
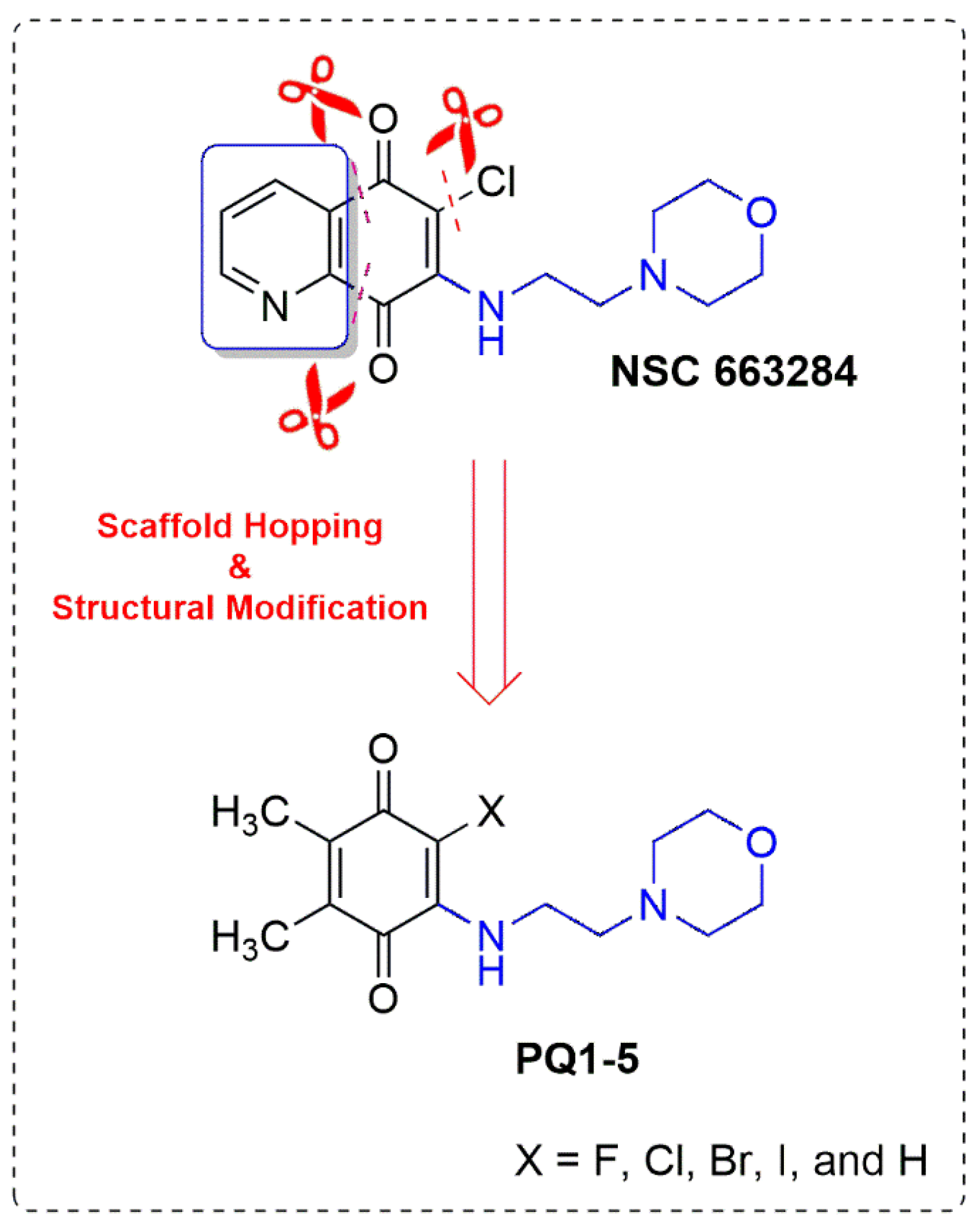
Scaffold Hopping and Structural Modification of NSC 663284: Discovery of Potent (Non)Halogenated Aminobenzoquinones

 ,
,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemistry
2.2. Synthesis of Halogenated 1,4-Benzoquinones (4–6)
2.2.1. Brominated 1,4-Benzoquinones (4)
2.2.2. Iodinated 1,4-Benzoquinones (5)
2.2.3. Chlorinated 1,4-Benzoquinones (6)
2.3. Synthesis of PQ Analogs
2.3.1. Procedure for the Preparation of PQ Analog (PQ1)
2,3-Dimethyl-5-((2-morpholinoethyl)amino)-1,4-benzoquinone (PQ1)
2.3.2. General Procedures for the Preparation of PQ Analogs (PQ2–4)
2-Bromo-5,6-dimethyl-3-((2-morpholinoethyl)amino)-1,4-benzoquinone (PQ2)
2-Iodo-5,6-dimethyl-3-((2-morpholinoethyl)amino)-1,4-benzoquinone (PQ3)
2-Chloro-5,6-dimethyl-3-((2-morpholinoethyl)amino)-1,4-benzoquinone (PQ4)
2.4. Biological Evaluation
2.4.1. In Vitro Single-Dose Anticancer Screening by NCI
2.4.2. In Vitro Five-Dose Anticancer Screening by NCI
2.4.3. Cell Culture, Drug Treatment, and MTT Assay
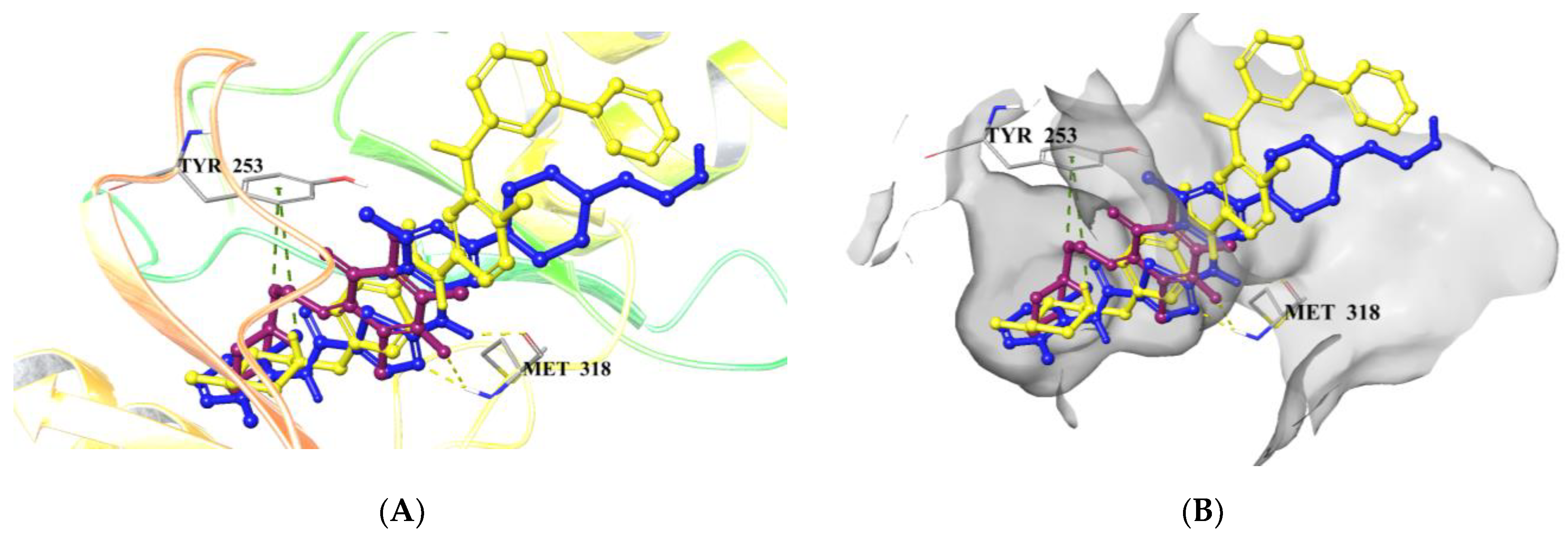
2.5. Computational Studies
2.5.1. Molecular Docking Studies
2.5.2. In-Silico ADME Estimation
3. Results
3.1. Design and Synthesis
3.2. Cell-Based Anticancer Screening for PQ Analogs
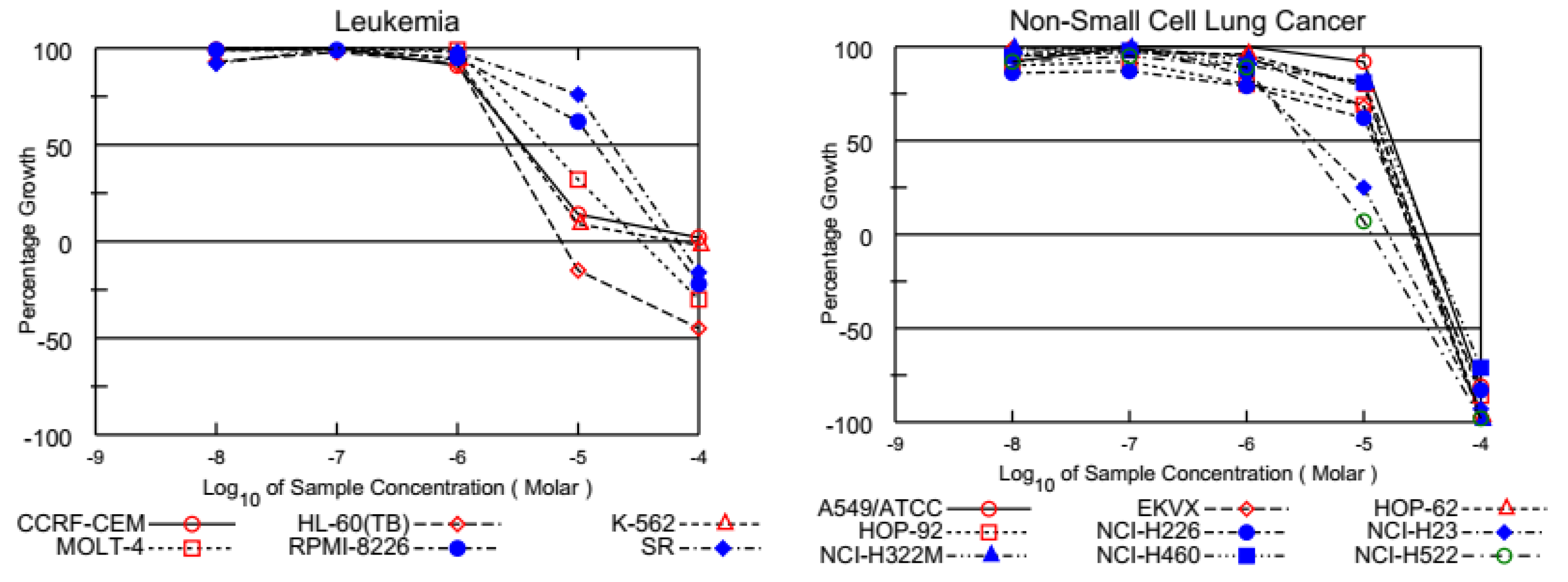
3.2.1. Single-Dose Screening on NCI 60 Cancer Cells
3.2.2. Five-Dose Screening on NCI 60 Cancer Cell Lines Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaitin, K.I. Deconstructing the Drug Development Process: The New Face of Innovation. Clin. Pharmacol. Ther. 2010, 87, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Dai, X.; Xu, Y.; Xing, G.; Liu, H.; Lu, T.; Chen, Y.; Zhang, Y. Drug repositioning: Progress and challenges in drug discovery for various diseases. Eur. J. Med. Chem. 2022, 234, 114239. [Google Scholar] [CrossRef] [PubMed]
- Callis, T.B.; Garrett, T.R.; Montgomery, A.P.; Danon, J.J.; Kassiou, M. Recent Scaffold Hopping Applications in Central Nervous System Drug Discovery. J. Med. Chem. 2022, 65, 13483–13504. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Stumpfe, D.; Bajorath, J. Recent Advances in Scaffold Hopping. J. Med. Chem. 2017, 60, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Fang, K.; Dong, G.; Chen, S.; Liu, N.; Miao, Z.; Yao, J.; Li, J.; Zhang, W.; Sheng, C. Scaffold Diversity Inspired by the Natural Product Evodiamine: Discovery of Highly Potent and Multitargeting Antitumor Agents. J. Med. Chem. 2015, 58, 6678–6696. [Google Scholar] [CrossRef] [PubMed]
- Abouelhassan, Y.; Garrison, A.T.; Yang, H.; Chávez-Riveros, A.; Burch, G.M.; Huigens, R.W., III. Recent Progress in Natural-Product-Inspired Programs Aimed To Address Antibiotic Resistance and Tolerance. J. Med. Chem. 2019, 62, 7618–7642. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Lu, J.; Wang, M.; Yang, C.-Y.; Wang, S. Discovery of SHP2-D26 as a First, Potent, and Effective PROTAC Degrader of SHP2 Protein. J. Med. Chem. 2020, 63, 7510–7528. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Schneider, G.; Fechner, U. Computer-based de novo design of drug-like molecules. Nat. Rev. Drug Discov. 2005, 4, 649–663. [Google Scholar] [CrossRef]
- Sadybekov, A.V.; Katritch, V. Computational approaches streamlining drug discovery. Nature 2023, 616, 673–685. [Google Scholar] [CrossRef]
