
Involvement of Lysophospholipids in Pulmonary Vascular Functions and Diseases


Abstract

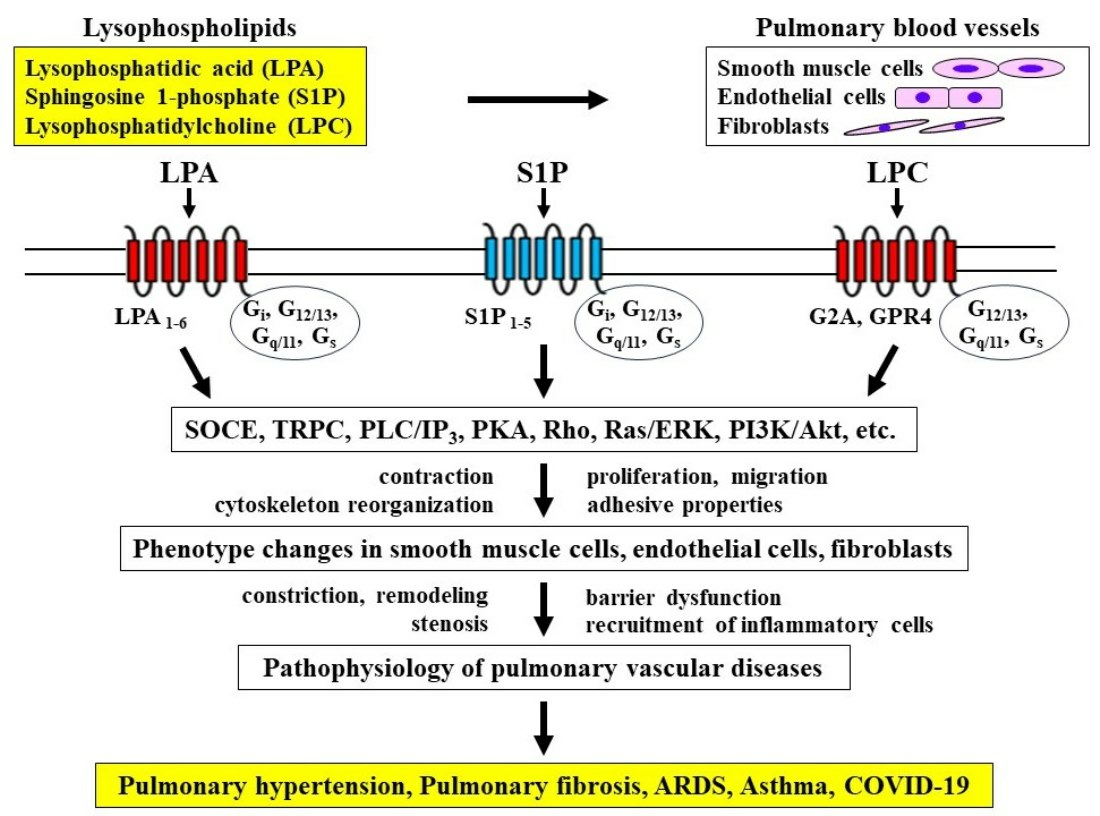{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Lysophospholipids
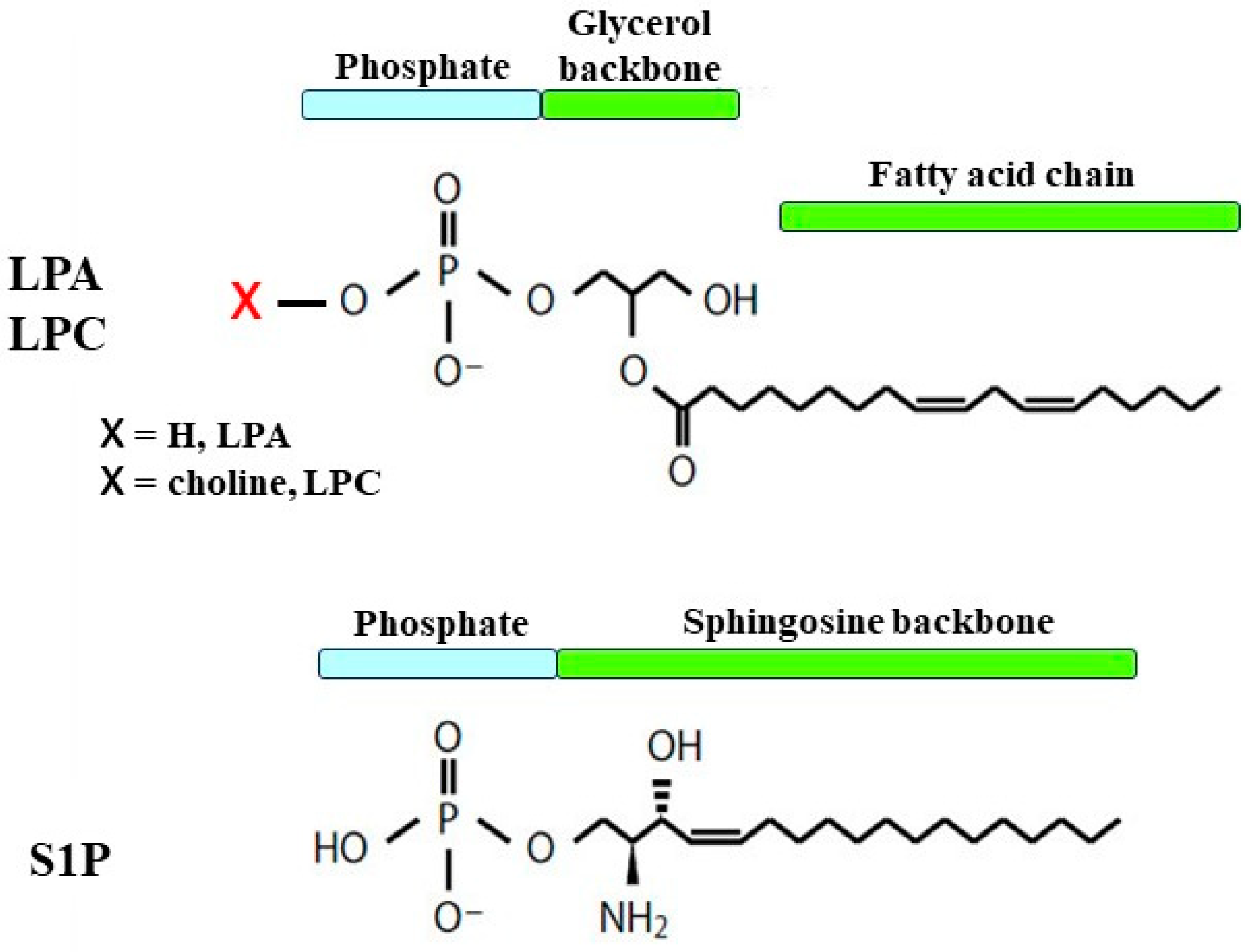
2.1. Structure
2.2. Physiological Activity

3. Lysophosphatidic Acid
3.1. Structure and Function
3.2. Effects on Smooth Muscle Cells
3.3. Effects on Fibroblasts
3.4. Effects on Endothelial Cells
3.5. Clinical Relevance
3.5.1. Pulmonary Hypertension
3.5.2. Pulmonary Fibrosis
3.5.3. Asthma
3.5.4. Acute Respiratory Distress Syndrome: COVID-19
4. Lysophosphatidylcholine
4.1. Structure and Function
4.2. Effects on Smooth Muscle
4.3. Effects of Endothelial Cells
4.4. Clinical Relevance
4.4.1. Atherosclerosis, Acute Respiratory Distress Syndrome
4.4.2. Asthma

4.4.3. Pulmonary Hypertension
5. Sphingosine 1-Phosphate
5.1. Structure and Function
5.2. Effects on Endothelial Cells
5.3. Effects of Smooth Muscle
5.4. Effects on Fibroblasts
5.5. Clinical Relevance
5.5.1. Pulmonary Hypertension
5.5.2. Acute Respiratory Distress Syndrome
5.5.3. Asthma
5.5.4. Pulmonary Fibrosis

6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rivera, R.; Chun, J. Biological effects of lysophospholipids. Rev. Physiol. Biochem. Pharmacol. 2008, 160, 25–46. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Leonarduzzi, G.; Biasi, F.; Chiarpotto, E. Oxidative stress and cell signalling. Curr. Med. Chem. 2004, 11, 1163–1182. [Google Scholar] [CrossRef] [PubMed]
- Ueda, N. A Rheostat of Ceramide and Sphingosine-1-Phosphate as a Determinant of Oxidative Stress-Mediated Kidney Injury. Int. J. Mol Sci. 2022, 23, 4010. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhu, W.; Chen, C.; Yan, B.; Zhu, L.; Chen, X.; Peng, C. The mechanisms of lysophosphatidylcholine in the development of diseases. Life Sci. 2020, 247, 117443. [Google Scholar] [CrossRef] [PubMed]
- Nikitopoulou, I.; Fanidis, D.; Ntatsoulis, K.; Moulos, P.; Mpekoulis, G.; Evangelidou, M.; Vassiliou, A.G.; Dimakopoulou, V.; Jahaj, E.; Tsipilis, S.; et al. Increased Autotaxin Levels in Severe COVID-19, Correlating with IL-6 Levels, Endothelial Dysfunction Biomarkers, and Impaired Functions of Dendritic Cells. Int. J. Mol. Sci. 2021, 22, 10006. [Google Scholar] [CrossRef] [PubMed]
- Ntatsoulis, K.; Karampitsakos, T.; Tsitoura, E.; Stylianaki, E.A.; Matralis, A.N.; Tzouvelekis, A.; Antoniou, K.; Aidinis, V. Commonalities Between ARDS, Pulmonary Fibrosis and COVID-19: The Potential of Autotaxin as a Therapeutic Target. Front. Immunol. 2021, 12, 687397. [Google Scholar] [CrossRef]
- Toebbe, J.T.; Genter, M.B. An Update on Sphingosine-1-Phosphate and Lysophosphatidic Acid Receptor Transcripts in Rodent Olfactory Mucosa. Int. J. Mol. Sci. 2022, 23, 4343. [Google Scholar] [CrossRef] [PubMed]
- Yaginuma, S.; Omi, J.; Kano, K.; Aoki, J. Lysophospholipids and their producing enzymes: Their pathological roles and potential as pathological biomarkers. Pharmacol. Ther. 2023, 246, 108415. [Google Scholar] [CrossRef]
- Georas, S.N.; Berdyshev, E.; Hubbard, W.; Gorshkova, I.A.; Usatyuk, P.V.; Saatian, B.; Myers, A.C.; Williams, M.A.; Xiao, H.Q.; Liu, M.; et al. Lysophosphatidic acid is detectable in human bronchoalveolar lavage fluids at baseline and increased after segmental allergen challenge. Clin. Exp. Allergy 2007, 37, 311–322. [Google Scholar] [CrossRef]
- Ammit, A.J.; Hastie, A.T.; Edsall, L.C.; Hoffman, R.K.; Amrani, Y.; Krymskaya, V.P.; Kane, S.A.; Peters, S.P.; Penn, R.B.; Spiegel, S.; et al. Sphingosine 1-phosphate modulates human airway smooth muscle cell functions that promote inflammation and airway remodeling in asthma. FASEB J. 2001, 15, 1212–1214. [Google Scholar] [CrossRef]
- Xu, Y.J.; Saini, H.K.; Cheema, S.K.; Dhalla, N.S. Mechanisms of lysophosphatidic acid-induced increase in intracellular calcium in vascular smooth muscle cells. Cell Calcium. 2005, 38, 569–579. [Google Scholar] [CrossRef]
- Kume, H.; Takeda, N.; Oguma, T.; Ito, S.; Kondo, M.; Ito, Y.; Shimokata, K. Sphingosine 1-phosphate causes airway hyper-reactivity by Rho-mediated myosin phosphatase inactivation. J. Pharmacol. Exp Ther. 2007, 320, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Sashio, T.; Kume, H.; Takeda, N.; Asano, T.; Tsuji, S.; Kondo, M.; Hasegawa, Y.; Shimokata, K. Possible Involvement of Sphingosine-1-Phosphate/G(i)/RhoA pathways in adherence of eosinophils to pulmonary endothelium. Allergol. Int. 2012, 61, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Shlyonsky, V.; Naeije, R.; Mies, F. Possible role of lysophosphatidic acid in rat model of hypoxic pulmonary vascular remodeling. Pulm. Circ. 2014, 4, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Tager, A.M.; LaCamera, P.; Shea, B.S.; Campanella, G.S.; Selman, M.; Zhao, Z.; Polosukhin, V.; Wain, J.; Karimi-Shah, B.A.; Kim, N.D.; et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 2008, 14, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, O.; Kume, H.; Kondo, M.; Ito, Y.; Ito, M.; Yamaki, K. Role of lysophosphatidylcholine in eosinophil infiltration and resistance in airways. Clin. Exp. Pharmacol. Physiol. 2004, 31, 179–184. [Google Scholar] [CrossRef]
