
Overcoming Chemotherapy Resistance in Metastatic Cancer: A Comprehensive Review



Abstract
1. Introduction
2. Molecular Mechanisms of Chemotherapy Resistance in Metastatic Cancer Cells
2.1. Genetic Alterations and Mutations Contributing to Chemotherapy Resistance
- DNA methylation inhibitors (DNMTi), including 5-azacitidine and 5-aza-2′-deoxycytidine (Decitabine; DAC; histone deacetylase inhibitors (iHDACs), such as Vorinostat, Belinostat, Romidepsin, and Panobinostat. Data have shown that combining conventional chemotherapeutics with epigenetic drugs, such as DAC, can overcome chemotherapy-resistant tumors. Even though DAC does not directly affect tumor growth, it inhibits DNA methylation, which sensitizes the tumor to other chemotherapeutics, including carboplatin, cisplatin, and 5-FU [11];
- Colorectal cancers have distinct epigenetics. While DNA methylation in MDF1, SSTR2, CMTM3, TGFB2, and NDRG4 genes is a potential marker for the detection of colorectal cancer in the early stages of its development, hypermethylation in the CLDN11 gene is associated with a metastasis characteristic and a poorer prognosis [12]. Silencing of tumor suppressor candidate 3 (TUSC3) mRNA expression by promoter hypermethylation induces upregulation of the epidermal growth factor receptor (EGDR), leading tumor cell resistance to apoptosis [12]. DNA methyltransferase inhibitors and drugs targeting histone deacetylases could potentially be a novel anticancer strategy in this model [13]. The latest data have demonstrated that CUDC-101 and CUDC-907, newly synthesized histone deacetylase/kinase inhibitors, showed therapeutic potential as anticancer agents in colon cancer [14,15].
2.2. Epigenetic Modifications and Their Impact on Drug Resistance
2.3. Altered Drug Metabolism and Efflux Mechanisms
2.4. Activation of Survival Pathways and Evasion of Cell Death
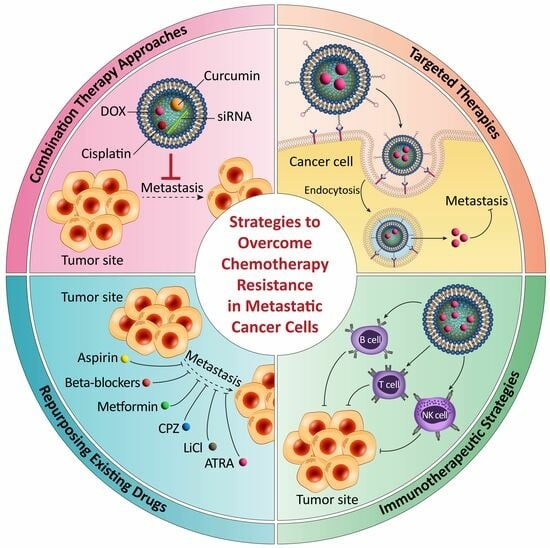
3. Strategies to Overcome Chemotherapy Resistance in Metastatic Cancer Cells
3.1. Combination Therapy Approaches and Rationale behind Their Effectiveness
3.2. Targeted Therapies and Their Potential in Overcoming Drug Resistance
3.3. Immunotherapeutic Strategies for Enhancing the Immune Response against Metastasis
3.4. Repurposing Existing Drugs and the Identification of New Therapeutic Targets
4. Prevention of Chemotherapy Resistance in Metastatic Cancer Cells
4.1. Strategies for Early Detection and Monitoring of Resistance Development
- (a)
- Fresh Tumor Cell Culture Assay (Tumor Chemosensitivity Assay): New tumor culture screening technology has been widely used for decades and good results have been achieved. However, its limitation is that it cannot predict the side effects of the drugs given to patients. Many randomized clinical trials and omics technologies, such as pharmacogenetics, have been proposed to solve this problem. This technology will be adapted to each patient’s needs and drug combination, providing a more in-depth understanding of the interaction between the patient’s genome and the drug used [127]. More than 50% of cancers are resistant to chemotherapy before chemotherapy is initiated. In additional cases, this resistance (so-called secondary resistance) develops after initiating treatment [120,127]. To obtain a new tumor, an oncologist must conduct a blood test, which requires proper planning for transferring the samples for a quick check. This method aims to obtain cancer cells from different tumor types that preserve their physiological properties [128]. Preparation methods will differ depending on the nature of the cancer cells; However, simple steps, such as cell extraction, incubation with antibodies, and cell viability assessment control, remain the same in all cell types. Many antibiotics are used because the primary purpose is to build the immune system. It is valuable to add that the gels used to treat the disease were also used in this experiment because the aim is to determine the anti-cancer effect. In each method, in addition to measuring cell viability, the molecular structure of tumor cells is also analyzed to indicate the growth or death of the cells and the hand activity level is also determined [127].Among the various pathways, thymidine incorporation into cellular DNA and the depletion of cellular ATP are the most commonly used mechanisms. The presence of protection can be confirmed by incorporating thymidine into cellular DNA or by the absence of a decrease in cellular ATP levels. The culture of new tumor cells is suitable for many types of cancer and, given their role in the cellular response, their predictive value can be measured as a precise measure of allergic reactions [128,129,130]. The advantage of this method is that it can be used not only in tumors (such as ovarian cancer, etc.) but also in hematological malignancies [130].
- (b)
- Cancer Biomarker Test: Biomarkers, such as DNA, RNA, peptides, genes, and proteins, can clearly understand a person’s cancer and its specific type. It is essential to understand this information because each person has a unique genome. Therefore, cancer treatment can be personalized according to the patient. This approach recognizes that chemotherapy resistance may vary from patient to patient, depending on the unique genome. Therefore, although the principle of cancer treatment remains the same, treatment details may differ due to genetic differences between people. These specific biomarkers provide essential information that can help physicians choose appropriate treatments, including using specific medications for cancer patients [129]. Cancer biomarkers also work as clinical tools that can measure the stage of cancer (blood in tissue) and predict, for example, a patient’s risk of developing cancer. They can also measure the resistance of cancer cells in the patient’s treatment. By following this approach, appropriate treatments can be selected for each specific cancer patient. This approach, with the help of omics technology, allows a better understanding of the needs of cancer patients and the use of different types of treatments. Thus, this approach may help increase the effectiveness of treatment and extend the patient’s life [6]. Two main groups of biomarkers are used to treat cancer patients: (1) anti-cancer biomarkers that help detect and treat cancer, in addition to diagnosing cancer and predicting the patient’s response to medications and (2) pharmacokinetic biomarkers that can help determine the optimal dose for cancer treatment. The biggest challenge facing these two biomarkers is that they are less helpful when applied to cancer than leukemia patients. This difference can be attributed to the occurrence of different types of cancer [128,129,130].In leukemia, many cancer cells can be easily found in the peripheral blood; thus, the use of anti-biomarkers is easier. In contrast, detecting these cells in the peripheral blood of cancer cells is more difficult because they can only follow the later stages of the disease. In this case, the only option is to have a biopsy or, in some cases, remove the tumor. However, analysis of these cells to determine the appropriate treatment often does not help diagnosis due to delays in diagnosis. Overall, each prognostic biomarker has advantages and disadvantages and is helpful for a particular stage. For example, genetic signatures are not valuable for cancer (due to difficulties in obtaining tissue) and do not serve as predictive disease biomarkers. On the other hand, tumor DNA genotyping appears to be more reliable as a predictive biomarker in these patients. Further research, especially in the field of predictive biomarkers, may help select the most appropriate treatment for cancer patients [130,131].
