang, K.C.H.; Citron, B.A. Biological links between traumatic brain injury and Parkinson’s disease. Acta Neuropathol. Commun. 2020, 8, 45. [Google Scholar] [CrossRef]
- Acosta, S.A.; Tajiri, N.; de la Pena, I.; Bastawrous, M.; Sanberg, P.R.; Kaneko, Y.; Borlongan, C.V. Alpha-synuclein as a pathological link between chronic traumatic brain injury and Parkinson’s disease. J. Cell. Physiol. 2015, 230, 1024–1032. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Axonal pathology in traumatic brain injury. Exp. Neurol. 2013, 246, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Biogen. 221AD301 Phase 3 Study of Aducanumab (BIIB037) in Early Alzheimer’s Disease (ENGAGE); United States National Library of Medicine at the National Institutes of Health: Bethesda, MD, USA, 2020.
- Biogen. 221AD302 Phase 3 Study of Aducanumab (BIIB037) in Early Alzheimer’s Disease (EMERGE); United States National Library of Medicine at the National Institutes of Health: Bethesda, MD, USA, 2020.
- Maegele, M.; Schochl, H.; Menovsky, T.; Marechal, H.; Marklund, N.; Buki, A.; Stanworth, S. Coagulopathy and haemorrhagic progression in traumatic brain injury: Advances in mechanisms, diagnosis, and management. Lancet Neurol. 2017, 16, 630–647. [Google Scholar] [CrossRef] [PubMed]
- Maegele, M. Coagulopathy after traumatic brain injury: Incidence, pathogenesis, and treatment options. Transfusion 2013, 53 (Suppl. S1), 28S–37S. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, F.; Dong, J.F. Coagulopathy induced by traumatic brain injury: Systemic manifestation of a localized injury. Blood 2018, 131, 2001–2006. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Wolburg, H.; Noell, S.; Mack, A.; Wolburg-Buchholz, K.; Fallier-Becker, P. Brain endothelial cells and the glio-vascular complex. Cell Tissue Res. 2009, 335, 75–96. [Google Scholar] [CrossRef]
- Lassmann, H.; Zimprich, F.; Vass, K.; Hickey, W.F. Microglial cells are a component of the perivascular glia limitans. J. Neurosci. Res. 1991, 28, 236–243. [Google Scholar] [CrossRef]
- Cash, A.; Theus, M.H. Mechanisms of Blood-Brain Barrier Dysfunction in Traumatic Brain Injury. Int. J. Mol. Sci. 2020, 21, 3344. [Google Scholar] [CrossRef]
- Kaur, P.; Sharma, S. Recent Advances in Pathophysiology of Traumatic Brain Injury. Curr. Neuropharmacol. 2018, 16, 1224–1238. [Google Scholar] [CrossRef]
- Werner, C.; Engelhard, K. Pathophysiology of traumatic brain injury. Br. J. Anaesth. 2007, 99, 4–9. [Google Scholar] [CrossRef]
- Jiang, X.; Andjelkovic, A.V.; Zhu, L.; Yang, T.; Bennett, M.V.L.; Chen, J.; Keep, R.F.; Shi, Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 2018, 163–164, 144–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Doerschuk, C.M. The signaling pathways induced by neutrophil-endothelial cell adhesion. Antioxid. Redox Signal 2002, 4, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Furie, B.; Furie, B.C. The molecular basis of platelet and endothelial cell interaction with neutrophils and monocytes: Role of P-selectin and the P-selectin ligand, PSGL-1. Thromb. Haemost. 1995, 74, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jiang, R.; Liu, L.; Watkins, T.; Zhang, F.; Dong, J.F. Traumatic brain injury-associated coagulopathy. J. Neurotrauma 2012, 29, 2597–2605. [Google Scholar] [CrossRef] [PubMed]
- Chodobski, A.; Chung, I.; Kozniewska, E.; Ivanenko, T.; Chang, W.; Harrington, J.F.; Duncan, J.A.; Szmydynger-Chodobska, J. Early neutrophilic expression of vascular endothelial growth factor after traumatic brain injury. Neuroscience 2003, 122, 853–867. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Olsson, A.K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling—In control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef]
- Vogel, C.; Bauer, A.; Wiesnet, M.; Preissner, K.T.; Schaper, W.; Marti, H.H.; Fischer, S. Flt-1, but not Flk-1 mediates hyperpermeability through activation of the PI3-K/Akt pathway. J. Cell. Physiol. 2007, 212, 236–243. [Google Scholar] [CrossRef]
- Argaw, A.T.; Gurfein, B.T.; Zhang, Y.; Zameer, A.; John, G.R. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc. Natl. Acad. Sci. USA 2009, 106, 1977–1982. [Google Scholar] [CrossRef]