- Hashimoto, T.; Yamashita, M.; Ohata, H.; Momose, K. Lysophosphatidic acid enhances in vivo infiltration and activation of guinea pig eosinophils and neutrophils via a Rho/Rho-associated protein kinase-mediated pathway. J. Pharmacol. Sci. 2003, 91, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Kume, H.; Ito, S.; Ito, Y.; Yamaki, K. Role of lysophosphatidylcholine in the desensitization of β-adrenergic receptors by Ca2+ sensitization in tracheal smooth muscle. Am. J. Respir. Cell Mol. Biol. 2001, 25, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.W.; Lagunoff, D. Interactions of lysophospholipids and mast cells. Nature 1979, 279, 250–252. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, Y. Lysophospholipids in Lung Inflammatory Diseases. Adv. Exp. Med. Biol. 2021, 1303, 373–391. [Google Scholar] [CrossRef]
- Kabarowski, J.H.; Feramisco, J.D.; Le, L.Q.; Gu, J.L.; Luoh, S.W.; Simon, M.I.; Witte, O.N. Direct genetic demonstration of G alpha 13 coupling to the orphan G protein-coupled receptor G2A leading to RhoA-dependent actin rearrangement. Proc. Natl. Acad. Sci. USA 2000, 97, 12109–12114. [Google Scholar] [CrossRef]
- Ludwig, M.G.; Vanek, M.; Guerini, D.; Gasser, J.A.; Jones, C.E.; Junker, U.; Hofstetter, H.; Wolf, R.M.; Seuwen, K. Proton-sensing G-protein-coupled receptors. Nature 2003, 425, 93–98. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, T.M.; Pontsler, A.V.; Silva, A.R.; St Hilaire, A.; Xu, Y.; Hinshaw, J.C.; Zimmerman, G.A.; Hama, K.; Aoki, J.; Arai, H.; et al. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARgamma agonist. Proc. Natl. Acad. Sci. USA 2003, 100, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Geraldo, L.H.M.; Spohr, T.C.L.S.; Amaral, R.F.D.; Fonseca, A.C.C.D.; Garcia, C.; Mendes, F.A.; Freitas, C.; dosSantos, M.F.; Lima, F.R.S. Role of lysophosphatidic acid and its receptors in health and disease: Novel therapeutic strategies. Signal Transduct. Target Ther. 2021, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Takabe, K.; Terracina, K.P.; Soma, D.; Hirose, Y.; Kobayashi, T.; Matsuda, Y.; Wakai, T. Sphingosine-1-phosphate transporters as targets for cancer therapy. Biomed. Res. Int. 2014, 2014, 651727. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Paknejad, N.; Zhu, L.; Kihara, Y.; Ray, M.; Chun, J.; Liu, W.; Hite, R.K.; Huang, X.Y. Differential activation mechanisms of lipid GPCRs by lysophosphatidic acid and sphingosine 1-phosphate. Nat. Commun. 2022, 13, 731. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; He, L.; Gan, B.; Ti, R.; Xiao, Q.; Hu, H.; Zhu, L.; Wang, S.; Ren, R. Structural insights into sphingosine-1-phosphate receptor activation. Proc. Natl. Acad. Sci. USA 2022, 119, e2117716119. [Google Scholar] [CrossRef]
- Taniguchi, R.; Inoue, A.; Sayama, M.; Uwamizu, A.; Yamashita, K.; Hirata, K.; Yoshida, M.; Tanaka, Y.; Kato, H.E.; Nakada-Nakura, Y.; et al. Structural insights into ligand recognition by the lysophosphatidic acid receptor LPA6. Nature 2017, 548, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Uehata, M.; Ishizaki, T.; Satoh, H.; Ono, T.; Kawahara, T.; Morishita, T.; Tamakawa, H.; Yamagami, K.; Inui, J.; Maekawa, M.; et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 1997, 389, 990–994. [Google Scholar] [CrossRef]
- Kume, H. RhoA/Rho-kinase as a therapeutic target in asthma. Curr. Med. Chem. 2008, 15, 2876–2885. [Google Scholar] [CrossRef]
- Dacheux, M.A.; Norman, D.D.; Tigyi, G.J.; Lee, S.C. Emerging roles of lysophosphatidic acid receptor subtype 5 (LPAR5) in inflammatory diseases and cancer. Pharmacol. Ther. 2023, 245, 108414. [Google Scholar] [CrossRef]
- Toews, M.L.; Ustinova, E.E.; Schultz, H.D. Lysophosphatidic acid enhances contractility of isolated airway smooth muscle. J. Appl. Physiol. 1997, 83, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- Dancs, P.T.; Ruisanchez, É.; Balogh, A.; Panta, C.R.; Miklós, Z.; Nüsing, R.M.; Aoki, J.; Chun, J.; Offermanns, S.; Tigyi, G.; et al. LPA1 receptor-mediated thromboxane A2 release is responsible for lysophosphatidic acid-induced vascular smooth muscle contraction. FASEB J. 2017, 31, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Boer, C.; van Nieuw Amerongen, G.P.; Groeneveld, A.B.; Scheffer, G.J.; de Lange, J.J.; Westerhof, N.; van Hinsbergh, V.W.; Sipkema, P. Smooth muscle F-actin disassembly and RhoA/Rho-kinase signaling during endotoxin-induced alterations in pulmonary arterial compliance. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L649–L655. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Keys, J.R.; Eckhart, A.D. Vascular smooth muscle migration and proliferation in response to lysophosphatidic acid (LPA) is mediated by LPA receptors coupling to Gq. Cell Signal. 2006, 18, 1695–1701. [Google Scholar] [CrossRef]