- Schneider, G.; Neidhart, W.; Giller, T.; Schmid, G. “Scaffold-Hopping” by Topological Pharmacophore Search: A Contribution to Virtual Screening. Angew. Chem. Int. Ed. 1999, 38, 2894–2896. [Google Scholar] [CrossRef]
- Sun, H.; Tawa, G.; Wallqvist, A. Classification of scaffold-hopping approaches. Drug Discov. Today 2012, 17, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, V.; Das, S.; Nayak, A.; Guchhait, S.K.; Kundu, C.N. Scaffold-hopping and hybridization based design and building block strategic synthesis of pyridine-annulated purines: Discovery of novel apoptotic anticancer agents. Rsc Adv. 2015, 5, 26051–26060. [Google Scholar] [CrossRef]
- Baviskar, A.T.; Madaan, C.; Preet, R.; Mohapatra, P.; Jain, V.; Agarwal, A.; Guchhait, S.K.; Kundu, C.N.; Banerjee, U.C.; Bharatam, P.V. N-Fused Imidazoles As Novel Anticancer Agents That Inhibit Catalytic Activity of Topoisomerase IIα and Induce Apoptosis in G1/S Phase. J. Med. Chem. 2011, 54, 5013–5030. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Das, C.; Acharya, A.; Bhal, S.; Joshi, M.; Kundu, C.N.; Choudhury, A.R.; Guchhait, S.K. Organocatalyzed umpolung addition for synthesis of heterocyclic-fused arylidene-imidazolones as anticancer agents. Bioorgan Med. Chem. 2022, 67, 116835. [Google Scholar] [CrossRef] [PubMed]
- Priyadarshani, G.; Amrutkar, S.; Nayak, A.; Banerjee, U.C.; Kundu, C.N.; Guchhait, S.K. Scaffold-hopping of bioactive flavonoids: Discovery of aryl-pyridopyrimidinones as potent anticancer agents that inhibit catalytic role of topoisomerase IIα. Eur. J. Med. Chem. 2016, 122, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Southall, N.T.; Ajay. Kinase Patent Space Visualization Using Chemical Replacements. J. Med. Chem. 2006, 49, 2103–2109. [Google Scholar] [CrossRef]
- Boström, J.; Berggren, K.; Elebring, T.; Greasley, P.J.; Wilstermann, M. Scaffold hopping, synthesis and structure–activity relationships of 5,6-diaryl-pyrazine-2-amide derivatives: A novel series of CB1 receptor antagonists. Bioorgan. Med. Chem. 2007, 15, 4077–4084. [Google Scholar] [CrossRef]
- Jiang, Z.; Liu, N.; Hu, D.; Dong, G.; Miao, Z.; Yao, J.; He, H.; Jiang, Y.; Zhang, W.; Wang, Y.; et al. The discovery of novel antifungal scaffolds by structural simplification of the natural product sampangine. Chem. Commun. 2015, 51, 14648–14651. [Google Scholar] [CrossRef]
- Chen, K.-L.; Gan, L.; Wu, Z.-H.; Qin, J.-F.; Liao, W.-X.; Tang, H. 4- Substituted sampangine derivatives: Novel acetylcholinesterase and β-myloid aggregation inhibitors. Int. J. Biol. Macromol. 2018, 107, 2725–2729. [Google Scholar] [CrossRef]
- Wellington, K.W. Understanding cancer and the anticancer activities of naphthoquinones—A review. Rsc Adv. 2015, 5, 20309–20338. [Google Scholar] [CrossRef]
- Jayasudha, P.; Manivannan, R.; Ciattini, S.; Chelazzi, L.; Elango, K.P. Selective sensing of cyanide in aqueous solution by quinone-indole ensembles—Quantitative effect of substituents on the HBD property of the receptor moiety. Sens. Actuat B Chem. 2017, 242, 736–745. [Google Scholar] [CrossRef]
- Agarwal, G.; Lande, D.N.; Chakrovarty, D.; Gejji, S.P.; Gosavi-Mirkute, P.; Patil, A.; Salunke-Gawali, S. Bromine substituted aminonaphthoquinones: Synthesis, characterization, DFT and metal ion binding studies. Rsc Adv. 2016, 6, 88010–88029. [Google Scholar] [CrossRef]