- (c)
- Positron Tomography: PET plays an essential role in the treatment strategy of cancer patients, especially in cancer treatment. This critical step allows clinicians to make informed results about the most appropriate treatment for individual patients. PET can help physicians make an accurate diagnosis of cancer by improving early detection or determining the stage of the disease. Therefore, PET scans may help select curative treatments for early-stage tumors or palliative approaches for invasive disease. In addition, since oncology treatment is complex and challenging, early diagnosis is essential in increasing the effectiveness of treatment and reducing financial costs for patients [6,129,130]. PET/CT imaging technology is beneficial in the early diagnosis of cancer. This method is based on the observation that cancer cells will absorb more radiation, resulting in a brighter image. This brightness can help identify cancer cells in the early stages of the disease. In addition, physicians can offer appropriate treatment to patients with cancer. This helps choose the proper treatment and reduces the risk of using anti-cancer drugs by avoiding inappropriate medication or dosage. Remember, although histopathology provides a reliable assessment of cancer treatment, only a smaller number of patients (20–40%) achieve a complete pathological response. Therefore, increasing accessibility to early diagnosis and treatment can improve the quality of treatment. PET/CT imaging is one of the methods that can help achieve this goal [130].
- (d)
- High Throughput Pharmacogenomic CRISPR Analysis: High throughput CRISPR technology is a promising new genomic approach for cancer research, especially in the summary of hematological malignancies. This technology can potentially be used in many types of cancer for fundamental purposes, such as identifying regulatory genes that can serve as biomarkers for malignant transformation and developing therapeutic targets and new drugs. It is an essential tool in biological research, especially in the biotechnology and pharmaceutical industries. It has also become routinely used to examine hematological cancers in recent years, making early cancer detection one of its main applications. CRISPR/Cas9 currently provides many genome editors; these include the CRISPR/Cas9 nucleotide sequence editor, CRISPR/Cas base editor (BE), CRISPR primer editor (PE), and CRISPR interference (CRISPRi) (such as CRISPRa, CRISPRa, and CRISPRr). They are also used in many biological sciences, such as early cancer detection, cancer diagnosis, and the development of new drugs to treat hemorrhagic cancer [118].
4.2. Rational Drug Design and Personalized Medicine Approach
4.3. The role of Predictive Biomarkers in Guiding Medical Decisions
4.4. Lifestyle Changes and Medical Support Improve Treatment Results
5. Clinical Importance and Future Direction
5.1. Clinical Research and Treatment Results of Metastatic Colorectal Cancer Cells
5.2. Challenges and Opportunities in Interpreting Clinical Trials
5.3. Potential Future Directions and New Therapeutic Approaches
- (a)
- The development of immunotherapy represents a promising future [154]. The combination of chemotherapy and radiation with immunotherapy is one of these methods. This approach is to reduce tumor cells, causing them to die while increasing the glucose level that natural killer cells need to kill cancer cells. Additionally, other methods include administering nutrients that can inhibit the glycolytic process of the immune system [155]. Epigenetic therapy also has the potential to find effective solutions to cancer [156]. Determining the strategy to improve the effect of epigenetic factors in these tumors is a good example in this field [154];
- (b)
- (c)
- Activity, especially exercise, has been shown to be beneficial to cancer patients. Exercising before surgery can increase the body’s strength, resulting in an overall improvement in the patient’s health before and after surgery [159]. There is also good evidence to support the use of exercise as a way to prevent cancer. Epidemiological studies have shown that exercise is effective in controlling symptoms and improving the quality of life in cancer patients, especially prostate cancer patients [160];
- (d)
- The emergence of multi-omics, a set of diagnostic tools that include genomic, epigenomic, transcriptomic, epitranscriptomic, and proteomic networks, has revolutionized cancer treatment. This technology has made it possible to diagnose and treat diseases such as cancer [161].
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACS | American Cancer Society |
| AMPK | Adenosine Monophosphate-activated Protein Kinase |
| ARSI | Androgen Receptor Signaling Inhibitor |
| BCRT | Breast Cancer C-terminal Domains |
| CSC | Cancer Stem Cell |
| CAR | Chimeric Antigen Receptor |
| COX | Cyclooxygenase |
| CPZ | Chlorpromazine |
| DDP | Diamminedichloroplatinum |
| DHFR | Dihydrofolate Reductase |
| DOX | Doxorubicin |
| ER | Estrogen Receptor |
| FLICE | FADD-like IL-1β-converting Enzyme |
| HER2 | Human Epidermal growth factor Receptor-related protein (HER2) |
| HIF | Hypoxia-inducible Factor |
| IAPs | Inhibitors of Apoptosis Proteins |
| iDNMT | DNA Methylation Inhibitors |
| IHC | Immunohistochemical |
| iHDACs | Histone Deacetylase Inhibitors |
| JNK | Jun N-terminal Kinase |
| MBZ | Mebendazole |
| MDM2 | Murine Double Minute 2 |
| MDR | Multidrug Resistance |
| MRP | Multidrug Resistance Protein |
| MTX | Methotrexate |
| PEI | Polyethyleneimine |
| PEO | Polyethylene Oxide |
| PET | Positron Emission Tomography |
| PHD | Prolyl Hydroxylase Domain |
| PLGA | Poly lactic-co-glycolic acid |
| PR | Progesterone Receptor |
| PTM | Posttranscriptional Modifications |
| RCC | Renal Cell Carcinoma |
| RES | Reticuloendothelial system |
| RFC | Reduced Folate Carrier |
| TAM | Tumor-associated Macrophages |
| TCTP | Translationally Controlled Tumor Protein |
| TME | Tumor Microenvironment |
| TRAIL | Tumor necrosis factor-Related Apoptosis-Inducing Ligand |
| TS | Thymidylate Synthase |
| VEGF | Vascular Endothelial Growth Factor |
References
- Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 4 January 2024).