- Gaaya, A.; Poirier, O.; Mougenot, N.; Hery, T.; Atassi, F.; Marchand, A.; Saulnier-Blache, J.S.; Amour, J.; Vogt, J.; Lompré, A.M.; et al. Plasticity-related gene-1 inhibits lysophosphatidic acid-induced vascular smooth muscle cell migration and proliferation and prevents neointima formation. Am. J. Physiol. Cell Physiol. 2012, 303, C1104–C1114. [Google Scholar] [CrossRef] [PubMed]
- Subedi, U.; Manikandan, S.; Bhattarai, S.; Sharma, P.; Sharma, S.; Sun, H.; Miriyala, S.; Panchatcharam, M. The Autotaxin-LPA Axis Emerges as a Novel Regulator of Smooth Muscle Cell Phenotypic Modulation during Intimal Hyperplasia. Int. J. Mol. Sci. 2023, 24, 2913. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, N.; Mouratis, M.A.; Tzouvelekis, A.; Kaffe, E.; Valavanis, C.; Vilaras, G.; Karameris, A.; Prestwich, G.D.; Bouros, D.; Aidinis, V. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 566–574. [Google Scholar] [CrossRef]
- Yukiura, H.; Hama, K.; Nakanaga, K.; Tanaka, M.; Asaoka, Y.; Okudaira, S.; Arima, N.; Inoue, A.; Hashimoto, T.; Arai, H.; et al. Autotaxin regulates vascular development via multiple lysophosphatidic acid (LPA) receptors in zebrafish. J. Biol. Chem. 2011, 286, 43972–43983. [Google Scholar] [CrossRef]
- Kazlauskas, A. Lysophosphatidic acid contributes to angiogenic homeostasis. Exp. Cell Res. 2015, 333, 166–170. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Panetti, T.S.; Nowlen, J.; Mosher, D.F. Sphingosine-1-phosphate and lysophosphatidic acid stimulate endothelial cell migration. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, F.; Ji, L.; Zhang, L.; Xu, Y.J.; Dhalla, N.S. Role of lysophosphatidic acid in vascular smooth muscle cell proliferation. Can. J. Physiol. Pharmacol. 2020, 98, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Little, P.J.; Ta, H.T.; Xu, S.; Kamato, D. Lysophosphatidic acid and its receptors: Pharmacology and therapeutic potential in atherosclerosis and vascular disease. Pharmacol. Ther. 2019, 204, 107404. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.S.; Fu, P.; Patel, P.; Harijith, A.; Sun, T.; Zhao, Y.; Garcia, J.G.; Chun, J.; Natarajan, V. Lysophosphatidic acid receptor-2 deficiency confers protection against bleomycin-induced lung injury and fibrosis in mice. Am. J. Respir. Cell Mol. Biol. 2013, 49, 912–922. [Google Scholar] [CrossRef] [PubMed]
- Park, G.Y.; Lee, Y.G.; Berdyshev, E.; Nyenhuis, S.; Du, J.; Fu, P.; Gorshkova, I.A.; Li, Y.; Chung, S.; Karpurapu, M.; et al. Autotaxin production of lysophosphatidic acid mediates allergic asthmatic inflammation. Am. J. Respir. Crit. Care Med. 2013, 188, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Kume, H.; Yamada, R.; Sato, Y.; Togawa, R. Airway Smooth Muscle Regulated by Oxidative Stress in COPD. Antioxidants 2023, 12, 142. [Google Scholar] [CrossRef]
- Gao, L.; Li, X.; Wang, H.; Liao, Y.; Zhou, Y.; Wang, K.; Hu, J.; Cheng, M.; Zeng, Z.; Wang, T.; et al. Autotaxin levels in serum and bronchoalveolar lavage fluid are associated with inflammatory and fibrotic biomarkers and the clinical outcome in patients with acute respiratory distress syndrome. J. Intensive Care 2021, 9, 44. [Google Scholar] [CrossRef]
- Kabarowski, J.H.; Zhu, K.; Le, L.Q.; Witte, O.N.; Xu, Y. Lysophosphatidylcholine as a ligand for the immunoregulatory receptor G2A. Science 2001, 293, 702–705. [Google Scholar] [CrossRef]
- Drzazga, A.; Okulus, M.; Rychlicka, M.; Biegała, Ł.; Gliszczyńska, A.; Gendaszewska-Darmach, E. Lysophosphatidylcholine Containing Anisic Acid Is Able to Stimulate Insulin Secretion Targeting G Protein Coupled Receptors. Nutrients 2020, 12, 1173. [Google Scholar] [CrossRef]
- Xu, Y. Sphingosylphosphorylcholine and lysophosphatidylcholine: G protein-coupled receptors and receptor-mediated signal transduction. Biochim. Biophys. Acta 2002, 1582, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Kume, N.; Cybulsky, M.I.; Gimbrone, M.A., Jr. Lysophosphatidylcholine, a component of atherogenic lipoproteins, induces mononuclear leukocyte adhesion molecules in cultured human and rabbit arterial endothelial cells. J. Clin. Investig. 1992, 90, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Kume, N.; Gimbrone, M.A., Jr. Lysophosphatidylcholine transcriptionally induces growth factor gene expression in cultured human endothelial cells. J. Clin. Investig. 1994, 93, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, W.; Liu, J.; Yang, H.; Hu, Y.; Zhang, M.; Bai, T.; Chang, F. Lysophosphatidylcholine promotes intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 expression in human umbilical vein endothelial cells via an orphan G protein receptor 2-mediated signaling pathway. Bioengineered 2021, 12, 4520–4535. [Google Scholar] [CrossRef] [PubMed]