- Wang, W.; Dentler, W.L.; Borchardt, R.T. VEGF increases BMEC monolayer permeability by affecting occludin expression and tight junction assembly. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H434–H440. [Google Scholar] [CrossRef]
- Murakami, T.; Felinski, E.A.; Antonetti, D.A. Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J. Biol. Chem. 2009, 284, 21036–21046. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.J.; Baker, T.L.; Wilson, C.T.; Brady, R.D.; Yamakawa, G.R.; Wright, D.K.; Mychasiuk, M.; Vo, A.; Wilson, T.; Allen, J.; et al. Treatment with the vascular endothelial growth factor-A antibody, bevacizumab, has sex-specific effects in a rat model of mild traumatic brain injury. J. Cereb. Blood Flow Metab. 2024, 44, 542–555. [Google Scholar] [CrossRef] [PubMed]
- Sims, N.R.; Zaidan, E. Biochemical changes associated with selective neuronal death following short-term cerebral ischaemia. Int. J. Biochem. Cell Biol. 1995, 27, 531–550. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.-H.; Hazell, A.S. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem. Int. 2006, 48, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W. Glutamate, a neurotoxic transmitter. J. Child Neurol. 1989, 4, 218–226. [Google Scholar] [CrossRef]
- Nicole, O.; Docagne, F.; Ali, C.; Margaill, I.; Carmeliet, P.; MacKenzie, E.T.; Vivien, D.; Buisson, A. The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat. Med. 2001, 7, 59–64. [Google Scholar] [CrossRef]
- Macrez, R.; Ortega, M.C.; Bardou, I.; Mehra, A.; Fournier, A.; Van der Pol, S.M.A.; Haelewyn, B.; Maubert, E.; Lesept, F.; Chevilley, A.; et al. Neuroendothelial NMDA receptors as therapeutic targets in experimental autoimmune encephalomyelitis. Brain 2016, 139, 2406–2419. [Google Scholar] [CrossRef]
- Macrez, R.; Obiang, P.; Gauberti, M.; Roussel, B.; Baron, A.; Parcq, J.; Cassé, F.; Hommet, Y.; Orset, C.; Agin, V.; et al. Antibodies preventing the interaction of tissue-type plasminogen activator with N-methyl-D-aspartate receptors reduce stroke damages and extend the therapeutic window of thrombolysis. Stroke 2011, 42, 2315–2322. [Google Scholar] [CrossRef]
- Rodriguez-Rodriguez, A.; Egea-Guerrero, J.J.; Murillo-Cabezas, F.; Carrillo-Vico, A. Oxidative stress in traumatic brain injury. Curr. Med. Chem. 2014, 21, 1201–1211. [Google Scholar] [CrossRef]
- Khatri, N.; Thakur, M.; Pareek, V.; Kumar, S.; Sharma, S.; Datusalia, A.K. Oxidative Stress: Major Threat in Traumatic Brain Injury. CNS Neurol. Disord. Drug Targets 2018, 17, 689–695. [Google Scholar] [CrossRef]
- Lewen, A.; Matz, P.; Chan, P.H. Free radical pathways in CNS injury. J. Neurotrauma 2000, 17, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, G.A.; Estrada, E.Y.; Dencoff, J.E. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke 1998, 29, 2189–2195. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Estrada, E.Y.; Thompson, J.F.; Liu, W.; Rosenberg, G.A. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J. Cereb. Blood Flow Metab. 2007, 27, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Gasche, Y.; Copin, J.C.; Sugawara, T.; Fujimura, M.; Chan, P.H. Matrix metalloproteinase inhibition prevents oxidative stress-associated blood-brain barrier disruption after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2001, 21, 1393–1400. [Google Scholar] [CrossRef]
- Cucullo, L.; Couraud, P.O.; Weksler, B.; Romero, I.A.; Hossain, M.; Rapp, E.; Janigro, D. Immortalized human brain endothelial cells and flow-based vascular modeling: A marriage of convenience for rational neurovascular studies. J. Cereb. Blood Flow Metab. 2008, 28, 312–328. [Google Scholar] [CrossRef]
- Haorah, J.; Ramirez, S.H.; Schall, K.; Smith, D.; Pandya, R.; Persidsky, Y. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. J. Neurochem. 2007, 101, 566–576. [Google Scholar] [CrossRef]
- Deem, T.L.; Cook-Mills, J.M. Vascular cell adhesion molecule 1 (VCAM-1) activation of endothelial cell matrix metalloproteinases: Role of reactive oxygen species. Blood 2004, 104, 2385–2393. [Google Scholar] [CrossRef]
- Alexander, J.S.; Elrod, J.W. Extracellular matrix, junctional integrity and matrix metalloproteinase interactions in endothelial permeability regulation. J. Anat. 2002, 200, 561–574. [Google Scholar] [CrossRef]
