

Hypertrophic Cardiomyopathy with Special Focus on Mavacamten and Its Future in Cardiology
 , , , ,
, , , ,  ,
, 
Abstract
1. Introduction
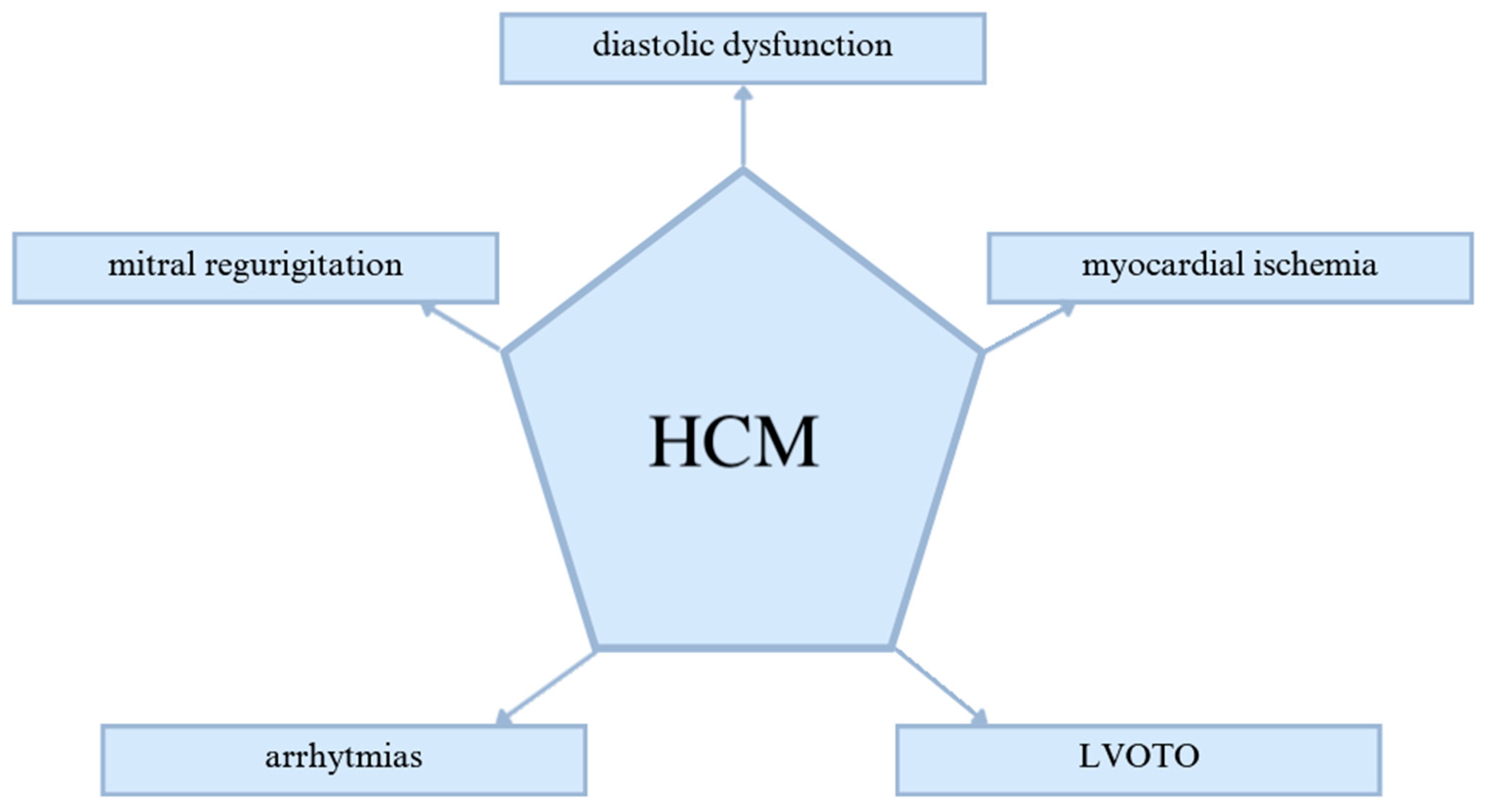
2. Pathogenesis of HCM
3. Epidemiology and Risk Factors
4. Diagnosis and Treatment
4.1. Advancements in Imaging Techniques
4.2. Genetic Testing
4.3. Biomarkers
4.4. Artificial Intelligence and Machine Learning
4.5. Multimodal Diagnostic Approach
4.6. Differential Diagnosis and Advanced Imaging Techniques
4.7. Contemporary Treatment Strategies
4.8. Challenges in Diagnosing and Managing HCM
4.9. Future Directions and Emerging Technologies

| Modality | Diagnostic Role in HCM | Strengths | Limitations |
|---|---|---|---|
| 3D Echocardiography | Measures LV wall thickness and geometry with high precision; essential for initial assessment of LV hypertrophy [61,62]. | Real-time imaging, optimal for initial hypertrophy assessment. | Limited spatial resolution, less effective for fibrosis detection. |
| Cardiac MRI (CMR) | Superior for detecting myocardial fibrosis and assessing LVOTO; CMR LGE patterns assist in stratifying risk of arrhythmia and SCD. | High spatial resolution, valuable for high-risk assessment with fibrosis quantification [64]. | Costly, requires specialized interpretation, limited access. |
| Positron Emission Tomography (PET) | Assesses metabolic and inflammatory activity in myocardium; differentiates HCM from infiltrative cardiomyopathies, such as amyloidosis. | Useful for assessing fibrosis extent and glucose metabolism in myocardium [86,87]. | High cost, limited availability, radiation exposure. |
| AI-Enhanced Echocardiography | Supports enhanced data analysis, identifies undiagnosed conditions (e.g., diabetes as a co-factor in HCM). | Increased diagnostic accuracy and disease progression prediction through large-scale data processing [78,79]. | Requires significant computational resources, ethical considerations. |
| Multimodal Approach | Combines echocardiography, CMR, genetic, and biomarker assessment to provide a comprehensive diagnosis. | Comprehensive and tailored diagnostic profile, crucial in ambiguous cases with strong family history [82,83]. | High cost, complex integration of data. |
5. Mavacamten—Characteristics and Scientific Studies
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
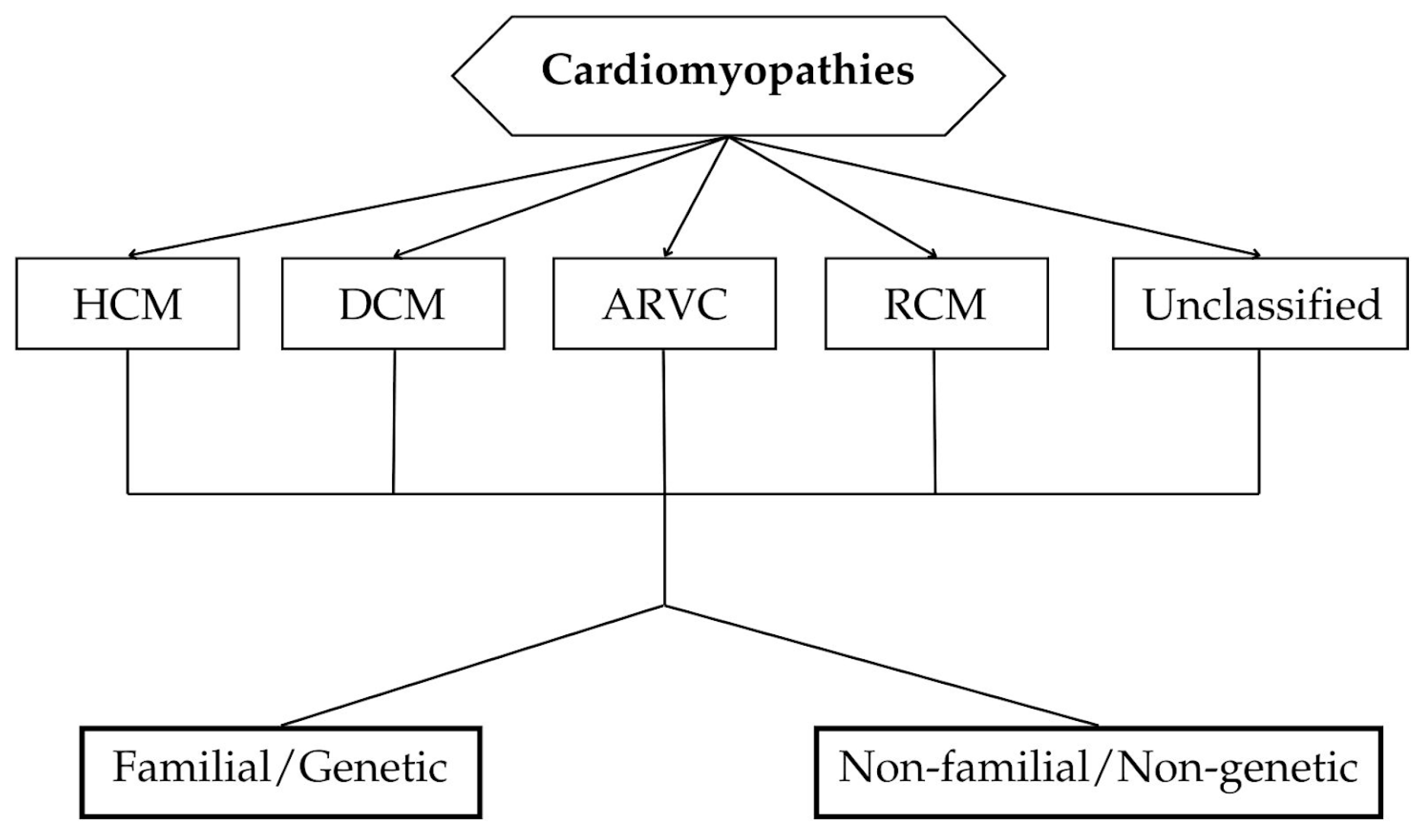
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B.; American Heart, A.; et al. Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006, 113, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- McKenna, W.J.; Maron, B.J.; Thiene, G. Classification, Epidemiology, and Global Burden of Cardiomyopathies. Circ. Res. 2017, 121, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kühl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Arbustini, E.; Narula, N.; Tavazzi, L.; Serio, A.; Grasso, M.; Favalli, V.; Bellazzi, R.; Tajik, J.A.; Bonow, R.O.; Fuster, V.; et al. The MOGE(S) classification of cardiomyopathy for clinicians. J. Am. Coll. Cardiol. 2014, 64, 304–318. [Google Scholar] [CrossRef] [PubMed]
- Geske, J.B.; Ommen, S.R.; Gersh, B.J. Hypertrophic Cardiomyopathy: Clinical Update. JACC Heart Fail. 2018, 6, 364–375. [Google Scholar] [CrossRef]
- Mazur, M.; Braksator, W.; Popjes, E. Hypertrophic Cardiomyopathy: From Medical Treatment to Advanced Heart Failure Therapies. Curr. Cardiol. Rep. 2024, 26, 985–994. [Google Scholar] [CrossRef]
- Maron, B.J.; Maron, M.S.; Semsarian, C. Genetics of hypertrophic cardiomyopathy after 20 years: Clinical perspectives. J. Am. Coll. Cardiol. 2012, 60, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Charron, P.; Richard, P.; Girolami, F.; Van Spaendonck-Zwarts, K.Y.; Pinto, Y. Genetic advances in sarcomeric cardiomyopathies: State of the art. Cardiovasc. Res. 2015, 105, 397–408. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Thierfelder, L.; Watkins, H.; MacRae, C.; Lamas, R.; McKenna, W.; Vosberg, H.P.; Seidman, J.G.; Seidman, C.E. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: A disease of the sarcomere. Cell 1994, 77, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Van Driest, S.L.; Ellsworth, E.G.; Ommen, S.R.; Tajik, A.J.; Gersh, B.J.; Ackerman, M.J. Prevalence and spectrum of thin filament mutations in an outpatient referral population with hypertrophic cardiomyopathy. Circulation 2003, 108, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Coppini, R.; Ho, C.Y.; Ashley, E.; Day, S.; Ferrantini, C.; Girolami, F.; Tomberli, B.; Bardi, S.; Torricelli, F.; Cecchi, F.; et al. Clinical phenotype and outcome of hypertrophic cardiomyopathy associated with thin-filament gene mutations. J. Am. Coll. Cardiol. 2014, 64, 2589–2600. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Van Driest, S.L.; Vasile, V.C.; Ommen, S.R.; Will, M.L.; Tajik, A.J.; Gersh, B.J.; Ackerman, M.J. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 1903–1910. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Witjas-Paalberends, E.R.; Piroddi, N.; Stam, K.; van Dijk, S.J.; Oliviera, V.S.; Ferrara, C.; Scellini, B.; Hazebroek, M.; ten Cate, F.J.; van Slegtenhorst, M.; et al. Mutations in MYH7 reduce the force generating capacity of sarcomeres in human familial hypertrophic cardiomyopathy. Cardiovasc. Res. 2013, 99, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Witjas-Paalberends, E.R.; Güçlü, A.; Germans, T.; Knaapen, P.; Harms, H.J.; Vermeer, A.M.; Christiaans, I.; Wilde, A.A.; Dos Remedios, C.; Lammertsma, A.A.; et al. Gene-specific increase in the energetic cost of contraction in hypertrophic cardiomyopathy caused by thick filament mutations. Cardiovasc. Res. 2014, 103, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Crilley, J.G.; Boehm, E.A.; Blair, E.; Rajagopalan, B.; Blamire, A.M.; Styles, P.; McKenna, W.J.; Ostman-Smith, I.; Clarke, K.; Watkins, H. Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J. Am. Coll. Cardiol. 2003, 41, 1776–1782. [Google Scholar] [CrossRef] [PubMed]