- Parthiban, C.; Elango, K.P. Selective and sensitive colorimetric detection of Hg(II) in aqueous solution by quinone-diimidazole ensemble with mimicking YES-OR-INHIBIT logic gate operation. Sens. Actuat B Chem. 2016, 237, 284–290. [Google Scholar] [CrossRef]
- El-Najjar, N.; Gali-Muhtasib, H.; Ketola, R.A.; Vuorela, P.; Urtti, A.; Vuorela, H. The chemical and biological activities of quinones: Overview and implications in analytical detection. Phytochem. Rev. 2011, 10, 353–370. [Google Scholar] [CrossRef]
- Asche, C. Antitumour quinones. Mini Rev. Med. Chem. 2005, 5, 449–467. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.M.; Lu, S.F. Plastoquinone and Ubiquinone in Plants: Biosynthesis, Physiological Function and Metabolic Engineering. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef]
- Parmar, S.S.; Jaiwal, A.; Dhankher, O.P.; Jaiwal, P.K. Coenzyme Q10 production in plants: Current status and future prospects. Crit. Rev. Biotechnol. 2015, 35, 152–164. [Google Scholar] [CrossRef]
- Ndikubwimana, J.D.; Lee, B.H. Enhanced production techniques, properties and uses of coenzyme Q10. Biotechnol. Lett. 2014, 36, 1917–1926. [Google Scholar] [CrossRef]
- Meier, T.; Buyse, G. Idebenone: An emerging therapy for Friedreich ataxia. J. Neurol. 2009, 256, 25–30. [Google Scholar] [CrossRef]
- Klotz, L.O.; Hou, X.Q.; Jacob, C. 1,4-Naphthoquinones: From Oxidative Damage to Cellular and Inter-Cellular Signaling. Molecules 2014, 19, 14902–14918. [Google Scholar] [CrossRef]
- Donohoe, T.J.; Jones, C.R.; Barbosa, L.C.A. Total Synthesis of (+/-)-Streptonigrin: De Novo Construction of a Pentasubstituted Pyridine using Ring-Closing Metathesis. J. Am. Chem. Soc. 2011, 133, 16418–16421. [Google Scholar] [CrossRef]
- Bolzan, A.D.; Bianchi, M.S. Genotoxicity of streptonigrin: A review. Mutat. Res. Rev. Mutat. 2001, 488, 25–37. [Google Scholar] [CrossRef]
- Pettit, G.R.; Knight, J.C.; Collins, J.C.; Herald, D.L.; Pettit, R.K.; Boyd, M.R.; Young, V.G. Antineoplastic agents 430. Isolation and structure of cribrostatins 3, 4, and 5 from the Republic of Maldives Cribrochalina species. J. Nat. Prod. 2000, 63, 793–798. [Google Scholar] [CrossRef]
- Hoyt, M.T.; Palchaudhuri, R.; Hergenrother, P.J. Cribrostatin 6 induces death in cancer cells through a reactive oxygen species (ROS)-mediated mechanism. Investig. New Drug 2011, 29, 562–573. [Google Scholar] [CrossRef]
- Cummings, J.; Spanswick, V.J.; Tomasz, M.; Smyth, J.F. Enzymology of mitomycin C metabolic activation in tumour tissue—Implications for enzyme-directed bioreductive drug development. Biochem. Pharmacol. 1998, 56, 405–414. [Google Scholar]
- Cummings, J.; Spanswick, V.J.; Ritchie, A.A.; Smyth, J.F. Pharmacological determinants of the antitumour activity of mitomycin C-implications for enzyme directed drug development. Ann. Oncol. 1998, 9, 134. [Google Scholar]
- Im, A.; Amjad, A.; Agha, M.; Raptis, A.; Hou, J.Z.; Farah, R.; Lim, S.; Sehgal, A.; Dorritie, K.A.; Redner, R.L.; et al. Mitoxantrone and Etoposide for the Treatment of Acute Myeloid Leukemia Patients in First Relapse. Oncol. Res. 2016, 24, 73–80. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Holmes, F.A.; Esparza, L.; Valero, V.; Buzdar, A.U.; Neidhart, J.A.; Hortobagyi, G.N. Phase I II trial of high dose mitoxantrone in metastatic breast cancer: The M.D. Anderson Cancer Center experience. Breast Cancer Res. Tr. 1999, 54, 225–233. [Google Scholar] [CrossRef]