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Carelle, N.; Piotto, E.; Bellanger, A.; Germanaud, J.; Thuillier, A.; Khayat, D. Changing patient perceptions of the side effects of cancer chemotherapy. Cancer 2002, 95, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, B.C.; Gillet, J.-P.; Patel, C.; Baer, M.R.; Bates, S.E.; Gottesman, M.M. Drug resistance: Still a daunting challenge to the successful treatment of AML. Drug Resist. Updates 2012, 15, 62–69. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Ahmad, A.; Azmi, A.S.; Kong, D.; Banerjee, S.; Sarkar, F.H. Targeting miRNAs involved in cancer stem cell and EMT regulation: An emerging concept in overcoming drug resistance. Drug Resist. Updates 2010, 13, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.; Johnston, P. Molecular mechanisms of drug resistance. J. Pathol. J. Pathol. Soc. Great Br. Irel. 2005, 205, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Shilatifard, A. Epigenetic modifications of histones in cancer. Genome Biol. 2019, 20, 245. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, R.; Gupta, S. Epigenetic modifications in cancer. Clin. Genet. 2012, 81, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, H.P.; Barbash, O.; Creasy, C.L. Targeting epigenetic modifications in cancer therapy: Erasing the roadmap to cancer. Nat. Med. 2019, 25, 403–418. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Shirjang, S.; Baradaran, B. Micro-RNAs: The new potential biomarkers in cancer diagnosis, prognosis and cancer therapy. Cell. Mol. Biol. 2015, 61, 1–10. [Google Scholar]
- Okabe, S.; Tanaka, Y.; Moriyama, M.; Gotoh, A. Effect of dual inhibition of histone deacetylase and phosphatidylinositol-3 kinase in Philadelphia chromosome-positive leukemia cells. Cancer Chemother. Pharmacol. 2020, 85, 401–412. [Google Scholar] [CrossRef]
- He, P.-F.; Zhou, J.-D.; Yao, D.-M.; Ma, J.-C.; Wen, X.-M.; Zhang, Z.-H.; Lian, X.-Y.; Xu, Z.-J.; Qian, J.; Lin, J. Efficacy and safety of decitabine in treatment of elderly patients with acute myeloid leukemia: A systematic review and meta-analysis. Oncotarget 2017, 8, 41498. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Luo, X.; Wu, Y.; Xia, D.; Chen, W.; Fang, Z.; Deng, J.; Hao, Y.; Yang, X.; Zhang, T.; et al. MicroRNA-34a attenuates paclitaxel resistance in prostate cancer cells via direct suppression of JAG1/Notch1 axis. Cell. Physiol. Biochem. 2018, 50, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Goey, A.K.; Sissung, T.M.; Peer, C.J.; Figg, W.D. Pharmacogenomics and histone deacetylase inhibitors. Pharmacogenomics 2016, 17, 1807–1815. [Google Scholar] [CrossRef]
- Patnaik, S.; Anupriya. Drugs targeting epigenetic modifications and plausible therapeutic strategies against colorectal cancer. Front. Pharmacol. 2019, 10, 588. [Google Scholar] [CrossRef] [PubMed]
- Si, W.; Shen, J.; Zheng, H.; Fan, W. The role and mechanisms of action of microRNAs in cancer drug resistance. Clin. Epigenet. 2019, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; LoRusso, P.M.; Papadopoulos, K.P.; Patnaik, A.; Beeram, M.; Smith, L.S.; Rasco, D.W.; Mays, T.-A.; Chambers, G.; Ma, A.; et al. Phase I first-in-human study of CUDC-101, a multitargeted inhibitor of HDACs, EGFR, and HER2 in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 5032–5040. [Google Scholar] [CrossRef]
- Kato, S.; Maeda, Y.; Sugiyama, D.; Watanabe, K.; Nishikawa, H.; Hinohara, K. The cancer epigenome: Non-cell autonomous player in tumor immunity. Cancer Sci. 2023, 114, 730–740. [Google Scholar] [CrossRef]
- Moore, P.C.; Henderson, K.W.; Classon, M. The epigenome and the many facets of cancer drug tolerance. Adv. Cancer Res. 2023, 158, 1–39. [Google Scholar]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef]
- Lowrence, R.C.; Subramaniapillai, S.G.; Ulaganathan, V.; Nagarajan, S. Tackling drug resistance with efflux pump inhibitors: From bacteria to cancerous cells. Crit. Rev. Microbiol. 2019, 45, 334–353. [Google Scholar] [CrossRef]
- Levy, S.B.; Marshall, B. Antibacterial resistance worldwide: Causes, challenges and responses. Nat. Med. 2004, 10 (Suppl. S12), S122–S129. [Google Scholar] [CrossRef] [PubMed]
- Persidis, A. Cancer multidrug resistance. Nat. Biotechnol. 1999, 17, 94–95. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.T.; Zhou, T.T.; Liu, B.; Bao, J.K. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef]
- Green, D.R.; Kroemer, G. The pathophysiology of mitochondrial cell death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.H.; Viacava Follis, A.; Kriwacki, R.W.; Moldoveanu, T. Discoveries and controversies in BCL-2 protein-mediated apoptosis. FEBS J. 2016, 283, 2690–2700. [Google Scholar] [CrossRef] [PubMed]
- Um, H.-D. Bcl-2 family proteins as regulators of cancer cell invasion and metastasis: A review focusing on mitochondrial respiration and reactive oxygen species. Oncotarget 2016, 7, 5193. [Google Scholar] [CrossRef] [PubMed]
- Schuyer, M.; Van der Burg, M.; Henzen-Logmans, S.; Fieret, J.; Klijn, J.; Look, M.; Foekens, J.-A.; Stoter, G.; Berns, E.-M. Reduced expression of BAX is associated with poor prognosis in patients with epithelial ovarian cancer: A multifactorial analysis of TP53, p21, BAX and BCL-2. Br. J. Cancer 2001, 85, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Sturm, I.; Stephan, C.; Gillissen, B.; Siebert, R.; Janz, M.; Radetzki, S.; Jung, K.; Loening, S.; Dörken, B.; Daniel, P.T. Loss of the tissue-specific proapoptotic BH3-only protein Nbk/Bik is a unifying feature of renal cell carcinoma. Cell Death Differ. 2006, 13, 619–627. [Google Scholar] [CrossRef]
- Fujimoto, A.; Takeuchi, H.; Taback, B.; Hsueh, E.C.; Elashoff, D.; Morton, D.L.; Hoon, D.-S. Allelic imbalance of 12q22–23 associated with APAF-1 locus correlates with poor disease outcome in cutaneous melanoma. Cancer Res. 2004, 64, 2245–2250. [Google Scholar] [CrossRef]
- Jung, J.; Kim, H.Y.; Maeng, J.; Kim, M.; Shin, D.H.; Lee, K. Interaction of translationally controlled tumor protein with Apaf-1 is involved in the development of chemoresistance in HeLa cells. BMC Cancer 2014, 14, 165. [Google Scholar] [CrossRef]