- Sedaghat-Hamedani, F.; Kayvanpour, E.; Tugrul, O.F.; Lai, A.; Amr, A.; Haas, J.; Proctor, T.; Ehlermann, P.; Jensen, K.; Katus, H.A.; et al. Clinical outcomes associated with sarcomere mutations in hypertrophic cardiomyopathy: A meta-analysis on 7675 individuals. Clin. Res. Cardiol. 2018, 107, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Li, R.K.; Li, G.; Mickle, D.A.; Weisel, R.D.; Merante, F.; Luss, H.; Rao, V.; Christakis, G.T.; Williams, W.G. Overexpression of transforming growth factor-beta1 and insulin-like growth factor-I in patients with idiopathic hypertrophic cardiomyopathy. Circulation 1997, 96, 874–881. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Lee, A.S.; Liang, P.; Sanchez-Freire, V.; Nguyen, P.K.; Wang, L.; Han, L.; Yen, M.; Wang, Y.; Sun, N.; et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell 2013, 12, 101–113. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Marian, A.J. Pathogenesis of diverse clinical and pathological phenotypes in hypertrophic cardiomyopathy. Lancet 2000, 355, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Jan, M.F.; Todaro, M.C.; Oreto, L.; Tajik, A.J. Apical hypertrophic cardiomyopathy: Present status. Int. J. Cardiol. 2016, 222, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Jan, M.F.; Tajik, A.J. Modern Imaging Techniques in Cardiomyopathies. Circ. Res. 2017, 121, 874–891. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Dhillon, A.; Popovic, Z.B.; Smedira, N.G.; Rizzo, J.; Thamilarasan, M.; Agler, D.; Lytle, B.W.; Lever, H.M.; Desai, M.Y. Left Ventricular Outflow Tract Obstruction in Hypertrophic Cardiomyopathy Patients Without Severe Septal Hypertrophy: Implications of Mitral Valve and Papillary Muscle Abnormalities Assessed Using Cardiac Magnetic Resonance and Echocardiography. Circ. Cardiovasc. Imaging 2015, 8, e003132. [Google Scholar] [CrossRef] [PubMed]
- Maron, M.S.; Olivotto, I.; Zenovich, A.G.; Link, M.S.; Pandian, N.G.; Kuvin, J.T.; Nistri, S.; Cecchi, F.; Udelson, J.E.; Maron, B.J. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006, 114, 2232–2239. [Google Scholar] [CrossRef] [PubMed]
- Maron, M.S.; Olivotto, I.; Betocchi, S.; Casey, S.A.; Lesser, J.R.; Losi, M.A.; Cecchi, F.; Maron, B.J. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N. Engl. J. Med. 2003, 348, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Hang, D.; Schaff, H.V.; Nishimura, R.A.; Lahr, B.D.; Abel, M.D.; Dearani, J.A.; Ommen, S.R. Accuracy of Jet Direction on Doppler Echocardiography in Identifying the Etiology of Mitral Regurgitation in Obstructive Hypertrophic Cardiomyopathy. J. Am. Soc. Echocardiogr. 2019, 32, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Spudich, J.A. Three perspectives on the molecular basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Pflug. Arch. 2019, 471, 701–717. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tardiff, J.C.; Carrier, L.; Bers, D.M.; Poggesi, C.; Ferrantini, C.; Coppini, R.; Maier, L.S.; Ashrafian, H.; Huke, S.; van der Velden, J. Targets for therapy in sarcomeric cardiomyopathies. Cardiovasc. Res. 2015, 105, 457–470. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Coppini, R.; Ferrantini, C.; Yao, L.; Fan, P.; Del Lungo, M.; Stillitano, F.; Sartiani, L.; Tosi, B.; Suffredini, S.; Tesi, C.; et al. Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation 2013, 127, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Kuppahally, S.S.; Akoum, N.; Burgon, N.S.; Badger, T.J.; Kholmovski, E.G.; Vijayakumar, S.; Rao, S.N.; Blauer, J.; Fish, E.N.; Dibella, E.V.; et al. Left atrial strain and strain rate in patients with paroxysmal and persistent atrial fibrillation: Relationship to left atrial structural remodeling detected by delayed-enhancement MRI. Circ. Cardiovasc. Imaging 2010, 3, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Park, K.M.; Im, S.I.; Kim, E.K.; Lee, S.C.; Park, S.J.; Kim, J.S.; On, Y.K. Atrial Fibrillation in Hypertrophic Cardiomyopathy: Is the Extent of Septal Hypertrophy Important? PLoS ONE 2016, 11, e0156410. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Olivotto, I.; Cecchi, F.; Casey, S.A.; Dolara, A.; Traverse, J.H.; Maron, B.J. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001, 104, 2517–2524. [Google Scholar] [CrossRef] [PubMed]