- Primeau, A.J.; Rendon, A.; Hedley, D.; Lilge, L.; Tannock, I.F. The distribution of the anticancer drug doxorubicin in relation to blood vessels in solid tumors. Clin. Cancer Res. 2005, 11, 8782–8788. [Google Scholar] [CrossRef]
- Braud, E.; Goddard, M.-L.; Kolb, S.; Brun, M.-P.; Mondésert, O.; Quaranta, M.; Gresh, N.; Ducommun, B.; Garbay, C. Novel naphthoquinone and quinolinedione inhibitors of CDC25 phosphatase activity with antiproliferative properties. Bioorgan. Med. Chem. 2008, 16, 9040–9049. [Google Scholar] [CrossRef]
- Jing, L.; Wu, G.; Hao, X.; Olotu, F.A.; Kang, D.; Chen, C.H.; Lee, K.-H.; Soliman, M.E.S.; Liu, X.; Song, Y.; et al. Identification of highly potent and selective Cdc25 protein phosphatases inhibitors from miniaturization click-chemistry-based combinatorial libraries. Eur. J. Med. Chem. 2019, 183, 111696. [Google Scholar] [CrossRef]
- Modranka, J.; Drogosz-Stachowicz, J.; Pietrzak, A.; Janecka, A.; Janecki, T. Synthesis and structure–activity relationship study of novel 3-diethoxyphosphorylfuroquinoline-4,9-diones with potent antitumor efficacy. Eur. J. Med. Chem. 2021, 219, 113429. [Google Scholar] [CrossRef]
- Rozanov, D.; Cheltsov, A.; Nilsen, A.; Boniface, C.; Forquer, I.; Korkola, J.; Gray, J.; Tyner, J.; Tognon, C.E.; Mills, G.B.; et al. Targeting mitochondria in cancer therapy could provide a basis for the selective anti-cancer activity. PLoS ONE 2019, 14, e0205623. [Google Scholar] [CrossRef]
- Pu, L.; Amoscato, A.A.; Bier, M.E.; Lazo, J.S. Dual G1 and G2 Phase Inhibition by a Novel, Selective Cdc25 Inhibitor 7-Chloro-6-(2-morpholin-4-ylethylamino)- quinoline-5,8-dione*. J. Biol. Chem. 2002, 277, 46877–46885. [Google Scholar] [CrossRef]
- Liu, J.C.; Granieri, L.; Shrestha, M.; Wang, D.-Y.; Vorobieva, I.; Rubie, E.A.; Jones, R.; Ju, Y.; Pellecchia, G.; Jiang, Z. Identification of CDC25 as a Common Therapeutic Target for Triple-Negative Breast Cancer. Cell Rep. 2018, 23, 112–126. [Google Scholar] [CrossRef]
- Bulut, G.; Hong, S.H.; Chen, K.; Beauchamp, E.M.; Rahim, S.; Kosturko, G.W.; Glasgow, E.; Dakshanamurthy, S.; Lee, H.S.; Daar, I.; et al. Small molecule inhibitors of ezrin inhibit the invasive phenotype of osteosarcoma cells. Oncogene 2012, 31, 269–281. [Google Scholar] [CrossRef]
- Tsuchiya, A.; Hirai, G.; Koyama, Y.; Oonuma, K.; Otani, Y.; Osada, H.; Sodeoka, M. Dual-Specificity Phosphatase CDC25A/B Inhibitor Identified from a Focused Library with Nonelectrophilic Core Structure. ACS Med. Chem. Lett. 2012, 3, 294–298. [Google Scholar] [CrossRef]
- Keinan, S.; Paquette, W.D.; Skoko, J.J.; Beratan, D.N.; Yang, W.; Shinde, S.; Johnston, P.A.; Lazo, J.S.; Wipf, P. Computational design, synthesis and biological evaluation of para-quinone-based inhibitors for redox regulation of the dual-specificity phosphatase Cdc25B. Org. Biomol. Chem. 2008, 6, 3256–3263. [Google Scholar] [CrossRef]
- Tang, H.; Yu, A.; Xing, L.; Chen, X.; Ding, H.; Yang, H.; Song, Z.; Shi, Q.; Geng, M.; Huang, X.; et al. Structural Modification and Pharmacological Evaluation of Substituted Quinoline-5,8-diones as Potent NSD2 Inhibitors. J. Med. Chem. 2023, 66, 1634–1651. [Google Scholar] [CrossRef]
- Narwanti, I.; Yu, Z.-Y.; Sethy, B.; Lai, M.-J.; Lee, H.-Y.; Olena, P.; Lee, S.-B.; Liou, J.-P. 6-Regioisomeric 5,8-quinolinediones as potent CDC25 inhibitors against colorectal cancers. Eur. J. Med. Chem. 2023, 258, 115505. [Google Scholar] [CrossRef] [PubMed]