- Jin, Z.; McDonald, E.R.; Dicker, D.T.; El-Deiry, W.S. Deficient tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor transport to the cell surface in human colon cancer cells selected for resistance to TRAIL-induced apoptosis. J. Biol. Chem. 2004, 279, 35829–35839. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, J.; Robbins, D.; Morris, K.; Sit, A.; Liu, Y.-Y.; Zhao, Y. Mutant p53 exhibits trivial effects on mitochondrial functions which can be reactivated by ellipticine in lymphoma cells. Apoptosis 2011, 16, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Juan, H.-C.; Tsai, H.-T.; Chang, P.-H.; Huang, C.-Y.F.; Hu, C.-P.; Wong, F.-H. Insulin-like growth factor 1 mediates 5-fluorouracil chemoresistance in esophageal carcinoma cells through increasing Survivin stability. Apoptosis 2011, 16, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Ganta, S.; Amiji, M. Coadministration of paclitaxel and curcumin in nanoemulsion formulations to overcome multidrug resistance in tumor cells. Mol. Pharm. 2009, 6, 928–939. [Google Scholar] [CrossRef]
- Amornwachirabodee, K.; Chiablaem, K.; Wacharasindhu, S.; Lirdprapamongkol, K.; Svasti, J.; Vchirawongkwin, V.; Wanichwecharungruang, S.-P. Paclitaxel delivery using carrier made from curcumin derivative: Synergism between carrier and the loaded drug for effective cancer treatment. J. Pharm. Sci. 2012, 101, 3779–3786. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, D.; Campbell, N.R.; Das, S.; Gupta, S.; Chenna, V.; Bisht, S.; Sysa-Shah, P.; Bedja, D.; Karikari, C.; Steenbergen, C.; et al. A composite polymer nanoparticle overcomes multidrug resistance and ameliorates doxorubicin-associated cardiomyopathy. Oncotarget 2012, 3, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Misra, R.; Sahoo, S.K. Coformulation of doxorubicin and curcumin in poly(D,L-lactide-co-glycolide) nanoparticles suppresses the development of multidrug resistance in K562 cells. Mol. Pharm. 2011, 8, 852–866. [Google Scholar] [CrossRef]
- Duan, J.; Mansour, H.M.; Zhang, Y.; Deng, X.; Chen, Y.; Wang, J.; Pan, Y.; Zhao, J. Reversion of multidrug resistance by co-encapsulation of doxorubicin and curcumin in chitosan/poly(butyl cyanoacrylate) nanoparticles. Int. J. Pharm. 2012, 426, 193–201. [Google Scholar] [CrossRef]
- Liscovitch, M.; Ravid, D. A case study in misidentification of cancer cell lines: MCF-7/AdrR cells (re-designated NCI/ADR-RES) are derived from OVCAR-8 human ovarian carcinoma cells. Cancer Lett. 2007, 245, 350–352. [Google Scholar] [CrossRef]
- Ye, M.X.; Zhao, Y.L.; Li, Y.; Miao, Q.; Li, Z.K.; Ren, X.L.; Song, L.Q.; Yin, H.; Zhang, J. Curcumin reverses cis-platin resistance and promotes human lung adenocarcinoma A549/DDP cell apoptosis through HIF-1α and caspase-3 mechanisms. Phytomedicine 2012, 19, 779–787. [Google Scholar] [CrossRef]
- Binkhathlan, Z.; Shayeganpour, A.; Brocks, D.R.; Lavasanifar, A. Encapsulation of P-glycoprotein inhibitors by polymeric micelles can reduce their pharmacokinetic interactions with doxorubicin. Eur. J. Pharm. Biopharm. 2012, 81, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.R.; Rathi, A.; Mongayt, D.; Torchilin, V.P. Reversal of multidrug resistance by co-delivery of tariquidar (XR9576) and paclitaxel using long-circulating liposomes. Int. J. Pharm. 2011, 416, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Prasad, P.; Shuhendler, A.; Cai, P.; Rauth, A.M.; Wu, X.Y. Doxorubicin and mitomycin C co-loaded polymer-lipid hybrid nanoparticles inhibit growth of sensitive and multidrug resistant human mammary tumor xenografts. Cancer Lett. 2013, 334, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Creixell, M.; Peppas, N.A. Co-delivery of siRNA and therapeutic agents using nanocarriers to overcome cancer resistance. Nano Today 2012, 7, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Sawant, R.R.; Biswas, S.; Essex, S.; Tros de Ilarduya, C.; Torchilin, V.P. P-glycoprotein silencing with siRNA delivered by DOPE-modified PEI overcomes doxorubicin resistance in breast cancer cells. Nanomedicine 2012, 7, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Su, W.P.; Cheng, F.Y.; Shieh, D.B.; Yeh, C.S.; Su, W.C. PLGA nanoparticles codeliver paclitaxel and Stat3 siRNA to overcome cellular resistance in lung cancer cells. Int. J. Nanomed. 2012, 7, 4269–4283. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Shen, J.; Chen, L.; Zhang, Z.; Gu, W.; Li, Y. Overcoming multidrug resistance by co-delivery of Mdr-1 and Survivin-targeting RNA with reduction-responsible cationic poly(β-amino esters). Biomaterials 2012, 33, 6495–6506. [Google Scholar] [CrossRef]
- Hao, Y.X.; He, Z.W.; Zhu, J.H.; Shen, Q.; Sun, J.Z.; Du, N.; Xiao, W.H. Reversal of multidrug resistance in renal cell carcinoma by short hairpin RNA targeting MDR1 gene. Chin. Med. J. 2012, 125, 2741–2745. [Google Scholar]
- Chen, A.M.; Zhang, M.; Wei, D.; Stueber, D.; Taratula, O.; Minko, T.; He, H. Co-delivery of doxorubicin and Bcl-2 siRNA by mesoporous silica nanoparticles enhances the efficacy of chemotherapy in multidrug-resistant cancer cells. Small 2009, 5, 2673–2677. [Google Scholar] [CrossRef]
- Dhule, S.S.; Penfornis, P.; Frazier, T.; Walker, R.; Feldman, J.; Tan, G.; He, J.; Alb, A.; John, V.; Pochampally, R. Curcumin-loaded γ-cyclodextrin liposomal nanoparticles as delivery vehicles for osteosarcoma. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 440–451. [Google Scholar] [CrossRef]
- Glienke, W.; Maute, L.; Wicht, J.; Bergmann, L. Wilms’ tumour gene 1 (WT1) as a target in curcumin treatment of pancreatic cancer cells. Eur. J. Cancer 2009, 45, 874–880. [Google Scholar] [CrossRef] [PubMed]
- Palakurthi, S.; Yellepeddi, V.K.; Vangara, K.K. Recent trends in cancer drug resistance reversal strategies using nanoparticles. Expert Opin. Drug Deliv. 2012, 9, 287–301. [Google Scholar] [CrossRef]
- Maeda, H. The enhanced permeability and retention (EPR) effect in tumor vasculature: The key role of tumor-selective macromolecular drug targeting. Adv. Enzym. Regul. 2001, 41, 189–207. [Google Scholar] [CrossRef]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Klibanov, A.L.; Maruyama, K.; Torchilin, V.P.; Huang, L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990, 268, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Zaki, N.M.; Tirelli, N. Gateways for the intracellular access of nanocarriers: A review of receptor-mediated endocytosis mechanisms and of strategies in receptor targeting. Expert Opin. Drug Deliv. 2010, 7, 895–913. [Google Scholar] [CrossRef]