- Masri, A.; Kanj, M.; Thamilarasan, M.; Wazni, O.; Smedira, N.G.; Lever, H.M.; Desai, M.Y. Outcomes in hypertrophic cardiomyopathy patients with and without atrial fibrillation: A survival meta-analysis. Cardiovasc. Diagn. Ther. 2017, 7, 36–44. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Boldt, A.; Wetzel, U.; Lauschke, J.; Weigl, J.; Gummert, J.; Hindricks, G.; Kottkamp, H.; Dhein, S. Fibrosis in left atrial tissue of patients with atrial fibrillation with and without underlying mitral valve disease. Heart 2004, 90, 400–405. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Debonnaire, P.; Joyce, E.; Hiemstra, Y.; Mertens, B.J.; Atsma, D.E.; Schalij, M.J.; Bax, J.J.; Delgado, V.; Marsan, N.A. Left Atrial Size and Function in Hypertrophic Cardiomyopathy Patients and Risk of New-Onset Atrial Fibrillation. Circ. Arrhythm. Electrophysiol. 2017, 10, e004052. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Desai, M.Y.; Nishimura, R.A.; Spirito, P.; Rakowski, H.; Towbin, J.A.; Rowin, E.J.; Maron, M.S.; Sherrid, M.V. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2022, 79, 372–389. [Google Scholar] [CrossRef] [PubMed]
- Cannon, R.O., 3rd; Rosing, D.R.; Maron, B.J.; Leon, M.B.; Bonow, R.O.; Watson, R.M.; Epstein, S.E. Myocardial ischemia in patients with hypertrophic cardiomyopathy: Contribution of inadequate vasodilator reserve and elevated left ventricular filling pressures. Circulation 1985, 71, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Schlittler, M.; Pramstaller, P.P.; Rossini, A.; De Bortoli, M. Myocardial Fibrosis in Hypertrophic Cardiomyopathy: A Perspective from Fibroblasts. Int. J. Mol. Sci. 2023, 24, 14845. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rowin, E.J.; Maron, B.J.; Haas, T.S.; Garberich, R.F.; Wang, W.; Link, M.S.; Maron, M.S. Hypertrophic Cardiomyopathy With Left Ventricular Apical Aneurysm: Implications for Risk Stratification and Management. J. Am. Coll. Cardiol. 2017, 69, 761–773, Erratum in: J. Am. Coll. Cardiol. 2017, 69, 1652. https://doi.org/10.1016/j.jacc.2017.02.014. [Google Scholar] [CrossRef] [PubMed]
- Harper, A.R.; Goel, A.; Grace, C.; Thomson, K.L.; Petersen, S.E.; Xu, X.; Waring, A.; Ormondroyd, E.; Kramer, C.M.; Ho, C.Y.; et al. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat. Genet. 2021, 53, 135–142. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Massera, D.; Sherrid, M.V.; Maron, M.S.; Rowin, E.J.; Maron, B.J. How common is hypertrophic cardiomyopathy… really?: Disease prevalence revisited 27 years after CARDIA. Int. J. Cardiol. 2023, 382, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Rowin, E.J.; Maron, M.S. Global Burden of Hypertrophic Cardiomyopathy. JACC Heart Fail 2018, 6, 376–378. [Google Scholar] [CrossRef] [PubMed]
- Semsarian, C.; Ingles, J.; Maron, M.S.; Maron, B.J. New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2015, 65, 1249–1254. [Google Scholar] [CrossRef] [PubMed]
- Siontis, K.C.; Ommen, S.R.; Geske, J.B. Sex, Survival, and Cardiomyopathy: Differences Between Men and Women With Hypertrophic Cardiomyopathy. J. Am. Heart Assoc. 2019, 8, e014448. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Maron, B.J.; Rowin, E.J.; Maron, M.S. Hypertrophic Cardiomyopathy: New Concepts and Therapies. Annu. Rev. Med. 2022, 73, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Arabadjian, M.; McCarthy, M.; Dickson, V.V. An Integrated Review of Hypertrophic Cardiomyopathy in Black Populations: Underrecognized and Understudied. J. Cardiovasc. Nurs. 2021, 36, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.A.; Wang, R.S.; Carnethon, M.R.; Rowin, E.J.; Loscalzo, J.; Maron, B.J.; Maron, M.S. What Causes Hypertrophic Cardiomyopathy? Am. J. Cardiol. 2022, 179, 74–82. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, G.; Su, L.; Lang, M. A systematic review and meta-analysis of sex differences in clinical outcomes of hypertrophic cardiomyopathy. Front. Cardiovasc. Med. 2023, 10, 1252266. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lakdawala, N.K.; Olivotto, I.; Day, S.M.; Han, L.; Ashley, E.A.; Michels, M.; Ingles, J.; Semsarian, C.; Jacoby, D.; Jefferies, J.L.; et al. Associations Between Female Sex, Sarcomere Variants, and Clinical Outcomes in Hypertrophic Cardiomyopathy. Circ. Genom. Precis Med. 2021, 14, e003062. [Google Scholar] [CrossRef] [PubMed]
- Butters, A.; Lakdawala, N.K.; Ingles, J. Sex Differences in Hypertrophic Cardiomyopathy: Interaction With Genetics and Environment. Curr. Heart Fail Rep. 2021, 18, 264–273. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zaromytidou, M.; Savvatis, K. The weight of obesity in hypertrophic cardiomyopathy. Clin. Med. 2023, 23, 357–363. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Javaheri, S.; Barbe, F.; Campos-Rodriguez, F.; Dempsey, J.A.; Khayat, R.; Javaheri, S.; Malhotra, A.; Martinez-Garcia, M.A.; Mehra, R.; Pack, A.I.; et al. Sleep Apnea: Types, Mechanisms, and Clinical Cardiovascular Consequences. J. Am. Coll. Cardiol. 2017, 69, 841–858. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jex, N.; Chowdhary, A.; Thirunavukarasu, S.; Procter, H.; Sengupta, A.; Natarajan, P.; Kotha, S.; Poenar, A.M.; Swoboda, P.; Xue, H.; et al. Coexistent Diabetes Is Associated with the Presence of Adverse Phenotypic Features in Patients with Hypertrophic Cardiomyopathy. Diabetes Care 2022, 45, 1852–1862. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, S.; Yang, L.; Sun, N.; Luo, X.; Li, P.; Wang, K.; Li, P.; Zhao, J.; Wang, Z.; Zhang, Q.; et al. Impact of coronary artery disease in patients with hypertrophic cardiomyopathy. Hell. J. Cardiol. 2024, 77, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Finocchiaro, G.; Magavern, E.; Sinagra, G.; Ashley, E.; Papadakis, M.; Tome-Esteban, M.; Sharma, S.; Olivotto, I. Impact of Demographic Features, Lifestyle, and Comorbidities on the Clinical Expression of Hypertrophic Cardiomyopathy. J. Am. Heart Assoc. 2017, 6, e007161. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hong, Y.; Su, W.W.; Li, X. Risk factors of sudden cardiac death in hypertrophic cardiomyopathy. Curr. Opin. Cardiol. 2022, 37, 15–21. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Maron, B.J.; McKenna, W.J.; Danielson, G.K.; Kappenberger, L.J.; Kuhn, H.J.; Seidman, C.E.; Shah, P.M.; Spencer, W.H., 3rd; Spirito, P.; Ten Cate, F.J.; et al. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J. Am. Coll. Cardiol. 2003, 42, 1687–1713. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Kado, Y.; Onoue, T.; Otani, K.; Nakazono, A.; Otsuji, Y.; Takeuchi, M. Impact of image quality on reliability of the measurements of left ventricular systolic function and global longitudinal strain in 2D echocardiography. Echo Res. Pract. 2018, 5, 28–39. [Google Scholar] [CrossRef]
- Gersh, B.J.; Maron, B.J.; Bonow, R.O.; Dearani, J.A.; Fifer, M.A.; Link, M.S.; Naidu, S.S.; Nishimura, R.A.; Ommen, S.R.; Rakowski, H.; et al. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Developed in collaboration with the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J. Am. Coll. Cardiol. 2011, 58, e212–e260. [Google Scholar] [CrossRef] [PubMed]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies: Developed by the task force on the management of cardiomyopathies of the European Society of Cardiology (ESC). Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef]
- Maron, B.J.; Ommen, S.R.; Semsarian, C.; Spirito, P.; Olivotto, I.; Maron, M.S. Hypertrophic cardiomyopathy: Present and future, with translation into contemporary cardiovascular medicine. J. Am. Coll. Cardiol. 2014, 64, 83–99, Erratum in: J. Am. Coll. Cardiol. 2014, 64, 1188. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, I.; Viegas, J.M.; Rosa, S.A.; Brás, P.G.; Grazina, A.; Cruz, I.; Branco, L.M.; Galrinho, A.; Fiarresga, A.; Lopes, L.R.; et al. Three-dimensional echocardiography for the evaluation of hypertrophic cardiomyopathy patients: Relation to symptoms and exercise capacity. Int. J. Cardiovasc. Imaging 2023, 39, 2475–2481. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Erden, M.; van Velzen, H.G.; Menting, M.E.; van den Bosch, A.E.; Ren, B.; Michels, M.; Vletter, W.B.; van Domburg, R.T.; Schinkel, A.F.L. Three-dimensional echocardiography for the assessment of left ventricular geometry and papillary muscle morphology in hypertrophic cardiomyopathy. J. Ultrasound. 2018, 21, 17–24. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Andrew, C.Y. To, Ashwat Dhillon, Milind Y. Desai, Cardiac Magnetic Resonance in Hypertrophic Cardiomyopathy. JACC Cardiovasc. Imaging 2011, 4, 1123–1137. [Google Scholar] [CrossRef]
- Moravsky, G.; Ofek, E.; Rakowski, H.; Butany, J.; Williams, L.; Ralph-Edwards, A.; Wintersperger, B.J.; Crean, A. Myocardial fibrosis in hypertrophic cardiomyopathy: Accurate reflection of histopathological findings by CMR. JACC Cardiovasc. Imaging 2013, 6, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Noureldin, R.A.; Liu, S.; Nacif, M.S.; Judge, D.P.; Halushka, M.K.; Abraham, T.P.; Ho, C.; Bluemke, D.A. The diagnosis of hypertrophic cardiomyopathy by cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reson. 2012, 14, 17. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dorobantu, D.M.; Wadey, C.A.; Amir, N.H.; Stuart, A.G.; Williams, C.A.; Pieles, G.E. The Role of Speckle Tracking Echocardiography in the Evaluation of Common Inherited Cardiomyopathies in Children and Adolescents: A Systematic Review. Diagnostics 2021, 11, 635. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Quintana, R.A.; Bui, L.P.; Moudgil, R.; Palaskas, N.; Hassan, S.; Abe, J.I.; Mouhayar, E.; Yusuf, S.W.; Hernandez, A.; Banchs, J. Speckle-Tracking Echocardiography in Cardio-Oncology and Beyond. Tex Heart Inst. J. 2020, 47, 96–107. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Biernacka, E.K.; Osadnik, T.; Bilińska, Z.T.; Krawczyński, M.; Latos-Bieleńska, A.; Łaczmańska, I.; Miszczak-Knecht, M.; Płoski, R.; Ponińska, J.K.; Prejbisz, A.; et al. A position statement of the Polish Cardiac Society endorsed by Polish Society of Human Genetics and Cardiovascular Patient Communities. Kardiol Pol. 2024, 82, 569–593. [Google Scholar] [CrossRef] [PubMed]
- Girolami, F.; Gozzini, A.; Pálinkás, E.D.; Ballerini, A.; Tomberli, A.; Baldini, K.; Marchi, A.; Zampieri, M.; Passantino, S.; Porcedda, G.; et al. Genetic Testing and Counselling in Hypertrophic Cardiomyopathy: Frequently Asked Questions. J. Clin. Med. 2023, 12, 2489. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tudurachi, B.S.; Zăvoi, A.; Leonte, A.; Țăpoi, L.; Ureche, C.; Bîrgoan, S.G.; Chiuariu, T.; Anghel, L.; Radu, R.; Sascău, R.A.; et al. An Update on MYBPC3 Gene Mutation in Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2023, 24, 10510. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lubitz, S.A.; Ellinor, P.T. Next-generation sequencing for the diagnosis of cardiac arrhythmia syndromes. Heart Rhythm. 2015, 12, 1062–1070. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Marian, A.J. Molecular Genetic Basis of Hypertrophic Cardiomyopathy. Circ. Res. 2021, 128, 1533–1553. [Google Scholar] [CrossRef] [PubMed]