- Bodige, S.G.; Mendez-Rojas, M.; Watson, W.H. 2-amino-1,2,3-triazole derivatives. J. Chem. Crystallogr. 1999, 29, 931–942. [Google Scholar] [CrossRef]
- Andrews, K.J.M.; Marrian, D.H.; Maxwell, D.R. 361. Potential radiosensitizers: Some quinones and related compounds. J. Chem. Soc. (Resumed), 1956; 1844–1854. [Google Scholar] [CrossRef]
- Ryu, C.K.; Lee, J.Y. Synthesis and antifungal activity of 6-hydroxycinnolines. Bioorg. Med. Chem. Lett. 2006, 16, 1850–1853. [Google Scholar] [CrossRef] [PubMed]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigrowolff, A.; et al. Feasibility of a High-Flux Anticancer Drug Screen Using a Diverse Panel of Cultured Human Tumor-Cell Lines. J. Natl. Cancer I 1991, 83, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Boyd, M.R.; Pauli, K.D. Some Practical Considerations and Applications of the National-Cancer-Institute in-Vitro Anticancer Drug Discovery Screen. Drug Dev. Res. 1995, 34, 91–109. [Google Scholar] [CrossRef]
- Grever, M.R.; Schepartz, S.A.; Chabner, B.A. The National-Cancer-Institute—Cancer Drug Discovery and Development Program. Semin. Oncol. 1992, 19, 622–638. [Google Scholar] [PubMed]
- Bayrak, N.; Ciftci, H.I.; Yildiz, M.; Yildirim, H.; Sever, B.; Tateishi, H.; Otsuka, M.; Fujita, M.; Tuyun, A.F. Structure based design, synthesis, and evaluation of anti-CML activity of the quinolinequinones as LY83583 analogs. Chem. Biol. Interact. 2021, 345, 109555. [Google Scholar] [CrossRef]
- Ciftci, H.I.; Bayrak, N.; Yildiz, M.; Yildirim, H.; Sever, B.; Tateishi, H.; Otsuka, M.; Fujita, M.; Tuyun, A.F. Design, synthesis and investigation of the mechanism of action underlying anti-leukemic effects of the quinolinequinones as LY83583 analogs. Bioorg. Chem. 2021, 114, 105160. [Google Scholar] [CrossRef]
- Tokarski, J.S.; Newitt, J.A.; Chang, C.Y.; Cheng, J.D.; Wittekind, M.; Kiefer, S.E.; Kish, K.; Lee, F.Y.; Borzillerri, R.; Lombardo, L.J.; et al. The structure of Dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants. Cancer Res. 2006, 66, 5790–5797. [Google Scholar] [CrossRef]
- Brogden, A.L.; Hopcroft, N.H.; Searcey, M.; Cardin, C.J. Ligand bridging of the DNA Holliday junction: Molecular recognition of a stacked-X four-way junction by a small molecule. Angew. Chem. Int. Ed. 2007, 46, 3850–3854. [Google Scholar] [CrossRef]
- Ciftci, H.; Sever, B.; Bayrak, N.; Tateishi, H.; Otsuka, M.; Fujita, M.; TuYuN, A.F.; Yildiz, M.; Yildirim, H. In Vitro Cytotoxicity Evaluation of Plastoquinone Analogues against Colorectal and Breast Cancers along with In Silico Insights. Pharmaceutilcals 2022, 15, 1266. [Google Scholar] [CrossRef] [PubMed]
- Ciftci, H.; Sever, B.; Ocak, F.; Bayrak, N.; Yıldız, M.; Yıldırım, H.; DeMirci, H.; Tateishi, H.; Otsuka, M.; Fujita, M.; et al. In Vitro and In Silico Study of Analogs of Plant Product Plastoquinone to Be Effective in Colorectal Cancer Treatment. Molecules 2022, 27, 693. [Google Scholar] [CrossRef] [PubMed]
- Ciftci, H.; Sever, B.; Kaya, N.; Bayrak, N.; Yildiz, M.; Yildirim, H.; Tateishi, H.; Otsuka, M.; Fujita, M.; TuYuN, A.F. Studies on 1,4-Quinone Derivatives Exhibiting Anti-Leukemic Activity along with Anti-Colorectal and Anti-Breast Cancer Effects. Molecules 2023, 28, 77. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2016-2: QikProp; Schrödinger, LLC: New York, NY, USA, 2016.