- Prabaharan, M.; Grailer, J.J.; Steeber, D.A.; Gong, S. Thermosensitive micelles based on folate-conjugated poly(N-vinylcaprolactam)-block-poly(ethylene glycol) for tumor-targeted drug delivery. Macromol. Biosci. 2009, 9, 744–753. [Google Scholar] [CrossRef]
- Byrne, J.D.; Betancourt, T.; Brannon-Peppas, L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1615–1626. [Google Scholar] [CrossRef]
- Davis, M.E.; Chen, Z.G.; Shin, D.M. Nanoparticle therapeutics: An emerging treatment modality for cancer. Nat. Rev. Drug Discov. 2008, 7, 771–782. [Google Scholar] [CrossRef]
- Wang, X.; Yang, L.; Chen, Z.G.; Shin, D.M. Application of nanotechnology in cancer therapy and imaging. CA Cancer J. Clin. 2008, 58, 97–110. [Google Scholar] [CrossRef]
- Wang, Y.J.; Pan, M.H.; Cheng, A.L.; Lin, L.I.; Ho, Y.S.; Hsieh, C.Y.; Lin, J.K. Stability of curcumin in buffer solutions and characterization of its degradation products. J. Pharm. Biomed. Anal. 1997, 15, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Cui, D. Cancer Nano-Immunoengineering: The Marriage of Immunoengineering and Nanotechnology for Cancer Therapy. Nano Biomed. Eng. 2016, 8, 105–107. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, J.; Xia, F.; Zhang, C.; Qian, Q.; Zhi, X.; Yue, C.; Sun, R.; Cheng, S.; Fang, S.; et al. Human CIK Cells Loaded with Au Nanorods as a Theranostic Platform for Targeted Photoacoustic Imaging and Enhanced Immunotherapy and Photothermal Therapy. Nanoscale Res. Lett. 2016, 11, 285. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Li, L.; Liu, Y.; Wang, Z.; Zhu, X.; Bai, X. Interleukin-1α induces immunosuppression by mesenchymal stem cells promoting the growth of prostate cancer cells. Mol. Med. Rep. 2012, 6, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Anguille, S.; Smits, E.L.; Lion, E.; van Tendeloo, V.F.; Berneman, Z.N. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014, 15, e257–e267. [Google Scholar] [CrossRef] [PubMed]
- Frey, A.B.; Monu, N. Effector-phase tolerance: Another mechanism of how cancer escapes antitumor immune response. J. Leukoc. Biol. 2006, 79, 652–662. [Google Scholar] [CrossRef]
- Cheng, M.; Chen, Y.; Xiao, W.; Sun, R.; Tian, Z. NK cell-based immunotherapy for malignant diseases. Cell. Mol. Immunol. 2013, 10, 230–252. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Tey, S.K.; Bollard, C.M.; Heslop, H.E. Adoptive T-cell transfer in cancer immunotherapy. Immunol. Cell Biol. 2006, 84, 281–289. [Google Scholar] [CrossRef]
- Dranoff, G. Cytokines in cancer pathogenesis and cancer therapy. Nat. Rev. Cancer 2004, 4, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Finn, O.J. Immuno-oncology: Understanding the function and dysfunction of the immune system in cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2012, 23 (Suppl. S8), viii6–viii9. [Google Scholar] [CrossRef]
- Ward, E.; Smith, R.; Branca, J.; Noakes, D.; Morucci, G.; Thyer, L. Clinical experience of cancer immunotherapy integrated with Oleic Acid complexed with de-glycosylated vitamin D binding protein. Am. J. Immunol. 2014, 10, 23–32. [Google Scholar] [CrossRef][Green Version]
- Boehm, A.L.; Higgins, J.; Franzusoff, A.; Schlom, J.; Hodge, J.W. Concurrent vaccination with two distinct vaccine platforms targeting the same antigen generates phenotypically and functionally distinct T-cell populations. Cancer Immunol. Immunother. CII 2010, 59, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Kuai, R.; Ochyl, L.J.; Bahjat, K.S.; Schwendeman, A.; Moon, J.J. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater. 2017, 16, 489–496. [Google Scholar] [CrossRef]
- Inada, A.; Nakanishi, T.; Konoshima, T.; Kozuka, M.; Tokuda, H.; Nishino, H.; Iwashima, A. Anti-tumor promoting activities of natural products. II. Inhibitory effects of digitoxin on two-stage carcinogenesis of mouse skin tumors and mouse pulmonary tumors. Biol. Pharm. Bull. 1993, 16, 930–931. [Google Scholar] [CrossRef][Green Version]
- Barry, W.H.; Hasin, Y.; Smith, T.W. Sodium pump inhibition, enhanced calcium influx via sodium-calcium exchange, and positive inotropic response in cultured heart cells. Circ. Res. 1985, 56, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Durlacher, C.T.; Chow, K.; Chen, X.W.; He, Z.X.; Zhang, X.; Yang, T.; Zhou, S.F. Targeting Na+/K+ -translocating adenosine triphosphatase in cancer treatment. Clin. Exp. Pharmacol. Physiol. 2015, 42, 427–443. [Google Scholar] [CrossRef]
- Bielawski, K.; Winnicka, K.; Bielawska, A. Inhibition of DNA topoisomerases I and II, and growth inhibition of breast cancer MCF-7 cells by ouabain, digoxin and proscillaridin A. Biol. Pharm. Bull. 2006, 29, 1493–1497. [Google Scholar] [CrossRef]
- Ishida, J.; Konishi, M.; Ebner, N.; Springer, J. Repurposing of approved cardiovascular drugs. J. Transl. Med. 2016, 14, 269. [Google Scholar] [CrossRef]
- Reimers, M.S.; Bastiaannet, E.; van Herk-Sukel, M.P.; Lemmens, V.E.; van den Broek, C.B.; van de Velde, C.J.; de Craen, A.J.; Liefers, G.J. Aspirin use after diagnosis improves survival in older adults with colon cancer: A retrospective cohort study. J. Am. Geriatr. Soc. 2012, 60, 2232–2236. [Google Scholar] [CrossRef] [PubMed]
- Goh, C.H.; Leong, W.Q.; Chew, M.H.; Pan, Y.S.; Tony, L.K.; Chew, L.; Tan, I.B.; Toh, H.C.; Tang, C.L.; Fu, W.P.; et al. Post-operative aspirin use and colorectal cancer-specific survival in patients with stage I-III colorectal cancer. Anticancer Res. 2014, 34, 7407–7414. [Google Scholar] [PubMed]
- Chin, C.C.; Li, J.M.; Lee, K.F.; Huang, Y.C.; Wang, K.C.; Lai, H.C.; Cheng, C.C.; Kuo, Y.H.; Shi, C.S. Selective β2-AR Blockage Suppresses Colorectal Cancer Growth Through Regulation of EGFR-Akt/ERK1/2 Signaling, G1-Phase Arrest, and Apoptosis. J. Cell. Physiol. 2016, 231, 459–472. [Google Scholar] [CrossRef]