- Huurman, R.; Bowen, D.J.; Mutluer, F.O.; Loff Barreto, B.; van Slegtenhorst, M.A.; Verhagen, J.M.A.; Hirsch, A.; van den Bosch, A.E.; Michels, M.; Schinkel, A.F.L. Prognostic significance of left atrial strain in sarcomere gene variant carriers without hypertrophic cardiomyopathy. Echocardiography 2022, 39, 1209–1218. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jansen, M.; Algül, S.; Bosman, L.P.; Michels, M.; van der Velden, J.; de Boer, R.A.; van Tintelen, J.P.; Asselbergs, F.W.; Baas, A.F. Blood-based biomarkers for the prediction of hypertrophic cardiomyopathy prognosis: A systematic review and meta-analysis. ESC Heart Fail 2022, 9, 3418–3434. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lim, L.J.; Tison, G.H.; Delling, F.N. Artificial Intelligence in Cardiovascular Imaging. Methodist Debakey Cardiovasc. J. 2020, 16, 138–145. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Farahani, N.Z.; Arunachalam, S.P.; Sundaram, D.S.B.; Pasupathy, K.; Enayati, M.; Arruda-Olson, A.M. Explanatory Analysis of a Machine Learning Model to Identify Hypertrophic Cardiomyopathy Patients from EHR Using Diagnostic Codes. In Proceedings of the 2020 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Seoul, Republic of Korea, 16–19 December 2020; Volume 2020, pp. 1932–1937. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, Y.-R.; Yang, K.; Wen, Y.; Wang, P.; Hu, Y.; Lai, Y.; Wang, Y.; Zhao, K.; Tang, S.; Zhang, A.; et al. Screening and diagnosis of cardiovascular disease using artificial intelligence-enabled cardiac magnetic resonance imaging. Nat. Med. 2024, 30, 1471–1480. [Google Scholar] [CrossRef]
- Krittanawong, C.; Johnson, K.W.; Choi, E.; Kaplin, S.; Venner, E.; Murugan, M.; Wang, Z.; Glicksberg, B.S.; Amos, C.I.; Schatz, M.C.; et al. Artificial Intelligence and Cardiovascular Genetics. Life 2022, 12, 279. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tower-Rader, A.; Kramer, C.M.; Neubauer, S.; Nagueh, S.F.; Desai, M.Y. Multimodality Imaging in Hypertrophic Cardiomyopathy for Risk Stratification. Circ. Cardiovasc. Imaging 2020, 13, e009026. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Monda, E.; Palmiero, G.; Lioncino, M.; Rubino, M.; Cirillo, A.; Fusco, A.; Caiazza, M.; Verrillo, F.; Diana, G.; Mauriello, A.; et al. Multimodality Imaging in Cardiomyopathies with Hypertrophic Phenotypes. J. Clin. Med. 2022, 11, 868. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Goldie, F.C.; Lee, M.M.Y.; Coats, C.J.; Nordin, S. Advances in Multi-Modality Imaging in Hypertrophic Cardiomyopathy. J. Clin. Med. 2024, 13, 842. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.R.; Abraham, T.P. Role of Imaging in the Diagnosis, Evaluation, and Management of Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2024, 212, S14–S32. [Google Scholar] [CrossRef] [PubMed]
- Calderon Martinez, E.; Ortiz-Garcia, N.Y.; Herrera Hernandez, D.A.; Arriaga Escamilla, D.; Diaz Mendoza, D.L.; Othon Martinez, D.; Ramirez, L.M.; Reyes-Rivera, J.; Choudhari, J.; Michel, G. Hypertrophic Cardiomyopathy Diagnosis and Treatment in High- and Low-Income Countries: A Narrative Review. Cureus 2023, 15, e46330. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fernandes, F.; Antunes, M.O.; Hotta, V.T.; Rochitte, C.E.; Mady, C. Deposit Diseases as Differential Diagnosis of Left Ventricular Hypertrophy in Patients with Heart Failure and Preserved Systolic Function. Arq. Bras. Cardiol. 2019, 113, 979–987. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ramchand, J.; Fava, A.M.; Chetrit, M.; Desai, M.Y. Advanced imaging for risk stratification of sudden death in hypertrophic cardiomyopathy. Heart 2020, 106, 793–801. [Google Scholar] [CrossRef] [PubMed]
- van der Velden, J.; Tocchetti, C.G.; Varricchi, G.; Bianco, A.; Sequeira, V.; Hilfiker-Kleiner, D.; Hamdani, N.; Leite-Moreira, A.F.; Mayr, M.; Falcão-Pires, I.; et al. Metabolic changes in hypertrophic cardiomyopathies: Scientific update from the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc. Res. 2018, 114, 1273–1280. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gersh, B.J.; Maron, B.J.; Dearani, J.A.; Fifer, M.A.; Link, M.S.; Naidu, S.S.; Nishimura, R.A.; Ommen, S.R.; Rakowski, H.; Seidman, C.E.; et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011, 124, e783–e831. [Google Scholar] [CrossRef]
- Zampieri, M.; Zampieri, M.; Berteotti, M.; Berteotti, M.; Ferrantini, C.; Ferrantini, C.; Tassetti, L.; Tassetti, L.; Gabriele, M.; Gabriele, M.; et al. Pathophysiology and Treatment of Hypertrophic Cardiomyopathy: New Perspectives. Curr. Heart Fail. Rep. 2021, 18, 169–179. [Google Scholar] [CrossRef]
- Maron, B.J. Clinical Course and Management of Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2018, 379, 655–668. [Google Scholar] [CrossRef]
- Dybro, A.M.; Rasmussen, T.B.; Nielsen, R.R.; Andersen, M.J.; Jensen, M.K.; Poulsen, S.H. Randomized Trial of Metoprolol in Patients with Obstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2021, 78, 2505–2517. [Google Scholar] [CrossRef]
- Wigle, E.D. Novel insights into the clinical manifestations and treatment of hypertrophic cardiomyopathy. Curr. Opin. Cardiol. 1995, 10, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; Kantor, P.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients with Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2020, 76, e159–e240. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Day, S.M.; Axelsson, A.; Russell, M.W.; Zahka, K.; Lever, H.M.; Pereira, A.C.; Colan, S.D.; Margossian, R.; Murphy, A.M.; et al. Valsartan in early-stage hypertrophic cardiomyopathy: A randomized phase 2 trial. Nat. Med. 2021, 27, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, R.A.; Holmes, D.R. Hypertrophic obstructive cardiomyopathy. N. Engl. J. Med. 2004, 350, 1320–1327. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, R.A.; Seggewiss, H.; Schaff, H.V. Hypertrophic Obstructive Cardiomyopathy: Surgical Myectomy and Septal Ablation. Circ. Res. 2017, 121, 771–783. [Google Scholar] [CrossRef]