- Jing, Q.; Xin, S.M.; Zhang, W.B.; Wang, P.; Qin, Y.W.; Pei, G. Lysophosphatidylcholine activates p38 and p42/44 mitogen-activated protein kinases in monocytic THP-1 cells, but only p38 activation is involved in its stimulated chemotaxis. Circ. Res. 2000, 87, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Homma, S. Vitamin D3 binding protein (group-specific component) is a precursor for the macrophage-activating signal factor from lysophosphatidylcholine-treated lymphocytes. Proc. Natl. Acad. Sci. USA 1991, 88, 8539–8543. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Kobayashi, T.; Kamata, K. Role of lysophosphatidylcholine (LPC) in atherosclerosis. Curr. Med. Chem. 2007, 14, 3209–3220. [Google Scholar] [CrossRef]
- Wolfram Kuhlmann, C.R.; Wiebke Lüdders, D.; Schaefer, C.A.; Kerstin Most, A.; Backenköhler, U.; Neumann, T.; Tillmanns, H.; Erdogan, A. Lysophosphatidylcholine-induced modulation of Ca2+-activated K+ channels contributes to ROS-dependent proliferation of cultured human endothelial cells. J. Mol. Cell Cardiol. 2004, 36, 675–682. [Google Scholar] [CrossRef]
- Suenaga, H.; Kamata, K. Marked dissociation between intracellular Ca2+ level and contraction on exposure of rat aorta to lysophosphatidylcholine. Eur. J. Pharmacol. 1999, 378, 177–186. [Google Scholar] [CrossRef]
- Ding, W.G.; Toyoda, F.; Ueyama, H.; Matsuura, H. Lysophosphatidylcholine enhances IKs currents in cardiac myocytes through activation of G protein, PKC and Rho signaling pathways. J. Mol. Cell Cardiol. 2011, 50, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Law, S.H.; Chan, M.L.; Marathe, G.K.; Parveen, F.; Chen, C.H.; Ke, L.Y. An Updated Review of Lysophosphatidylcholine Metabolism in Human Diseases. Int. J. Mol. Sci. 2019, 20, 1149. [Google Scholar] [CrossRef] [PubMed]
- Dudek, R.; Conforto, A.; Bing, R.J. Lysophosphatidylcholine-induced vascular relaxation and production of cGMP are mediated by endothelium-derived relaxing factor. Proc. Soc. Exp. Biol. Med. 1993, 203, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Kugiyama, K.; Kerns, S.A.; Morrisett, J.D.; Roberts, R.; Henry, P.D. Impairment of endothelium-dependent arterial relaxation by lysolecithin in modified low-density lipoproteins. Nature 1990, 344, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Galle, J.; Mameghani, A.; Bolz, S.S.; Gambaryan, S.; Görg, M.; Quaschning, T.; Raff, U.; Barth, H.; Seibold, S.; Wanner, C.; et al. Oxidized LDL and its compound lysophosphatidylcholine potentiate AngII-induced vasoconstriction by stimulation of RhoA. J. Am. Soc. Nephrol. 2003, 14, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Oguma, T.; Kume, H.; Ito, S.; Takeda, N.; Honjo, H.; Kodama, I.; Shimokata, K.; Kamiya, K. Involvement of reduced sensitivity to Ca2+ in β-adrenergic action on airway smooth muscle. Clin. Exp. Allergy 2006, 36, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, K.; Kume, H.; Oguma, T.; Shigemori, W.; Tohda, Y.; Ogawa, E.; Nakano, Y. Involvement of Ca2+ Signaling in the Synergistic Effects between Muscarinic Receptor Antagonists and β₂-Adrenoceptor Agonists in Airway Smooth Muscle. Int. J. Mol. Sci. 2016, 17, 1590. [Google Scholar] [CrossRef]
- Yamakawa, T.; Eguchi, S.; Yamakawa, Y.; Motley, E.D.; Numaguchi, K.; Utsunomiya, H.; Inagami, T. Lysophosphatidylcholine stimulates MAP kinase activity in rat vascular smooth muscle cells. Hypertension 1998, 31 Pt 2, 248–253. [Google Scholar] [CrossRef]
- Yamakawa, T.; Tanaka, S.; Kamei, J.; Kadonosono, K.; Okuda, K. Pitavastatin inhibits vascular smooth muscle cell proliferation by inactivating extracellular signal-regulated kinases 1/2. J. Atheroscler. Thromb. 2003, 10, 37–42. [Google Scholar] [CrossRef]
- Chai, Y.C.; Binion, D.G.; Macklis, R.; Chisolm, G.M., III. Smooth muscle cell proliferation induced by oxidized LDL-borne lysophosphatidylcholine. Evidence for FGF-2 release from cells not extracellular matrix. Vascul. Pharmacol. 2002, 38, 229–237. [Google Scholar] [CrossRef]