- SwissADME. Available online: http://www.swissadme.ch (accessed on 30 August 2023).
- Goler, A.M.Y.; Jannuzzi, A.T.; Bayrak, N.; Yildiz, M.; Yildirim, H.; Otsuka, M.; Fujita, M.; Radwan, M.O.; TuYuN, A.F. In Vitro and In Silico Study to Assess Toxic Mechanisms of Hybrid Molecules of Quinone-Benzocaine as Plastoquinone Analogues in Breast Cancer Cells. Acs Omega 2022, 7, 30250–30264. [Google Scholar] [CrossRef] [PubMed]
- Jannuzzi, A.T.; Yildiz, M.; Bayrak, N.; Yildirim, H.; Shilkar, D.; Jayaprakash, V.; TuYuN, A.F. Anticancer agents based on Plastoquinone analogs with N-phenylpiperazine: Structure-activity relationship and mechanism of action in breast cancer cells. Chem. Biol. Interact. 2021, 349, 109673. [Google Scholar] [CrossRef] [PubMed]
- Bayrak, N.; Yildirim, H.; Yildiz, M.; Radwan, M.O.; Otsuka, M.; Fujita, M.; Ciftci, H.I.; Tuyun, A.F. A novel series of chlorinated plastoquinone analogs: Design, synthesis, and evaluation of anticancer activity. Chem. Biol. Drug Des. 2020, 95, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Bayrak, N.; Yildirim, H.; Yildiz, M.; Radwan, M.O.; Otsuka, M.; Fujita, M.; Tuyun, A.F.; Ciftci, H.I. Design, synthesis, and biological activity of Plastoquinone analogs as a new class of anticancer agents. Bioorg. Chem. 2019, 92, 103255. [Google Scholar] [CrossRef]
- Ciftci, H.I.; Bayrak, N.; Yildirim, H.; Yildiz, M.; Radwan, M.O.; Otsuka, M.; Fujita, M.; Tuyun, A.F. Discovery and structure-activity relationship of plastoquinone analogs as anticancer agents against chronic myelogenous leukemia cells. Arch. Pharm. 2019, 352, 1900170. [Google Scholar] [CrossRef]
- Ikeda, T.; Wakabayashi, H.; Nakane, M. Benzoquinone Antiallergy and Antiinflammatory Agents. U.S. Patent 5,104,874, 14 April 1992. [Google Scholar]
- Alvarado, Y.; Apostolidou, E.; Swords, R.; Giles, F.J. Emerging therapeutic options for Philadelphia-positive acute lymphocytic leukemia. Expert. Opin. Emerg. Drugs 2007, 12, 165–179. [Google Scholar] [CrossRef]
- Li, S. Src kinase signaling in leukaemia. Int. J. Biochem. Cell Biol. 2007, 39, 1483–1488. [Google Scholar] [CrossRef][Green Version]
- Ulusoy, N.G.; Emirdağ, S.; Sözer, E.; Radwan, M.O.; Çiftçi, H.; Aksel, M.; Bölükbaşı, S.; Özmen, A.; Yaylı, N.; Karayıldırım, T.; et al. Design, semi-synthesis and examination of new gypsogenin derivatives against leukemia via Abl tyrosine kinase inhibition and apoptosis induction. Int. J. Biol. Macromol. 2022, 222 Pt A, 1487–1499. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Cell Lines | PQ1 (D-838754/1) | PQ2 (D-837715/1) | PQ3 (D-838755/1) | PQ4 (D-838758/1) | |
|---|---|---|---|---|---|
| Leukemia | |||||
| GP | CCRF-CEM | 98.14 | −21.05 | 73.74 | 26.16 |
| HL-60(TB) | 96.71 | −24.93 | 10.26 | 68.81 | |
| K-562 | 97.40 | 0.99 | 5.24 | 56.63 | |
| MOLT-4 | 104.00 | 0.28 | 87.07 | 58.71 | |
| RPMI-8226 | 92.90 | 58.59 | 88.36 | 85.40 | |
| SR | 96.65 | 55.91 | 99.69 | 99.98 | |
| Non-Small Cell Lung Cancer | |||||
| GP | A549/ATCC | 110.25 | 87.57 | 102.61 | 104.88 |
| EKVX | 95.10 | 46.44 | 58.55 | 75.59 | |
| HOP-62 | 103.37 | 87.21 | 68.69 | 104.49 | |
| HOP-92 | 98.81 | 38.69 | 114.18 | 107.70 | |
| NCI-H226 | 97.55 | 67.08 | 27.88 | 94.68 | |
| NCI-H23 | 95.02 | −0.61 | 8.14 | 52.57 | |
| NCI-H322M | 101.36 | 91.51 | 103.91 | 98.10 | |
| NCI-H460 | 103.89 | 97.97 | 102.17 | 103.40 | |
| NCI-H522 | 92.26 | −18.44 | −39.17 | 96.16 | |
| Colon Cancer | |||||
| GP | COLO 205 | 111.45 | 129.82 | −75.81 | 120.20 |
| HCC-2998 | 107.00 | 101.25 | 117.04 | 121.61 | |