- Hansel, T.T.; Kropshofer, H.; Singer, T.; Mitchell, J.A.; George, A.J. The safety and side effects of monoclonal antibodies. Nat. Rev. Drug Discov. 2010, 9, 325–338. [Google Scholar] [CrossRef]
- Presta, L.G. Engineering of therapeutic antibodies to minimize immunogenicity and optimize function. Adv. Drug Deliv. Rev. 2006, 58, 640–656. [Google Scholar] [CrossRef]
- Boucher, B.A.; Hanes, S.D. Pharmacokinetic alterations after severe head injury. Clinical relevance. Clin. Pharmacokinet. 1998, 35, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Udy, A.; Boots, R.; Senthuran, S.; Stuart, J.; Deans, R.; Lassig-Smith, M.; Lipman, J. Augmented creatinine clearance in traumatic brain injury. Anesth. Analg. 2010, 111, 1505–1510. [Google Scholar] [CrossRef] [PubMed]
- Singh Gautam, A.; Pandey, S.K.; Lasure, V.; Dubey, S.; Singh, R.K. Monoclonal antibodies for the management of central nervous system diseases: Clinical success and future strategies. Expert Opin. Biol. Ther. 2023, 23, 603–618. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| mAb | Targeted Secondary Injury Mechanism | Stage of Development | Effects/Indications | Reference |
|---|---|---|---|---|
| HMGB1 | Inflammation | Pre-clinical | Reduce microglial activation and neuronal death | [30,31] |
| CD11/CD18 | Inflammation | Pre-clinical | Reduce brain edema and microglial activation | [32,33,34,35,36] |
| Tocilizumab, Sarilumab Siltuximab | Inflammation (IL-6/IL-6R) | FDA-approved | Rheumatoid arthritis Castleman’s disease; ovarian, prostate, and lung cancers Multiple myeloma | [19] [37,38] |
| Canakinumab | Inflammation (IL-1β) | FDA-approved | Cryopyrin-associated disorders | [39] |
| Cis-P-tau | Neurodegeneration (P-tau) | Pre-clinical | Block cis-P-tau pathology and restore neuronal dysfunction | [40] |
| Acetylated-tau | Neurodegeneration (acetylated-tau) | Pre-clinical | Reduce tau pathology and glia activation Improve neurobehavioral impairment | [41] |
| Aducanumab Leqembi Donanemab | Neurodegeneration (amyloid-beta) | FDA-approved | Alzheimer’s disease | [42] [43] [44] |
| Remternetug | Neurodegeneration (amyloid-beta) | Phase III (Clinical Trial #:NCT05463731) | Alzheimer’s disease | [45,46,47] |
| α-synuclein | Neurodegeneration | Pre-clinical | Parkinson’s disease | [48,49,50,51] |
| Abciximab | Coagulopathy (glycoprotein IIb/IIIa) | FDA-approved | Coronary artery procedures | [52] |
| Caplacizumab | Coagulopathy (glycoprotein IIb/IIIa) | FDA-approved | Thrombotic thrombocytopenic purpura | [53] |
| Crizanlizuman | Vascular function (P-selectin) | FDA-approved | Sickle cell disease | [54] |
| Bevacizumab Brolucizumab | Vascular function (VEGF-A) | FDA-approved | Colorectal cancer Macular edema | [55] [56] |
| Ramucirumab | Vascular function (VEGFR) | FDA-approved | Gastric cancer | [57] |
| NMDA receptor | Excitotoxicity | Pre-clinical | Multiple sclerosis | [58] |
| MMP-9 | Oxidative stress | Pre-clinical | Reduce fibrosis, oxidative stress, and BBB dysfunction | [59,60] |
| Andecaliximab | Oxidative stress (MMP-9) | Phase III (Clinical Trial #: NCT02545504) | Gastric cancer | [61] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, P.; Okada-Rising, S.; Scultetus, A.H.; Bailey, Z.S. The Relevance and Implications of Monoclonal Antibody Therapies on Traumatic Brain Injury Pathologies. Biomedicines 2024, 12, 2698. https://doi.org/10.3390/biomedicines12122698
Wang P, Okada-Rising S, Scultetus AH, Bailey ZS. The Relevance and Implications of Monoclonal Antibody Therapies on Traumatic Brain Injury Pathologies. Biomedicines. 2024; 12(12):2698. https://doi.org/10.3390/biomedicines12122698
Chicago/Turabian StyleWang, Ping, Starlyn Okada-Rising, Anke H. Scultetus, and Zachary S. Bailey. 2024. "The Relevance and Implications of Monoclonal Antibody Therapies on Traumatic Brain Injury Pathologies" Biomedicines 12, no. 12: 2698. https://doi.org/10.3390/biomedicines12122698
APA StyleWang, P., Okada-Rising, S., Scultetus, A. H., & Bailey, Z. S. (2024). The Relevance and Implications of Monoclonal Antibody Therapies on Traumatic Brain Injury Pathologies. Biomedicines, 12(12), 2698. https://doi.org/10.3390/biomedicines12122698