- Watanabe, T.; Pakala, R.; Katagiri, T.; Benedict, C.R. Lysophosphatidylcholine is a major contributor to the synergistic effect of mildly oxidized low-density lipoprotein with endothelin-1 on vascular smooth muscle cell proliferation. J. Cardiovasc. Pharmacol. 2002, 39, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Raines, E.W.; Abraham, J.A.; Klagsbrun, M.; Ross, R. Lysophosphatidylcholine upregulates the level of heparin-binding epidermal growth factor-like growth factor mRNA in human monocytes. Proc. Natl. Acad. Sci. USA 1994, 91, 1069–1073. [Google Scholar] [CrossRef] [PubMed]
- Chisolm, G.M., III; Chai, Y. Regulation of cell growth by oxidized LDL. Free Radic. Biol. Med. 2000, 28, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Kohno, M.; Yokokawa, K.; Yasunari, K.; Minami, M.; Kano, H.; Hanehira, T.; Yoshikawa, J. Induction by lysophosphatidylcholine, a major phospholipid component of atherogenic lipoproteins, of human coronary artery smooth muscle cell migration. Circulation 1998, 98, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Yasunari, K.; Maeda, K.; Minami, M.; Yoshikawa, J. HMG-CoA reductase inhibitors prevent migration of human coronary smooth muscle cells through suppression of increase in oxidative stress. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Qi, J.; Wang, Y.W.; Xi, Q.; Tserennadmid, T.; Zhao, P.F.; Qi, J.; Damirin, A. The atherogenic actions of LPC on vascular smooth muscle cells and its LPA receptor mediated mechanism. Biochem. Biophys. Res. Commun. 2018, 503, 1911–1918. [Google Scholar] [CrossRef]
- Jung, H.-J.; Im, S.-S.; Song, D.-K.; Bae, J.-H. Effects of chlorogenic acid on intracellular calcium regulation in lysophosphatidylcholine-treated endothelial cells. BMB Rep. 2017, 50, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Kume, H.; Takai, A.; Tokuno, H.; Tomita, T. Regulation of Ca2+-dependent K+-channel activity in tracheal myocytes by phosphorylation. Nature 1989, 341, 152–154. [Google Scholar] [CrossRef]
- Kume, H.; Graziano, M.P.; Kotlikoff, M.I. Stimulatory and inhibitory regulation of calcium-activated potassium channels by guanine nucleotide-binding proteins. Proc. Natl. Acad. Sci. USA 1992, 89, 11051–11055. [Google Scholar] [CrossRef]
- Kume, H.; Hall, I.P.; Washabau, R.J.; Takagi, K.; Kotlikoff, M.I. β-Adrenergic agonists regulate KCa channels in airway smooth muscle by cAMP-dependent and -independent mechanisms. J. Clin. Investig. 1994, 93, 371–379. [Google Scholar] [CrossRef]
- Kume, H. Role of Airway Smooth Muscle in Inflammation Related to Asthma and COPD. Adv. Exp. Med. Biol. 2021, 1303, 139–172. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Kitayama, J.; Springer, T.A. Differential regulation of beta 1 and beta 2 integrin avidity by chemoattractants in eosinophils. Proc. Natl. Acad. Sci. USA 1996, 93, 10939–10944. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Learoyd, J.; Butt, S.; Zhu, L.; Usatyuk, P.V.; Natarajan, V.; Munoz, N.M.; Leff, A.R. Regulation of eosinophil adhesion by lysophosphatidylcholine via a non-store-operated Ca2+ channel. Am. J. Respir. Cell Mol. Biol. 2007, 36, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Kim, C.H.; Chung, J.H.; Kim, J.Y.; Chung, S.W.; Kim, M.K.; Im, D.S.; Lee, J.; Yu, B.P.; Chung, H.Y. Upregulation of endothelial adhesion molecules by lysophosphatidylcholine. Involvement of G protein-coupled receptor GPR4. FEBS J. 2007, 274, 2573–2584. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shao, Y.; Sha, X.; Fang, P.; Kuo, Y.M.; Andrews, A.J.; Li, Y.; Yang, W.Y.; Maddaloni, M.; Pascual, D.W.; et al. IL-35 (Interleukin-35) Suppresses Endothelial Cell Activation by Inhibiting Mitochondrial Reactive Oxygen Species-Mediated Site-Specific Acetylation of H3K14 (Histone 3 Lysine 14). Arterioscler. Thromb. Vasc. Biol. 2018, 38, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Subbaiah, P.V.; Holian, O.; Zhang, J.; Johnson, A.; Gertzberg, N.; Lum, H. Lysophosphatidylcholine increases endothelial permeability: Role of PKCa and RhoA cross talk. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, L176–L185. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Huang, F.; Naikawadi, R.P.; Kim, K.S.; Said, T.; Lum, H. Lysophosphatidylcholine impairs endothelial barrier function through the G protein-coupled receptor GPR4. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 291, L91–L101. [Google Scholar] [CrossRef]
- Yokoyama, M.; Inoue, N.; Kawashima, S. Role of the vascular NADH/NADPH oxidase system in atherosclerosis. Ann. N. Y. Acad. Sci. 2000, 902, 241–247; discussion 247–248. [Google Scholar] [CrossRef]
- Bansal, P.; Gaur, S.N.; Arora, N. Lysophosphatidylcholine plays critical role in allergic airway disease manifestation. Sci. Rep. 2016, 6, 27430. [Google Scholar] [CrossRef]
- Mehta, D.; Gupta, S.; Gaur, S.N.; Gangal, S.V.; Agrawal, K.P. Increased leukocyte phospholipase A2 activity and plasma lysophosphatidylcholine levels in asthma and rhinitis and their relationship to airway sensitivity to histamine. Am. Rev. Respir. Dis. 1990, 142, 157–161. [Google Scholar] [CrossRef]