- Sloan, E.K.; Priceman, S.J.; Cox, B.F.; Yu, S.; Pimentel, M.A.; Tangkanangnukul, V.; Arevalo, J.M.; Morizono, K.; Karanikolas, B.D.; Wu, L.; et al. The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res. 2010, 70, 7042–7052. [Google Scholar] [CrossRef]
- Pollak, M. Overcoming Drug Development Bottlenecks with Repurposing: Repurposing biguanides to target energy metabolism for cancer treatment. Nat. Med. 2014, 20, 591–593. [Google Scholar] [CrossRef] [PubMed]
- He, X.X.; Tu, S.M.; Lee, M.H.; Yeung, S.J. Thiazolidinediones and metformin associated with improved survival of diabetic prostate cancer patients. Ann. Oncol. 2011, 22, 2640–2645. [Google Scholar] [CrossRef]
- He, X.; Esteva, F.J.; Ensor, J.; Hortobagyi, G.N.; Lee, M.H.; Yeung, S.C. Metformin and thiazolidinediones are associated with improved breast cancer-specific survival of diabetic women with HER2+ breast cancer. Ann. Oncol. 2012, 23, 1771–1780. [Google Scholar] [CrossRef]
- Horman, S.; Browne, G.; Krause, U.; Patel, J.; Vertommen, D.; Bertrand, L.; Lavoinne, A.; Hue, L.; Proud, C.; Rider, M. Activation of AMP-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Curr. Biol. CB 2002, 12, 1419–1423. [Google Scholar] [CrossRef]
- Laskov, I.; Drudi, L.; Beauchamp, M.C.; Yasmeen, A.; Ferenczy, A.; Pollak, M.; Gotlieb, W.H. Anti-diabetic doses of metformin decrease proliferation markers in tumors of patients with endometrial cancer. Gynecol. Oncol. 2014, 134, 607–614. [Google Scholar] [CrossRef]
- Rothermundt, C.; Hayoz, S.; Templeton, A.J.; Winterhalder, R.; Strebel, R.T.; Bärtschi, D.; Pollak, M.; Lui, L.; Endt, K.; Schiess, R.; et al. Metformin in chemotherapy-naive castration-resistant prostate cancer: A multicenter phase 2 trial (SAKK 08/09). Eur. Urol. 2014, 66, 468–474. [Google Scholar] [CrossRef]
- Chen, M.H.; Yang, W.L.; Lin, K.T.; Liu, C.H.; Liu, Y.W.; Huang, K.W.; Chang, P.M.; Lai, J.M.; Hsu, C.N.; Chao, K.M.; et al. Gene expression-based chemical genomics identifies potential therapeutic drugs in hepatocellular carcinoma. PLoS ONE 2011, 6, e27186. [Google Scholar] [CrossRef] [PubMed]
- Pulkoski-Gross, A.; Li, J.; Zheng, C.; Li, Y.; Ouyang, N.; Rigas, B.; Zucker, S.; Cao, J. Repurposing the antipsychotic trifluoperazine as an antimetastasis agent. Mol. Pharmacol. 2015, 87, 501–512. [Google Scholar] [CrossRef]
- Daley, E.; Wilkie, D.; Loesch, A.; Hargreaves, I.P.; Kendall, D.A.; Pilkington, G.J.; Bates, T.E. Chlorimipramine: A novel anticancer agent with a mitochondrial target. Biochem. Biophys. Res. Commun. 2005, 328, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Phiel, C.J.; Klein, P.S. Molecular targets of lithium action. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 789–813. [Google Scholar] [CrossRef] [PubMed]
- Cordero, M.D.; Sánchez-Alcázar, J.A.; Bautista-Ferrufino, M.R.; Carmona-López, M.I.; Illanes, M.; Ríos, M.J.; Garrido-Maraver, J.; Alcudia, A.; Navas, P.; de Miguel, M. Acute oxidant damage promoted on cancer cells by amitriptyline in comparison with some common chemotherapeutic drugs. Anti-Cancer Drugs 2010, 21, 932–944. [Google Scholar] [CrossRef] [PubMed]
- Maeng, Y.-S.; Lee, R.; Lee, B.; Choi, S.-I.; Kim, E.K. Lithium inhibits tumor lymphangiogenesis and metastasis through the inhibition of TGFBIp expression in cancer cells. Sci. Rep. 2016, 6, 20739. [Google Scholar] [CrossRef]
- Lubner, S.J.; Kunnimalaiyaan, M.; Holen, K.D.; Ning, L.; Ndiaye, M.; Loconte, N.K.; Mulkerin, D.L.; Schelman, W.R.; Chen, H. A preclinical and clinical study of lithium in low-grade neuroendocrine tumors. Oncologist 2011, 16, 452–457. [Google Scholar] [CrossRef]
- Cui, L.; Su, X.Z. Discovery, mechanisms of action and combination therapy of artemisinin. Expert Rev. Anti-Infect. Ther. 2009, 7, 999–1013. [Google Scholar] [CrossRef]
- Kundu, C.N.; Das, S.; Nayak, A.; Satapathy, S.R.; Das, D.; Siddharth, S. Anti-malarials are anti-cancers and vice versa—One arrow two sparrows. Acta Trop. 2015, 149, 113–127. [Google Scholar] [CrossRef]
- Holien, T.; Olsen, O.E.; Misund, K.; Hella, H.; Waage, A.; Rø, T.B.; Sundan, A. Lymphoma and myeloma cells are highly sensitive to growth arrest and apoptosis induced by artesunate. Eur. J. Haematol. 2013, 91, 339–346. [Google Scholar] [CrossRef]
- Dobrosotskaya, I.Y.; Hammer, G.D.; Schteingart, D.E.; Maturen, K.E.; Worden, F.P. Mebendazole monotherapy and long-term disease control in metastatic adrenocortical carcinoma. Endocr. Pract. 2011, 17, e59–e62. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.J.; Wang, W.Q.; Wu, G.D.; Lee, J.; Li, A. Artesunate inhibits angiogenesis and downregulates vascular endothelial growth factor expression in chronic myeloid leukemia K562 cells. Vasc. Pharmacol. 2007, 47, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.Y.; Staedtke, V.; Aprhys, C.M.; Gallia, G.L.; Riggins, G.J. Antiparasitic mebendazole shows survival benefit in 2 preclinical models of glioblastoma multiforme. Neuro-Oncology 2011, 13, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Nacev, B.A.; Grassi, P.; Dell, A.; Haslam, S.M.; Liu, J.O. The antifungal drug itraconazole inhibits vascular endothelial growth factor receptor 2 (VEGFR2) glycosylation, trafficking, and signaling in endothelial cells. J. Biol. Chem. 2011, 286, 44045–44056. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Varona-Santos, J.; Singh, S.; Robbins, D.J.; Savaraj, N.; Nguyen, D.M. Targeting of the Hedgehog signal transduction pathway suppresses survival of malignant pleural mesothelioma cells in vitro. J. Thorac. Cardiovasc. Surg. 2014, 147, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Adekola, K.U.; Dalva Aydemir, S.; Ma, S.; Zhou, Z.; Rosen, S.T.; Shanmugam, M. Investigating and targeting chronic lymphocytic leukemia metabolism with the human immunodeficiency virus protease inhibitor ritonavir and metformin. Leuk. Lymphoma 2015, 56, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Dalva-Aydemir, S.; Bajpai, R.; Martinez, M.; Adekola, K.U.; Kandela, I.; Wei, C.; Singhal, S.; Koblinski, J.E.; Raje, N.S.; Rosen, S.T.; et al. Targeting the metabolic plasticity of multiple myeloma with FDA-approved ritonavir and metformin. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1161–1171. [Google Scholar] [CrossRef]