- Dearani, J.A.; Ommen, S.R.; Gersh, B.J.; Schaff, H.V.; Danielson, G.K. Surgery insight: Septal myectomy for obstructive hypertrophic cardiomyopathy--the Mayo Clinic experience. Nature clinical practice. Cardiovasc. Med. 2007, 4, 503–512. [Google Scholar] [CrossRef]
- Kimmelstiel, C.; Zisa, D.C.; Kuttab, J.S.; Wells, S.; Udelson, J.E.; Wessler, B.S.; Rastegar, H.; Kapur, N.K.; Weintraub, A.R.; Maron, B.J.; et al. Guideline-Based Referral for Septal Reduction Therapy in Obstructive Hypertrophic Cardiomyopathy Is Associated With Excellent Clinical Outcomes. Circulation. Cardiovasc. Interv. 2019, 12, e007673. [Google Scholar] [CrossRef]
- Ottaviani, A.; Mansour, D.; Molinari, L.V.; Galanti, K.; Mantini, C.; Khanji, M.Y.; Chahal, A.A.; Zimarino, M.; Renda, G.; Sciarra, L.; et al. Revisiting Diagnosis and Treatment of Hypertrophic Cardiomyopathy: Current Practice and Novel Perspectives. J. Clin. Med. 2023, 12, 5710. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mistrulli, R.; Ferrera, A.; Salerno, L.; Vannini, F.; Guida, L.; Corradetti, S.; Addeo, L.; Valcher, S.; Di Gioia, G.; Spera, F.R.; et al. Cardiomyopathy and Sudden Cardiac Death: Bridging Clinical Practice with Cutting-Edge Research. Biomedicines 2024, 12, 1602. [Google Scholar] [CrossRef]
- Losi, M.A.; Nistri, S.; Galderisi, M.; Betocchi, S.; Cecchi, F.; Olivotto, I.; Agricola, E.; Ballo, P.; Buralli, S.; D’Andrea, A.; et al. Echocardiography in patients with hypertrophic cardiomyopathy: Usefulness of old and new techniques in the diagnosis and pathophysiological assessment. Cardiovasc. Ultrasound. 2010, 8, 7. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sipola, P.; Magga, J.; Husso, M.; Jääskeläinen, P.; Peuhkurinen, K.; Kuusisto, J. Cardiac MRI assessed left ventricular hypertrophy in differentiating hypertensive heart disease from hypertrophic cardiomyopathy attributable to a sarcomeric gene mutation. Eur. Radiol. 2011, 21, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- De Marco, F.; Ferrucci, F.; Risi, M.; Tortora, G. Classification of QRS complexes to detect Premature Ventricular Contraction using machine learning techniques. PLoS ONE 2022, 17, e0268555. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- De Marco, F.; Di Biasi, L.; Auriemma Citarella, A.; Tortora, G. Improving PVC Detection in ECG Signals: A Recurrent Neural Network Approach. In Artificial Life and Evolutionary Computation; WIVACE 2023. Communications in Computer and Information Science; Villani, M., Cagnoni, S., Serra, R., Eds.; Springer: Cham, Switzerland, 2024; Volume 1977. [Google Scholar] [CrossRef]
- Kujime, K.; Seno, H.; Nakajima, K.; Yamazaki, M.; Sakuma, I.; Yamagata, K.; Kusano, K.; Tomii, N. Explainable localization of premature ventricular contraction using deep learning-based semantic segmentation of 12-lead electrocardiogram. J. Arrhythm. 2024, 40, 948–957. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bonaventura, J.; Polakova, E.; Vejtasova, V.; Veselka, J. Genetic Testing in Patients with Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2021, 22, 10401. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tfelt-Hansen, J.; Garcia, R.; Albert, C.; Merino, J.; Krahn, A.; Marijon, E.; Basso, C.; Wilde, A.A.M.; Haugaa, K.H. Risk stratification of sudden cardiac death: A review. Europace 2023, 25, euad203. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vehmeijer, J.T.; Christiaans, I.; van Langen, I.M.; Birnie, E.; Bonsel, G.J.; Smets, E.M.; Wilde, A.A. Risk stratification for sudden cardiac death in hypertrophic cardiomyopathy: Dutch cardiologists and the care of mutation carriers. Neth. Heart J. 2009, 17, 464–469. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Maron, M.S.; Rowin, E.J.; Olivotto, I.; Casey, S.A.; Arretini, A.; Tomberli, B.; Garberich, R.F.; Link, M.S.; Chan, R.H.M.; Lesser, J.R.; et al. Contemporary Natural History and Management of Nonobstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, P.F.; Kilner, P.J.; McGill, L.A.; Nielles-Vallespin, S.; Scott, A.D.; Ho, S.Y.; McCarthy, K.P.; Haba, M.M.; Ismail, T.F.; Gatehouse, P.D.; et al. In vivo cardiovascular magnetic resonance diffusion tensor imaging shows evidence of abnormal myocardial laminar orientations and mobility in hypertrophic cardiomyopathy. J. Cardiovasc. Magn. Reson. 2014, 16, 87. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bhaltadak, V.; Ghewade, B.; Yelne, S. A Comprehensive Review on Advancements in Wearable Technologies: Revolutionizing Cardiovascular Medicine. Cureus 2024, 16, e61312. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Abbas, M.T.; Baba Ali, N.; Farina, J.M.; Mahmoud, A.K.; Pereyra, M.; Scalia, I.G.; Kamel, M.A.; Barry, T.; Lester, S.J.; Cannan, C.R.; et al. Role of Genetics in Diagnosis and Management of Hypertrophic Cardiomyopathy: A Glimpse into the Future. Biomedicines 2024, 12, 682. [Google Scholar] [CrossRef]