- Rice, K.L.; Duane, P.G.; Niewoehner, D.E. Lysophosphatidylcholine augments elastase-induced alveolar epithelial permeability and emphysema in the hamster. Am. Rev. Respir. Dis. 1987, 136, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Tanosaki, T.; Mikami, Y.; Shindou, H.; Suzuki, T.; Hashidate-Yoshida, T.; Hosoki, K.; Kagawa, S.; Miyata, J.; Kabata, H.; Masaki, K.; et al. Lysophosphatidylcholine Acyltransferase 1 Deficiency Promotes Pulmonary Emphysema via Apoptosis of Alveolar Epithelial Cells. Inflammation 2022, 45, 1765–1779. [Google Scholar] [CrossRef] [PubMed]
- Blaho, V.A.; Galvani, S.; Engelbrecht, E.; Liu, C.; Swendeman, S.L.; Kono, M.; Proia, R.L.; Steinman, L.; Han, M.H.; Hla, T. HDL-bound sphingosine-1-phosphate restrains lymphopoiesis and neuroinflammation. Nature 2015, 523, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.M.; Ishizu, A.N.; Foo, J.C.; Toh, X.R.; Zhang, F.; Whee, D.M.; Torta, F.; Cazenave-Gassiot, A.; Matsumura, T.; Kim, S.; et al. Mfsd2b is essential for the sphingosine-1-phosphate export in erythrocytes and platelets. Nature 2017, 550, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, S.; Simmons, S.; Kawamura, S.; Inoue, A.; Orba, Y.; Tokudome, T.; Sunden, Y.; Arai, Y.; Moriwaki, K.; Ishida, J.; et al. The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J. Clin. Investig. 2012, 122, 1416–1426. [Google Scholar] [CrossRef] [PubMed]
- Sadahira, Y.; Ruan, F.; Hakomori, S.; Igarashi, Y. Sphingosine 1-phosphate, a specific endogenous signaling molecule controlling cell motility and tumor cell invasiveness. Proc. Natl. Acad. Sci. USA 1992, 89, 9686–9690. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.G.; Liu, F.; Verin, A.D.; Birukova, A.; Dechert, M.A.; Gerthoffer, W.T.; Bamberg, J.R.; English, D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J. Clin. Investig. 2001, 108, 689–701. [Google Scholar] [CrossRef]
- Xiong, Y.; Yang, P.; Proia, R.L.; Hla, T. Erythrocyte-derived sphingosine 1-phosphate is essential for vascular development. J. Clin. Investig. 2014, 124, 4823–4828. [Google Scholar] [CrossRef]
- Venkataraman, K.; Thangada, S.; Michaud, J.; Oo, M.L.; Ai, Y.; Lee, Y.M.; Wu, M.; Parikh, N.S.; Khan, F.; Proia, R.L.; et al. Extracellular export of sphingosine kinase-1a contributes to the vascular S1P gradient. Biochem. J. 2006, 397, 461–471. [Google Scholar] [CrossRef]
- Olivera, A.; Allende, M.L.; Proia, R.L. Shaping the landscape: Metabolic regulation of S1P gradients. Biochim. Biophys. Acta 2013, 1831, 193–202. [Google Scholar] [CrossRef]
- Proia, R.L.; Hla, T. Emerging biology of sphingosine-1-phosphate: Its role in pathogenesis and therapy. J. Clin. Investig. 2015, 125, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Ebenezer, D.L.; Fu, P.; Natarajan, V. Targeting sphingosine-1-phosphate signaling in lung diseases. Pharmacol. Ther. 2016, 168, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Camerer, E.; Regard, J.B.; Cornelissen, I.; Srinivasan, Y.; Duong, D.N.; Palmer, D.; Pham, T.H.; Wong, J.S.; Pappu, R.; Coughlin, S.R. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J. Clin. Investig. 2009, 119, 1871–1879. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Tomura, H.; Mogi, C.; Kuwabara, A.; Ishiwara, M.; Shibasawa, K.; Sato, K.; Ohwada, S.; Im, D.S.; Kurose, H.; et al. Sphingosine 1-phosphate receptors mediate stimulatory and inhibitory signalings for expression of adhesion molecules in endothelial cells. Cell Signal. 2006, 18, 841–850. [Google Scholar] [CrossRef]
- Muraki, K.; Imaizumi, Y. A novel function of sphingosine-1-phosphate to activate a non-selective cation channel in human endothelial cells. J. Physiol. 2001, 537 Pt 2, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Suki, B.; Kume, H.; Numaguchi, Y.; Ishii, M.; Iwaki, M.; Kondo, M.; Naruse, K.; Hasegawa, Y.; Sokabe, M. Actin cytoskeleton regulates stretch-activated Ca2+ influx in human pulmonary microvascular endothelial cells. Am. J. Respir. Cell Mol. Biol. 2010, 43, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Panta, C.R.; Ruisanchez, É.; Móré, D.; Dancs, P.T.; Balogh, A.; Fülöp, Á.; Kerék, M.; Proia, R.L.; Offermanns, S.; Tigyi, G.J.; et al. Sphingosine-1-Phosphate Enhances α1-Adrenergic Vasoconstriction via S1P2-G12/13-ROCK Mediated Signaling. Int. J. Mol. Sci. 2019, 20, 6361. [Google Scholar] [CrossRef] [PubMed]
- Murakami, A.; Takasugi, H.; Ohnuma, S.; Koide, Y.; Sakurai, A.; Takeda, S.; Hasegawa, T.; Sasamori, J.; Konno, T.; Hayashi, K.; et al. Sphingosine 1-phosphate (S1P) regulates vascular contraction via S1P3 receptor: Investigation based on a new S1P3 receptor antagonist. Mol. Pharmacol. 2010, 77, 704–713. [Google Scholar] [CrossRef]