| HCT-116 | 107.87 | −75.55 | 14.55 | 37.97 | |
| HCT-15 | 108.15 | 90.72 | 97.45 | 107.40 | |
| HT29 | 110.66 | 93.70 | 15.50 | 117.75 | |
| KM12 | 99.66 | 94.18 | 99.89 | 100.91 | |
| SW-620 | 95.25 | 32.71 | −67.98 | 97.86 | |
| CNS Cancer | |||||
| GP | SF-268 | 96.23 | 63.68 | 111.69 | 105.60 |
| SF-295 | 105.00 | 103.06 | 91.72 | 99.71 | |
| SF-539 | 95.10 | 32.06 | 106.82 | 104.23 | |
| SNB-19 | 96.86 | 91.26 | 92.47 | 95.16 | |
| SNB-75 | nd | 105.87 | nd | nd | |
| U251 | 104.89 | 44.78 | 36.73 | 77.91 | |
| Melanoma | |||||
| GP | LOX IMVI | 88.51 | −28.50 | −84.32 | 23.24 |
| MALME-3M | 98.09 | −68.96 | −83.36 | 97.42 | |
| M14 | 102.24 | 37.33 | 40.42 | 82.94 | |
| MDA-MB-435 | 103.36 | −87.50 | −92.95 | 97.62 | |
| SK-MEL-2 | nd | 68.89 | nd | nd | |
| SK-MEL-28 | 109.52 | 75.37 | 51.44 | 119.00 | |
| SK-MEL-5 | 94.12 | 62.08 | 84.40 | 75.65 | |
| UACC-257 | 101.01 | −13.26 | −50.56 | 74.16 | |
| UACC-62 | 94.97 | 39.37 | −27.50 | 79.11 | |
| Ovarian Cancer | |||||
| GP | IGROV1 | 107.81 | 31.40 | 95.36 | 108.74 |
| OVCAR-3 | 100.23 | −26.94 | 1.12 | 100.55 | |
| OVCAR-4 | 100.84 | −67.38 | 34.55 | 99.26 | |
| OVCAR-5 | 111.14 | 4.00 | 5.49 | 113.98 | |
| OVCAR-8 | 99.72 | 16.31 | 82.87 | 88.94 | |
| NCI/ADR-RES | 97.77 | 86.39 | 89.02 | 78.01 | |
| SK-OV-3 | 94.20 | 87.54 | 83.08 | 92.47 | |
| Renal Cancer | |||||
| GP | 786-0 | 107.58 | 100.01 | 104.36 | 106.45 |
| A498 | 117.40 | 123.55 | 139.17 | 138.52 | |
| ACHN | 98.19 | 62.27 | 83.07 | 93.00 | |
| CAKI-1 | 92.79 | 45.31 | 85.40 | 94.84 | |
| RXF 393 | 127.93 | 122.04 | 111.04 | 124.75 | |
| SN12C | 95.55 | 51.99 | 61.82 | 96.11 | |
| TK-10 | 116.06 | 149.92 | 106.85 | 138.28 | |
| UO-31 | nd | 44.75 | nd | nd | |
| Prostate Cancer | |||||
| GP | PC-3 | 93.40 | 61.79 | 82.26 | 83.31 |
| DU-145 | 107.42 | −76.86 | 94.31 | 113.09 | |
| Breast Cancer | |||||
| GP | MCF7 | 97.84 | 10.33 | −55.70 | 82.24 |
| MDA-MB-231/ATCC | nd | −6.27 | nd | nd | |
| HS 578T | 99.52 | 60.36 | 98.54 | 100.32 | |
| BT-549 | 121.24 | 87.43 | 105.05 | 112.78 | |
| T-47D | 95.48 | −58.60 | 44.01 | −47.43 | |
| MDA-MB-468 | 106.65 | −96.19 | −83.56 | −89.03 | |
| Molecule | PQ2 (D-837715/1) | PQ3 (D-838755/1) | ||||
|---|---|---|---|---|---|---|
| Panel/Cell Line | GI50 | TGI | LC50 | GI50 | TGI | LC50 |
| Leukemia | ||||||
| CCRF-CEM | 3.44 | >100 | >100 | 19.8 | 53.6 | >100 |
| HL-60(TB) | 2.46 | 7.24 | >100 | 3.76 | 16.6 | 76.1 |
| K-562 | 3.30 | 61.9 | >100 | 4.25 | 60.8 | >100 |
| MOLT-4 | 5.35 | 32.8 | >100 | 15.4 | 39.5 | >100 |
| RPMI-8226 | 13.9 | 55.0 | >100 | 15.1 | 38.2 | 96.3 |
| SR | 19.2 | 66.7 | >100 | 22.8 | 58.3 | >100 |
| Non-Small Cell Lung Cancer | ||||||
| A549/ATCC | 17.4 | 34.0 | 66.3 | 23.2 | 53.3 | >100 |
| EKVX | 12.8 | 25.9 | 52.0 | 14.3 | 28.0 | 54.9 |
| HOP-62 | 14.6 | 28.1 | 54.0 | 14.6 | 27.9 | 53.5 |
| HOP-92 | 13.3 | 28.0 | 58.9 | 13.3 | 29.1 | 63.7 |
| NCI-H226 | 12.1 | 26.7 | 59.2 | 15.6 | 33.6 | 72.4 |
| NCI-H23 | 3.84 | 16.3 | 43.2 | 10.5 | 25.8 | 63.1 |
| NCI-H322M | 14.8 | 28.1 | 53.3 | 14.2 | 27.2 | 52.3 |
| NCI-H460 | 16.1 | 34.2 | 72.6 | 17.1 | 36.1 | 76.2 |
| NCI-H522 | 3.01 | 11.7 | 34.7 | 5.30 | 19.4 | 50.3 |
| Colon Cancer | ||||||
| COLO 205 | 17.9 | 33.6 | 63.4 | 17.6 | 31.9 | 57.8 |
| HCC-2998 | 15.3 | 28.7 | 54.1 | 16.1 | 31.6 | 61.7 |
| HCT-116 | 2.93 | 10.9 | 33.7 | 7.58 | 24.3 | 65.7 |
| HCT-15 | 15.9 | 30.9 | 59.9 | 17.0 | 37.7 | 83.9 |
| HT29 | 16.0 | 31.3 | 61.0 | 21.8 | 46.3 | 98.3 |
| KM12 | 16.7 | 39.6 | 94.0 | 18.3 | 41.2 | 92.8 |
| SW-620 | 2.94 | 14.0 | 52.3 | 13.9 | 29.4 | 62.1 |