- Dong, T.; Alencherry, B.; Ospina, S.; Desai, M.Y. Review of Mavacamten for Obstructive Hypertrophic Cardiomyopathy and Future Directions. Drug Des. Devel Ther. 2023, 17, 1097–1106. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zatorski, N.; Sobie, E.A.; Schlessinger, A. Mavacamten improves symptoms in obstructive hypertrophic cardiomyopathy patients. Trends Pharmacol. Sci. 2023, 44, 318–319. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Braunwald, E.; Saberi, S.; Abraham, T.P.; Elliott, P.M.; Olivotto, I. Mavacamten: A first-in-class myosin inhibitor for obstructive hypertrophic cardiomyopathy. Eur. Heart J. 2023, 44, 4622–4633, Erratum in: Eur. Heart J. 2024, 45, 286. https://doi.org/10.1093/eurheartj/ehad854. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rangwala, H.S.; Fatima, H.; Ali, M.; Ahmed, S.T.; Rangwala, B.S.; Abbas, S.R. Analyzing safety and effectiveness of Mavacamten in comparison with placebo for managing hypertrophic cardiomyopathy: A systemic review and meta-analysis. Egypt Heart J. 2023, 75, 99. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ammirati, E.; Gallone, G. Mavacamten: Practical Answers for the Clinician and New Questions From the MAVA-Long-Term Extension Study. JACC Heart Fail 2024, 12, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Woodland, M.; Al-Horani, R.A. New Era: Mavacamten for Obstructive Hypertrophic Cardiomyopathy. Cardiovasc. Hematol. Agents Med. Chem. 2023, 21, 78–83. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Keam, S.J. Mavacamten: First Approval. Drugs 2022, 82, 1127–1135, Erratum in: Drugs 2022, 82, 1235. https://doi.org/10.1007/s40265-022-01758-4. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bello, J.; Pellegrini, M.V. Mavacamten. 2024 Aug 21. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar] [PubMed]
- Nag, S.; Gollapudi, S.K.; Del Rio, C.L.; Spudich, J.A.; McDowell, R. Mavacamten, a precision medicine for hypertrophic cardiomyopathy: From a motor protein to patients. Sci. Adv. 2023, 9, eabo7622. [Google Scholar] [CrossRef] [PubMed]
- Ismayl, M.; Abbasi, M.A.; Marar, R.; Geske, J.B.; Gersh, B.J.; Anavekar, N.S. Mavacamten Treatment for Hypertrophic Cardiomyopathy: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Curr. Probl. Cardiol. 2023, 48, 101429. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, M.; Argirò, A.; Marchi, A.; Berteotti, M.; Targetti, M.; Fornaro, A.; Tomberli, A.; Stefàno, P.; Marchionni, N.; Olivotto, I. Mavacamten, a Novel Therapeutic Strategy for Obstructive Hypertrophic Cardiomyopathy. Curr. Cardiol. Rep. 2021, 23, 79. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, T.; Martins, E. Mavacamten, a novel revolutionizing therapy in hypertrophic obstructive cardiomyopathy: A literature review. Rev. Port. Cardiol. 2022, 41, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, M.T.; Jacoby, D.; Elliott, P.M.; Saberi, S.; Hegde, S.M.; Lakdawala, N.K.; Myers, J.; Sehnert, A.J.; Edelberg, J.M.; Li, W.; et al. Effect of beta-blocker therapy on the response to mavacamten in patients with symptomatic obstructive hypertrophic cardiomyopathy. Eur. J. Heart Fail 2023, 25, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Flenner, F.; Geertz, B.; Reischmann-Düsener, S.; Weinberger, F.; Eschenhagen, T.; Carrier, L.; Friedrich, F.W. Diltiazem prevents stress-induced contractile deficits in cardiomyocytes, but does not reverse the cardiomyopathy phenotype in Mybpc3-knock-in mice. J. Physiol. 2017, 595, 3987–3999. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dominguez, F.; Cabrera, E. Mavacamten in obstructive hypertrophic cardiomyopathy—Are beta-blockers blocking part of its shine? Eur. J. Heart Fail 2023, 25, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Bishev, D.; Fabara, S.; Loseke, I.; Alok, A.; Al-Ani, H.; Bazikian, Y. Efficacy and Safety of Mavacamten in the Treatment of Hypertrophic Cardiomyopathy: A Systematic Review. Heart Lung Circ. 2023, 32, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Rabiee Rad, M.; Ghasempour Dabaghi, G.; Habibi, D. Safety and efficacy of mavacamten for treatment of hypertrophic cardiomyopathy: A systematic review and meta-analysis of randomized clinical trials. Egypt Heart J. 2023, 75, 4. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Olivotto, I.; Oreziak, A.; Barriales-Villa, R.; Abraham, T.P.; Masri, A.; Garcia-Pavia, P.; Saberi, S.; Lakdawala, N.K.; Wheeler, M.T.; Owens, A.; et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 396, 759–769, Erratum in: Lancet 2020, 396, 758. https://doi.org/10.1016/S0140-6736(20)31872-9. [Google Scholar] [CrossRef] [PubMed]
- Rader, F.; Oręziak, A.; Choudhury, L.; Saberi, S.; Fermin, D.; Wheeler, M.T.; Abraham, T.P.; Garcia-Pavia, P.; Zwas, D.R.; Masri, A.; et al. Mavacamten Treatment for Symptomatic Obstructive Hypertrophic Cardiomyopathy: Interim Results From the MAVA-LTE Study, EXPLORER-LTE Cohort. JACC Heart Fail 2024, 12, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Olivotto, I.; Jacoby, D.; Lester, S.J.; Roe, M.; Wang, A.; Waldman, C.B.; Zhang, D.; Sehnert, A.J.; Heitner, S.B. Study Design and Rationale of EXPLORER-HCM: Evaluation of Mavacamten in Adults with Symptomatic Obstructive Hypertrophic Cardiomyopathy. Circ. Heart Fail 2020, 13, e006853. [Google Scholar] [CrossRef] [PubMed]