- Sato, A.; Asano, T.; Isono, M.; Ito, K.; Asano, T. Ritonavir acts synergistically with panobinostat to enhance histone acetylation and inhibit renal cancer growth. Mol. Clin. Oncol. 2014, 2, 1016–1022. [Google Scholar] [CrossRef]
- Pore, N.; Gupta, A.K.; Cerniglia, G.J.; Jiang, Z.; Bernhard, E.J.; Evans, S.M.; Koch, C.J.; Hahn, S.M.; Maity, A. Nelfinavir down-regulates hypoxia-inducible factor 1alpha and VEGF expression and increases tumor oxygenation: Implications for radiotherapy. Cancer Res. 2006, 66, 9252–9259. [Google Scholar] [CrossRef]
- Pantziarka, P.; Sukhatme, V.; Bouche, G.; Meheus, L.; Sukhatme, V.P. Repurposing Drugs in Oncology (ReDO)-diclofenac as an anti-cancer agent. Ecancermedicalscience 2016, 10, 610. [Google Scholar] [CrossRef]
- Driessen, C.; Kraus, M.; Joerger, M.; Rosing, H.; Bader, J.; Hitz, F.; Berset, C.; Xyrafas, A.; Hawle, H.; Berthod, G.; et al. Treatment with the HIV protease inhibitor nelfinavir triggers the unfolded protein response and may overcome proteasome inhibitor resistance of multiple myeloma in combination with bortezomib: A phase I trial (SAKK 65/08). Haematologica 2016, 101, 346–355. [Google Scholar] [CrossRef]
- Chen, C.; Shen, G.; Hebbar, V.; Hu, R.; Owuor, E.D.; Kong, A.N. Epigallocatechin-3-gallate-induced stress signals in HT-29 human colon adenocarcinoma cells. Carcinogenesis 2003, 24, 1369–1378. [Google Scholar] [CrossRef]
- Suh, N.; Reddy, B.S.; DeCastro, A.; Paul, S.; Lee, H.J.; Smolarek, A.K.; So, J.Y.; Simi, B.; Wang, C.X.; Janakiram, N.B.; et al. Combination of atorvastatin with sulindac or naproxen profoundly inhibits colonic adenocarcinomas by suppressing the p65/β-catenin/cyclin D1 signaling pathway in rats. Cancer Prev. Res. 2011, 4, 1895–1902. [Google Scholar] [CrossRef]
- Ringshausen, I.; Oelsner, M.; Bogner, C.; Peschel, C.; Decker, T. The immunomodulatory drug Leflunomide inhibits cell cycle progression of B-CLL cells. Leukemia 2008, 22, 635–638. [Google Scholar] [CrossRef]
- Wang, J.; Sun, J.; Hu, J.; Wang, C.; Prinz, R.A.; Peng, D.; Liu, X.; Xu, X. A77 1726, the active metabolite of the anti-rheumatoid arthritis drug leflunomide, inhibits influenza A virus replication in vitro and in vivo by inhibiting the activity of Janus kinases. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 10132–10145. [Google Scholar] [CrossRef]
- Cox, G.; Wright, G.D. Intrinsic antibiotic resistance: Mechanisms, origins, challenges and solutions. Int. J. Med. Microbiol. IJMM 2013, 303, 287–292. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, B. A review on CRISPR/Cas: A versatile tool for cancer screening, diagnosis, and clinic treatment. Funct. Integr. Genom. 2023, 23, 182. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug 2019, 2, 141–160. [Google Scholar] [CrossRef]
- Emran, T.B.; Shahriar, A.; Mahmud, A.R.; Rahman, T.; Abir, M.H.; Siddiquee, M.F.; Ahmed, H.; Rahman, N.; Nainu, F.; Wahyudin, E.; et al. Multidrug Resistance in Cancer: Understanding Molecular Mechanisms, Immunoprevention and Therapeutic Approaches. Front. Oncol. 2022, 12, 891652. [Google Scholar] [CrossRef]
- Pan, S.T.; Li, Z.L.; He, Z.X.; Qiu, J.X.; Zhou, S.F. Molecular mechanisms for tumour resistance to chemotherapy. Clin. Exp. Pharmacol. Physiol. 2016, 43, 723–737. [Google Scholar] [CrossRef]
- Tobore, T.O. On the need for the development of a cancer early detection, diagnostic, prognosis, and treatment response system. Future Sci. OA 2019, 6, Fso439. [Google Scholar] [CrossRef]
- Ramos, A.; Sadeghi, S.; Tabatabaeian, H. Battling Chemoresistance in Cancer: Root Causes and Strategies to Uproot Them. Int. J. Mol. Sci. 2021, 22, 9451. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Gole, J.; Gore, A.; He, Q.; Lu, M.; Min, J.; Yuan, Z.; Yang, X.; Jiang, Y.; Zhang, T.; et al. Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat. Commun. 2020, 11, 3475. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Crosby, D.; Bhatia, S.; Brindle, K.M.; Coussens, L.M.; Dive, C.; Emberton, M.; Esener, S.; Fitzgerald, R.C.; Gambhir, S.S.; Kuhn, P.; et al. Early detection of cancer. Science 2022, 375, eaay9040. [Google Scholar] [CrossRef] [PubMed]
- Lippert, T.H.; Ruoff, H.J.; Volm, M. Current status of methods to assess cancer drug resistance. Int. J. Med. Sci. 2011, 8, 245–253. [Google Scholar] [CrossRef]
- Kartal-Yandim, M.; Adan-Gokbulut, A.; Baran, Y. Molecular mechanisms of drug resistance and its reversal in cancer. Crit. Rev. Biotechnol. 2016, 36, 716–726. [Google Scholar] [CrossRef]
- Goossens, N.; Nakagawa, S.; Sun, X.; Hoshida, Y. Cancer biomarker discovery and validation. Transl. Cancer Res. 2015, 4, 256–269. [Google Scholar]
- Cucchi, D.G.J.; Groen, R.W.J.; Janssen, J.; Cloos, J. Ex vivo cultures and drug testing of primary acute myeloid leukemia samples: Current techniques and implications for experimental design and outcome. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer. Chemother. 2020, 53, 100730. [Google Scholar] [CrossRef]
- Cree, I.A.; Glaysher, S.; Harvey, A.L. Efficacy of anti-cancer agents in cell lines versus human primary tumour tissue. Curr. Opin. Pharmacol. 2010, 10, 375–379. [Google Scholar] [CrossRef]
- Gallamini, A.; Zwarthoed, C.; Borra, A. Positron Emission Tomography (PET) in Oncology. Cancers 2014, 6, 1821–1889. [Google Scholar] [CrossRef]
- Doytchinova, I. Drug Design-Past, Present, Future. Molecules 2022, 27, 1496. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F.; Zhong, W.Z. Drug Design and Discovery: Principles and Applications. Molecules 2017, 22, 279. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Meng, Q.; Yang, M.; Liu, D.; Hou, X.; Tang, L.; Wang, X.; Lyu, Y.; Chen, X.; Liu, K.; et al. Current trends in drug metabolism and pharmacokinetics. Acta Pharm. Sin. B 2019, 9, 1113–1144. [Google Scholar] [CrossRef]
- Jackson, S.E.; Chester, J.D. Personalised cancer medicine. Int. J. Cancer 2015, 137, 262–266. [Google Scholar] [CrossRef]
- Braig, Z.V. Personalized medicine: From diagnostic to adaptive. Biomed. J. 2022, 45, 132–142. [Google Scholar] [CrossRef]
- Available online: https://www.atlasantibodies.com/primary-antibodies/ (accessed on 4 January 2024).