- Hopson, K.P.; Truelove, J.; Chun, J.; Wang, Y.; Waeber, C. S1P activates store-operated calcium entry via receptor- and non-receptor-mediated pathways in vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2011, 300, C919–C926. [Google Scholar] [CrossRef]
- Li, F.; Wang, J.; Zhu, Y.; Liu, L.; Feng, W.; Shi, W.; Wang, Q.; Zhang, Q.; Chai, L.; Li, M. SphK1/S1P Mediates PDGF-Induced Pulmonary Arterial Smooth Muscle Cell Proliferation via miR-21/BMPRII/Id1 Signaling Pathway. Cell Physiol. Biochem. 2018, 51, 487–500. [Google Scholar] [CrossRef]
- Wang, J.; Feng, W.; Li, F.; Shi, W.; Zhai, C.; Li, S.; Zhu, Y.; Yan, X.; Wang, Q.; Liu, L.; et al. SphK1/S1P mediates TGF-β1-induced proliferation of pulmonary artery smooth muscle cells and its potential mechanisms. Pulm Circ. 2019, 9, 2045894018816977. [Google Scholar] [CrossRef]
- Xu, S.Z.; Muraki, K.; Zeng, F.; Li, J.; Sukumar, P.; Shah, S.; Dedman, A.M.; Flemming, P.K.; McHugh, D.; Naylor, J.; et al. A sphingosine-1-phosphate-activated calcium channel controlling vascular smooth muscle cell motility. Circ. Res. 2006, 98, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.A.; Welch, Z.; Sliva, D.; Siddiqui, R.A. Role of Rho kinase in sphingosine 1-phosphate-mediated endothelial and smooth muscle cell migration and differentiation. Mol. Cell Biochem. 2010, 342, 7–19. [Google Scholar] [CrossRef]
- Shi, W.; Wang, Q.; Wang, J.; Yan, X.; Feng, W.; Zhang, Q.; Zhai, C.; Chai, L.; Li, S.; Xie, X.; et al. Activation of yes-associated protein mediates sphingosine-1-phosphate-induced proliferation and migration of pulmonary artery smooth muscle cells and its potential mechanisms. J. Cell Physiol. 2021, 236, 4694–4708. [Google Scholar] [CrossRef] [PubMed]
- Roztocil, E.; Nicholl, S.M.; Davies, M.G. Sphingosine-1-phosphate-induced oxygen free radical generation in smooth muscle cell migration requires Galpha12/13 protein-mediated phospholipase C activation. J. Vasc. Surg. 2007, 46, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Roztocil, E.; Nicholl, S.M.; Davies, M.G. Mechaisms of sphingosine-1-phosphate-induced akt-dependent smooth muscle cell migration. Surgery 2009, 145, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Wang, X.; Mao, L.; Kobayashi, T.; Kawasaki, S.; Mori, N.; Toews, M.L.; Kim, H.J.; Cerutis, D.R.; Liu, X.; et al. Sphingosine 1-phosphate potentiates human lung fibroblast chemotaxis through the S1P2 receptor. Am. J. Respir. Cell Mol. Biol. 2008, 39, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Sobel, K.; Menyhart, K.; Killer, N.; Renault, B.; Bauer, Y.; Studer, R.; Steiner, B.; Bolli, M.H.; Nayler, O.; Gatfield, J. Sphingosine 1-phosphate (S1P) receptor agonists mediate pro-fibrotic responses in normal human lung fibroblasts via S1P2 and S1P3 receptors and Smad-independent signaling. J. Biol. Chem. 2013, 288, 14839–14851. [Google Scholar] [CrossRef]
- Zhang, X.; Cai, Y.; Zhang, W.; Chen, X. Quercetin ameliorates pulmonary fibrosis by inhibiting SphK1/S1P signaling. Biochem. Cell Biol. 2018, 96, 742–751. [Google Scholar] [CrossRef]
- Takuwa, Y.; Ikeda, H.; Okamoto, Y.; Takuwa, N.; Yoshioka, K. Sphingosine-1-phosphate as a mediator involved in development of fibrotic diseases. Biochim. Biophys. Acta 2013, 1831, 185–192. [Google Scholar] [CrossRef]
- Suryadevara, V.; Ramchandran, R.; Kamp, D.W.; Natarajan, V. Lipid Mediators Regulate Pulmonary Fibrosis: Potential Mechanisms and Signaling Pathways. Int. J. Mol. Sci. 2020, 21, 4257. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.S.; Sudhadevi, T.; Fu, P.; Punathil-Kannan, P.K.; Ebenezer, D.L.; Ramchandran, R.; Putherickal, V.; Cheresh, P.; Zhou, G.; Ha, A.W.; et al. Sphingosine Kinase 1/S1P Signaling Contributes to Pulmonary Fibrosis by Activating Hippo/YAP Pathway and Mitochondrial Reactive Oxygen Species in Lung Fibroblasts. Int. J. Mol. Sci. 2020, 21, 2064. [Google Scholar] [CrossRef] [PubMed]
- Gairhe, S.; Joshi, S.R.; Bastola, M.M.; McLendon, J.M.; Oka, M.; Fagan, K.A.; McMurtry, I.F. Sphingosine-1-phosphate is involved in the occlusive arteriopathy of pulmonary arterial hypertension. Pulm. Circ. 2016, 6, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Tabeling, C.; Yu, H.; Wang, L.; Ranke, H.; Goldenberg, N.M.; Zabini, D.; Noe, E.; Krauszman, A.; Gutbier, B.; Yin, J.; et al. CFTR and sphingolipids mediate hypoxic pulmonary vasoconstriction. Proc. Natl. Acad. Sci. USA 2015, 112, E1614–E1623. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tang, H.; Sysol, J.R.; Moreno-Vinasco, L.; Shioura, K.M.; Chen, T.; Gorshkova, I.; Wang, L.; Huang, L.S.; Usatyuk, P.V.; et al. The sphingosine kinase 1/sphingosine-1-phosphate pathway in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2014, 190, 1032–1043. [Google Scholar] [CrossRef]