- Spertus, J.A.; Fine, J.T.; Elliott, P.; Ho, C.Y.; Olivotto, I.; Saberi, S.; Li, W.; Dolan, C.; Reaney, M.; Sehnert, A.J.; et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): Health status analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2021, 397, 2467–2475. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.Y.; Owens, A.; Wolski, K.; Geske, J.B.; Saberi, S.; Wang, A.; Sherrid, M.; Cremer, P.C.; Lakdawala, N.K.; Tower-Rader, A.; et al. Mavacamten in Patients with Hypertrophic Cardiomyopathy Referred for Septal Reduction: Week 56 Results From the VALOR-HCM Randomized Clinical Trial. JAMA Cardiol. 2023, 8, 968–977. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Desai, M.Y.; Owens, A.; Geske, J.B.; Wolski, K.; Naidu, S.S.; Smedira, N.G.; Cremer, P.C.; Schaff, H.; McErlean, E.; Sewell, C.; et al. Myosin Inhibition in Patients with Obstructive Hypertrophic Cardiomyopathy Referred for Septal Reduction Therapy. J. Am. Coll. Cardiol. 2022, 80, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Cremer, P.C.; Geske, J.B.; Owens, A.; Jaber, W.A.; Harb, S.C.; Saberi, S.; Wang, A.; Sherrid, M.; Naidu, S.S.; Schaff, H.; et al. Myosin Inhibition and Left Ventricular Diastolic Function in Patients with Obstructive Hypertrophic Cardiomyopathy Referred for Septal Reduction Therapy: Insights From the VALOR-HCM Study. Circ. Cardiovasc. Imaging 2022, 15, e014986. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.Y.; Okushi, Y.; Wolski, K.; Geske, J.B.; Owens, A.; Saberi, S.; Wang, A.; Cremer, P.C.; Sherrid, M.; Lakdawala, N.K.; et al. Mavacamten-Associated Temporal Changes in Left Atrial Function in Obstructive HCM: Insights from the VALOR-HCM Trial. JACC Cardiovasc. Imaging, 2024; ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.Y.; Owens, A.; Geske, J.B.; Wolski, K.; Saberi, S.; Wang, A.; Sherrid, M.; Cremer, P.C.; Naidu, S.S.; Smedira, N.G.; et al. Dose-Blinded Myosin Inhibition in Patients With Obstructive Hypertrophic Cardiomyopathy Referred for Septal Reduction Therapy: Outcomes Through 32 Weeks. Circulation 2023, 147, 850–863. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, J.E.; McNally, E.M. Lessons From MAVERICK-HCM: The Need for Less Speed. J. Am. Coll. Cardiol. 2020, 75, 2661–2663. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ho, C.Y.; Mealiffe, M.E.; Bach, R.G.; Bhattacharya, M.; Choudhury, L.; Edelberg, J.M.; Hegde, S.M.; Jacoby, D.; Lakdawala, N.K.; Lester, S.J.; et al. Evaluation of Mavacamten in Symptomatic Patients With Nonobstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2020, 75, 2649–2660. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.L.; Liang, Y.; Liang, B. Evaluation of mavacamten in patients with hypertrophic cardiomyopathy. J. Cardiovasc. Med. 2024, 25, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Scholtz, S.; Rudolph, V.; Reil, J.C. Alcohol Septal Ablation or Mavacamten for Obstructive Hypertrophic Cardiomyopathy. J. Clin. Med. 2023, 12, 6628. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Garcia-Pavia, P.; Oręziak, A.; Masri, A.; Barriales-Villa, R.; Abraham, T.P.; Owens, A.T.; Jensen, M.K.; Wojakowski, W.; Seidler, T.; Hagege, A.; et al. Long-term effect of mavacamten in obstructive hypertrophic cardiomyopathy. Eur. Heart J. 2024; ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Masri, A.; Lester, S.J.; Stendahl, J.C.; Hegde, S.M.; Sehnert, A.J.; Balaratnam, G.; Shah, A.; Fox, S.; Wang, A. Long-Term Safety and Efficacy of Mavacamten in Symptomatic Obstructive Hypertrophic Cardiomyopathy: Interim Results of the PIONEER-OLE Study. J. Am. Heart Assoc. 2024, 13, e030607. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tuohy, C.V.; Kaul, S.; Song, H.K.; Nazer, B.; Heitner, S.B. Hypertrophic cardiomyopathy: The future of treatment. Eur. J. Heart Fail 2020, 22, 228–240. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Outcome Measure | Mavacamten Group (n = 123) | Placebo Group (n = 128) |
|---|---|---|
| Primary Endpoint Achievement | 37% | 17% |
| NYHA Class Improvement (≥1 Class) | 65% | 31% |
| Peak post-exercise reduction in LVOT Gradient (mmHg) | 48 mmHg | 11 mmHg |
| At least a 3 mL/kg/min increase in pVO2 and + at least one class improvement in NYHA class | 20% | 8% |
| KCCQ Quality of Life Score | +9 points more than in placebo | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Młynarska, E.; Radzioch, E.; Dąbek, B.; Leszto, K.; Witkowska, A.; Czarnik, W.; Jędraszak, W.; Rysz, J.; Franczyk, B. Hypertrophic Cardiomyopathy with Special Focus on Mavacamten and Its Future in Cardiology. Biomedicines 2024, 12, 2675. https://doi.org/10.3390/biomedicines12122675
Młynarska E, Radzioch E, Dąbek B, Leszto K, Witkowska A, Czarnik W, Jędraszak W, Rysz J, Franczyk B. Hypertrophic Cardiomyopathy with Special Focus on Mavacamten and Its Future in Cardiology. Biomedicines. 2024; 12(12):2675. https://doi.org/10.3390/biomedicines12122675
Chicago/Turabian StyleMłynarska, Ewelina, Ewa Radzioch, Bartłomiej Dąbek, Klaudia Leszto, Alicja Witkowska, Witold Czarnik, Weronika Jędraszak, Jacek Rysz, and Beata Franczyk. 2024. "Hypertrophic Cardiomyopathy with Special Focus on Mavacamten and Its Future in Cardiology" Biomedicines 12, no. 12: 2675. https://doi.org/10.3390/biomedicines12122675
APA StyleMłynarska, E., Radzioch, E., Dąbek, B., Leszto, K., Witkowska, A., Czarnik, W., Jędraszak, W., Rysz, J., & Franczyk, B. (2024). Hypertrophic Cardiomyopathy with Special Focus on Mavacamten and Its Future in Cardiology. Biomedicines, 12(12), 2675. https://doi.org/10.3390/biomedicines12122675