| CNS Cancer | ||||||
| SF-268 | 12.8 | 32.0 | 80.2 | 17.1 | 31.9 | 59.8 |
| SF-295 | 17.7 | 31.9 | 57.4 | 12.8 | 25.6 | 51.1 |
| SF-539 | 17.0 | 31.5 | 58.2 | 11.9 | 24.5 | 50.4 |
| SNB-19 | 16.3 | 30.5 | 57.2 | 16.0 | 29.7 | 54.9 |
| SNB-75 | 18.5 | 34.2 | 63.0 | 11.0 | 23.0 | 48.2 |
| U251 | 12.0 | 25.2 | 52.7 | 15.6 | 45.0 | >100 |
| Melanoma | ||||||
| LOX IMVI | 2.33 | 8.36 | 32.0 | 2.56 | 11.5 | 35.4 |
| MALME-3M | 2.25 | 5.37 | 17.2 | 1.71 | 3.31 | 6.41 |
| M14 | 13.6 | 26.7 | 52.5 | 14.8 | 28.4 | 54.3 |
| MDA-MB-435 | 1.81 | 3.35 | 6.21 | 1.72 | 3.18 | 5.88 |
| SK-MEL-2 | 11.7 | 24.2 | 50.1 | 12.1 | 26.6 | 58.4 |
| SK-MEL-28 | 14.5 | 28.0 | 54.0 | 8.19 | 20.9 | 46.7 |
| SK-MEL-5 | 11.6 | 23.8 | 49.1 | 12.3 | 24.8 | 50.1 |
| UACC-257 | 2.55 | 7.00 | 25.0 | 2.33 | 5.09 | 14.7 |
| UACC-62 | 5.78 | 18.4 | 43.8 | 14.4 | 27.6 | 52.9 |
| Ovarian Cancer | ||||||
| IGROV1 | 10.5 | 22.9 | 49.9 | 12.7 | 26.3 | 54.2 |
| OVCAR-3 | 2.10 | 7.16 | 29.1 | 2.06 | 6.51 | 24.8 |
| OVCAR-4 | 5.02 | 17.6 | 42.7 | 8.18 | 20.9 | 46.3 |
| OVCAR-5 | 6.43 | 19.3 | 44.8 | 3.65 | 14.7 | 43.7 |
| OVCAR-8 | 3.84 | 15.8 | 48.0 | 18.4 | 40.3 | 88.4 |
| NCI/ADR-RES | - | - | - | 17.6 | 38.2 | 82.9 |
| SK-OV-3 | 12.5 | 25.2 | 50.8 | 14.8 | 31.7 | 67.8 |
| Renal Cancer | ||||||
| 786-0 | 17.8 | 32.1 | 58.1 | 18.3 | 32.8 | 58.8 |
| A498 | 19.2 | 34.0 | 60.4 | 18.8 | 33.5 | 59.5 |
| ACHN | 13.8 | 26.8 | 51.8 | 12.5 | 25.1 | 50.2 |
| CAKI-1 | 10.7 | 23.3 | 50.4 | 11.9 | 24.5 | 50.3 |
| RXF 393 | 12.7 | 25.3 | 50.5 | 17.2 | 31.2 | 56.7 |
| SN12C | 14.9 | 28.3 | 53.5 | - | - | - |
| TK-10 | 19.2 | 33.3 | 57.8 | 22.6 | 37.9 | 63.8 |
| UO-31 | 11.0 | 23.2 | 49.1 | 8.31 | 21.7 | 48.0 |
| Prostate Cancer | ||||||
| PC-3 | 16.9 | 50.6 | >100 | 16.5 | 66.4 | >100 |
| DU-145 | 12.7 | 25.7 | 52.0 | 2.31 | 5.67 | 18.6 |
| Breast Cancer | ||||||
| MCF7 | 3.66 | 15.2 | 50.9 | 1.80 | 3.91 | 8.53 |
| MDA-MB-231/ATCC | 5.65 | 18.7 | 47.1 | 13.9 | 32.0 | 73.80 |
| HS 578T | 9.04 | 34.6 | >100 | 20.0 | 62.8 | >100 |
| BT-549 | 13.7 | 26.6 | 51.8 | 14.5 | 27.8 | 53.3 |
| T-47D | 2.01 | 4.74 | 69.7 | 12.3 | 33.4 | 91.3 |
| MDA-MB-468 | 0.222 | 0.885 | 3.25 | 1.21 | 2.88 | 6.83 |
| Compound | Cell Type (IC50, μM) | SI * | |
|---|---|---|---|
| Jurkat | PBMC | ||
| PQ2 | 2.94 ± 1.72 | 12.62 ± 4.07 | 4.29 |
| Imatinib | 7.11 ± 1.18 | 25.14 ± 6.41 | 3.54 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bayrak, N.; Sever, B.; Ciftci, H.; Otsuka, M.; Fujita, M.; TuYuN, A.F. Scaffold Hopping and Structural Modification of NSC 663284: Discovery of Potent (Non)Halogenated Aminobenzoquinones. Biomedicines 2024, 12, 50. https://doi.org/10.3390/biomedicines12010050
Bayrak N, Sever B, Ciftci H, Otsuka M, Fujita M, TuYuN AF. Scaffold Hopping and Structural Modification of NSC 663284: Discovery of Potent (Non)Halogenated Aminobenzoquinones. Biomedicines. 2024; 12(1):50. https://doi.org/10.3390/biomedicines12010050
Chicago/Turabian StyleBayrak, Nilüfer, Belgin Sever, Halilibrahim Ciftci, Masami Otsuka, Mikako Fujita, and Amaç Fatih TuYuN. 2024. "Scaffold Hopping and Structural Modification of NSC 663284: Discovery of Potent (Non)Halogenated Aminobenzoquinones" Biomedicines 12, no. 1: 50. https://doi.org/10.3390/biomedicines12010050
APA StyleBayrak, N., Sever, B., Ciftci, H., Otsuka, M., Fujita, M., & TuYuN, A. F. (2024). Scaffold Hopping and Structural Modification of NSC 663284: Discovery of Potent (Non)Halogenated Aminobenzoquinones. Biomedicines, 12(1), 50. https://doi.org/10.3390/biomedicines12010050