- Ding, S.; Hsu, C.; Wang, Z.; Natesh, N.R.; Millen, R.; Negrete, M.; Giroux, N.; Rivera, G.O.; Dohlman, A.; Bose, S.; et al. Patient-derived micro-organospheres enable clinical precision oncology. Cell Stem Cell 2022, 29, 905–917.e6. [Google Scholar] [CrossRef] [PubMed]
- Krzyszczyk, P.; Acevedo, A.; Davidoff, E.J.; Timmins, L.M.; Marrero-Berrios, I.; Patel, M.; White, C.; Lowe, C.; Sherba, J.J.; Hartmanshenn, C.; et al. The growing role of precision and personalized medicine for cancer treatment. Technology 2018, 6, 79–100. [Google Scholar] [CrossRef]
- Chatterjee, S.K.; Zetter, B.R. Cancer biomarkers: Knowing the present and predicting the future. Future Oncol. 2005, 1, 37–50. [Google Scholar] [CrossRef]
- Van der Jeught, K.; Xu, H.C.; Li, Y.J.; Lu, X.B.; Ji, G. Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol. 2018, 24, 3834–3848. [Google Scholar] [CrossRef]
- Bai, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Cancer stem cell in breast cancer therapeutic resistance. Cancer Treat. Rev. 2018, 69, 152–163. [Google Scholar] [CrossRef]
- Posdzich, P.; Darr, C.; Hilser, T.; Wahl, M.; Herrmann, K.; Hadaschik, B.; Grünwald, V. Metastatic Prostate Cancer-A Review of Current Treatment Options and Promising New Approaches. Cancers 2023, 15, 461. [Google Scholar] [CrossRef] [PubMed]
- Palomeras, S.; Ruiz-Martínez, S.; Puig, T. Targeting Breast Cancer Stem Cells to Overcome Treatment Resistance. Molecules 2018, 23, 2193. [Google Scholar] [CrossRef] [PubMed]
- Borovski, T.; De Sousa, E.M.F.; Vermeulen, L.; Medema, J.P. Cancer stem cell niche: The place to be. Cancer Res 2011, 71, 634–639. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Taghizadeh-Hesary, F.; Akbari, H.; Bahadori, M.; Behnam, B. Targeted Anti-Mitochondrial Therapy: The Future of Oncology. Genes 2022, 13, 1728. [Google Scholar] [CrossRef] [PubMed]
- Behnam, B.; Taghizadeh-Hesary, F. Mitochondrial Metabolism: A New Dimension of Personalized Oncology. Cancers 2023, 15, 4058. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Fuller, M. Stem cells and cancer: Two faces of eve. Cell 2006, 124, 1111–1115. [Google Scholar] [CrossRef]
- Dick, J.E. Stem cell concepts renew cancer research. Blood 2008, 112, 4793–4807. [Google Scholar] [CrossRef]
- Behnam, B.; Fazilaty, H.; Ghadyani, M.; Fadavi, P.; Taghizadeh-Hesary, F. Ciliated, Mitochondria-Rich Postmitotic Cells are Immune-privileged, and Mimic Immunosuppressive Microenvironment of Tumor-Initiating Stem Cells: From Molecular Anatomy to Molecular Pathway. Front. Biosci.-Landmark 2023, 28, 261. [Google Scholar] [CrossRef]
- Nowak, M.; Klink, M. The Role of Tumor-Associated Macrophages in the Progression and Chemoresistance of Ovarian Cancer. Cells 2020, 9, 1299. [Google Scholar] [CrossRef]
- Abu-Odah, H.; Said, N.; Nair, S.; Allsop, M.; Currow, D.; Salah, M.; Hamad, B.A.; Elessi, K.; Alkhatib, A.; ElMokhallalati, Y. Identifying barriers and facilitators of translating research evidence into clinical practice: A systematic review of reviews. Health Soc. Care Community 2022, 30, e3265–e3276. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, X.; Chen, D.; Yu, J. Radiotherapy combined with immunotherapy: The dawn of cancer treatment. Signal Transduct. Target. Ther. 2022, 7, 258. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhang, T.; Zheng, L.; Liu, H.; Song, W.; Liu, D.; Li, Z.; Pan, C. Combination strategies to maximize the benefits of cancer immunotherapy. J. Hematol. Oncol. 2021, 14, 156. [Google Scholar] [CrossRef]
- Bates, S.E. Epigenetic Therapies for Cancer. N. Engl. J. Med. 2020, 383, 650–663. [Google Scholar] [CrossRef]
- Jin, N.; George, T.L.; Otterson, G.A.; Verschraegen, C.; Wen, H.; Carbone, D.; Herman, J.; Bertino, E.M.; He, K. Advances in epigenetic therapeutics with focus on solid tumors. Clin. Epigenet. 2021, 13, 83. [Google Scholar] [CrossRef] [PubMed]
- Cormie, P.; Trevaskis, M.; Thornton-Benko, E.; Zopf, E.M. Exercise medicine in cancer care. Aust. J. Gen. Pract. 2020, 49, 169–174. [Google Scholar] [CrossRef]
- Wang, Q.; Zhou, W. Roles and molecular mechanisms of physical exercise in cancer prevention and treatment. J. Sport Health Sci. 2021, 10, 201–210. [Google Scholar] [CrossRef]
- Chakraborty, S.; Hosen, M.I.; Ahmed, M.; Shekhar, H.U. Onco-Multi-OMICS Approach: A New Frontier in Cancer Research. BioMed Res. Int. 2018, 2018, 9836256. [Google Scholar] [CrossRef]

{kind=link}
| Number | Type | Mode of Action | Advantages | Limitation | References |
|---|---|---|---|---|---|
| 1 | Fresh Tumor Cell Culture Assay (Tumor Chemosensitivity Assay) | To collect cancer cells from fresh cell types that preserve their physiological properties. | Good results over the last decades. Simple steps for all cancer cell types. Can be used for all cancer cell types. | Lack of predicting the drug side effects given to the patients. Preparation method steps vary depending on different cancer cell types. | [120,127,128] |
| 2 | Cancer Biomarker Tests | To measure biomarkers, such as DNA, RNA, peptides, genes, and proteins. | Clinical biomarkers for predicting cancer stage (e.g., blood in tissue) and to predict a patient’s risk of cancer development. To measure the chemoresistance of cancer cells to drugs. Combining the first two above approaches with the assistance of omics technologies can increase each patient’s life survival. | Detecting cancer cell types is not easy in the peripheral blood of cancer cells, unless with biopsy or removal of the tumor; therefore, just the opposite of leukemia, the other cancers are detected in late stages. | [6,129] |
| 3 | Positron Emission Tomography (PET) | This method is based on cancer cells absorbing more radiation, resulting in a brighter image. This leads to more accurate, reliable, and early detection of cancer in patients. | To help clinicians to make an accurate diagnosis. To determine the stage of cancer. To help choose the most appropriate curative therapy for early-stage tumors. To help palliative methods for invasive disease. To reduce the cost of therapy by choosing the most accurate therapy method. | Higher costs of this diagnostics method. | [6,129,130] |
| 4 | High Throughput Pharmacogenomic CRISPER Analysis | Novel genomic method for cancer research. | To detect regulatory genes that can act as biomarkers for malignant transformation. Developing therapeutic targets of new drugs. Early detection of hematological cancers. | Higher costs of this novel research method. | [118] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eslami, M.; Memarsadeghi, O.; Davarpanah, A.; Arti, A.; Nayernia, K.; Behnam, B. Overcoming Chemotherapy Resistance in Metastatic Cancer: A Comprehensive Review. Biomedicines 2024, 12, 183. https://doi.org/10.3390/biomedicines12010183
Eslami M, Memarsadeghi O, Davarpanah A, Arti A, Nayernia K, Behnam B. Overcoming Chemotherapy Resistance in Metastatic Cancer: A Comprehensive Review. Biomedicines. 2024; 12(1):183. https://doi.org/10.3390/biomedicines12010183
Chicago/Turabian StyleEslami, Maryam, Omid Memarsadeghi, Ali Davarpanah, Afshin Arti, Karim Nayernia, and Babak Behnam. 2024. "Overcoming Chemotherapy Resistance in Metastatic Cancer: A Comprehensive Review" Biomedicines 12, no. 1: 183. https://doi.org/10.3390/biomedicines12010183
APA StyleEslami, M., Memarsadeghi, O., Davarpanah, A., Arti, A., Nayernia, K., & Behnam, B. (2024). Overcoming Chemotherapy Resistance in Metastatic Cancer: A Comprehensive Review. Biomedicines, 12(1), 183. https://doi.org/10.3390/biomedicines12010183