- Chen, J.; Lockett, A.; Zhao, S.; Huang, L.S.; Wang, Y.; Wu, W.; Tang, M.; Haider, S.; Velez Rendon, D.; Khan, R.; et al. Sphingosine Kinase 1 Deficiency in Smooth Muscle Cells Protects against Hypoxia-Mediated Pulmonary Hypertension via YAP1 Signaling. Int. J. Mol. Sci. 2022, 23, 14516. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Ware, L.B.; Zimmerman, G.A. The acute respiratory distress syndrome. J. Clin. Investig. 2012, 122, 2731–2740. [Google Scholar] [CrossRef] [PubMed]
- McVerry, B.J.; Peng, X.; Hassoun, P.M.; Sammani, S.; Simon, B.A.; Garcia, J.G. Sphingosine 1-phosphate reduces vascular leak in murine and canine models of acute lung injury. Am. J. Respir. Crit. Care Med. 2004, 170, 987–993. [Google Scholar] [CrossRef]
- Wadgaonkar, R.; Patel, V.; Grinkina, N.; Romano, C.; Liu, J.; Zhao, Y.; Sammani, S.; Garcia, J.G.; Natarajan, V. Differential regulation of sphingosine kinases 1 and 2 in lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 296, L603–L613. [Google Scholar] [CrossRef]
- Sammani, S.; Moreno-Vinasco, L.; Mirzapoiazova, T.; Singleton, P.A.; Chiang, E.T.; Evenoski, C.L.; Wang, T.; Mathew, B.; Husain, A.; Moitra, J.; et al. Differential effects of sphingosine 1-phosphate receptors on airway and vascular barrier function in the murine lung. Am. J. Respir. Cell Mol. Biol. 2010, 43, 394–402. [Google Scholar] [CrossRef]
- Olivera, A.; Eisner, C.; Kitamura, Y.; Dillahunt, S.; Allende, L.; Tuymetova, G.; Watford, W.; Meylan, F.; Diesner, S.C.; Li, L.; et al. Sphingosine kinase 1 and sphingosine-1-phosphate receptor 2 are vital to recovery from anaphylactic shock in mice. J. Clin. Investig. 2010, 120, 1429–1440. [Google Scholar] [CrossRef]
- Jolly, P.S.; Rosenfeldt, H.M.; Milstien, S.; Spiegel, S. The roles of sphingosine-1-phosphate in asthma. Mol. Immunol. 2002, 38, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Oskeritzian, C.A.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate in allergic responses, asthma and anaphylaxis. Pharmacol. Ther. 2007, 115, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Choi, O.H.; Kim, J.H.; Kinet, J.P. Calcium mobilization via sphingosine kinase in signalling by the Fc epsilon RI antigen receptor. Nature 1996, 380, 634–636. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Shim, K.; Jeoung, D. The Crosstalk between FcεRI and Sphingosine Signaling in Allergic Inflammation. Int. J. Mol. Sci. 2022, 23, 13892. [Google Scholar] [CrossRef] [PubMed]
- Roviezzo, F.; Del Galdo, F.; Abbate, G.; Bucci, M.; D’Agostino, B.; Antunes, E.; De Dominicis, G.; Parente, L.; Rossi, F.; Cirino, G.; et al. Human eosinophil chemotaxis and selective in vivo recruitment by sphingosine 1-phosphate. Proc. Natl. Acad. Sci. USA 2004, 101, 11170–11175. [Google Scholar] [CrossRef]
- Trifilieff, A.; Fozard, J.R. Sphingosine-1-phosphate-induced airway hyper-reactivity in rodents is mediated by the sphingosine-1-phosphate type 3 receptor. J. Pharmacol. Exp. Ther. 2012, 342, 399–406. [Google Scholar] [CrossRef]
- Price, M.M.; Oskeritzian, C.A.; Falanga, Y.T.; Harikumar, K.B.; Allegood, J.C.; Alvarez, S.E.; Conrad, D.; Ryan, J.J.; Milstien, S.; Spiegel, S. A specific sphingosine kinase 1 inhibitor attenuates airway hyperresponsiveness and inflammation in a mast cell-dependent murine model of allergic asthma. J. Allergy Clin. Immunol. 2013, 131, 501–511.e1. [Google Scholar] [CrossRef]
- Milara, J.; Navarro, R.; Juan, G.; Peiró, T.; Serrano, A.; Ramón, M.; Morcillo, E.; Cortijo, J. Sphingosine-1-phosphate is increased in patients with idiopathic pulmonary fibrosis and mediates epithelial to mesenchymal transition. Thorax 2012, 67, 147–156. [Google Scholar] [CrossRef]
- Huang, L.S.; Natarajan, V. Sphingolipids in pulmonary fibrosis. Adv. Biol. Regul. 2015, 57, 55–63. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kume, H.; Harigane, R.; Rikimaru, M. Involvement of Lysophospholipids in Pulmonary Vascular Functions and Diseases. Biomedicines 2024, 12, 124. https://doi.org/10.3390/biomedicines12010124
Kume H, Harigane R, Rikimaru M. Involvement of Lysophospholipids in Pulmonary Vascular Functions and Diseases. Biomedicines. 2024; 12(1):124. https://doi.org/10.3390/biomedicines12010124
Chicago/Turabian StyleKume, Hiroaki, Rina Harigane, and Mami Rikimaru. 2024. "Involvement of Lysophospholipids in Pulmonary Vascular Functions and Diseases" Biomedicines 12, no. 1: 124. https://doi.org/10.3390/biomedicines12010124
APA StyleKume, H., Harigane, R., & Rikimaru, M. (2024). Involvement of Lysophospholipids in Pulmonary Vascular Functions and Diseases. Biomedicines, 12(1), 124. https://doi.org/10.3390/biomedicines12010124