
Mitochondria and Brain Disease: A Comprehensive Review of Pathological Mechanisms and Therapeutic Opportunities

 , ,
, ,  ,
,  ,
,  ,
,  , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
Methodology of Search
2. Molecular Mechanisms in the Development of Diseases
2.1. Mitochondria Bioenergetics
2.2. Calcium Signalling
2.3. Cell Death
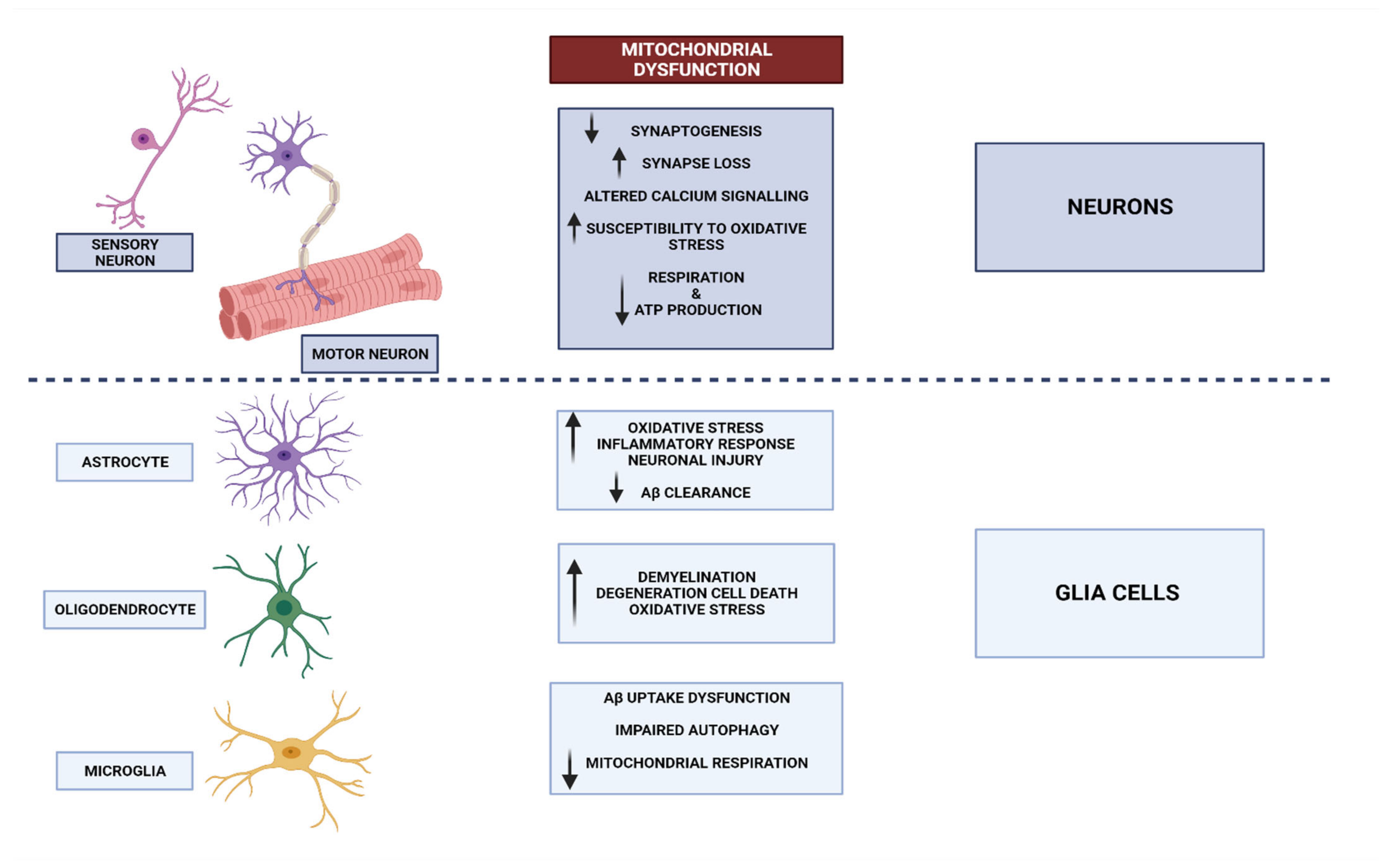
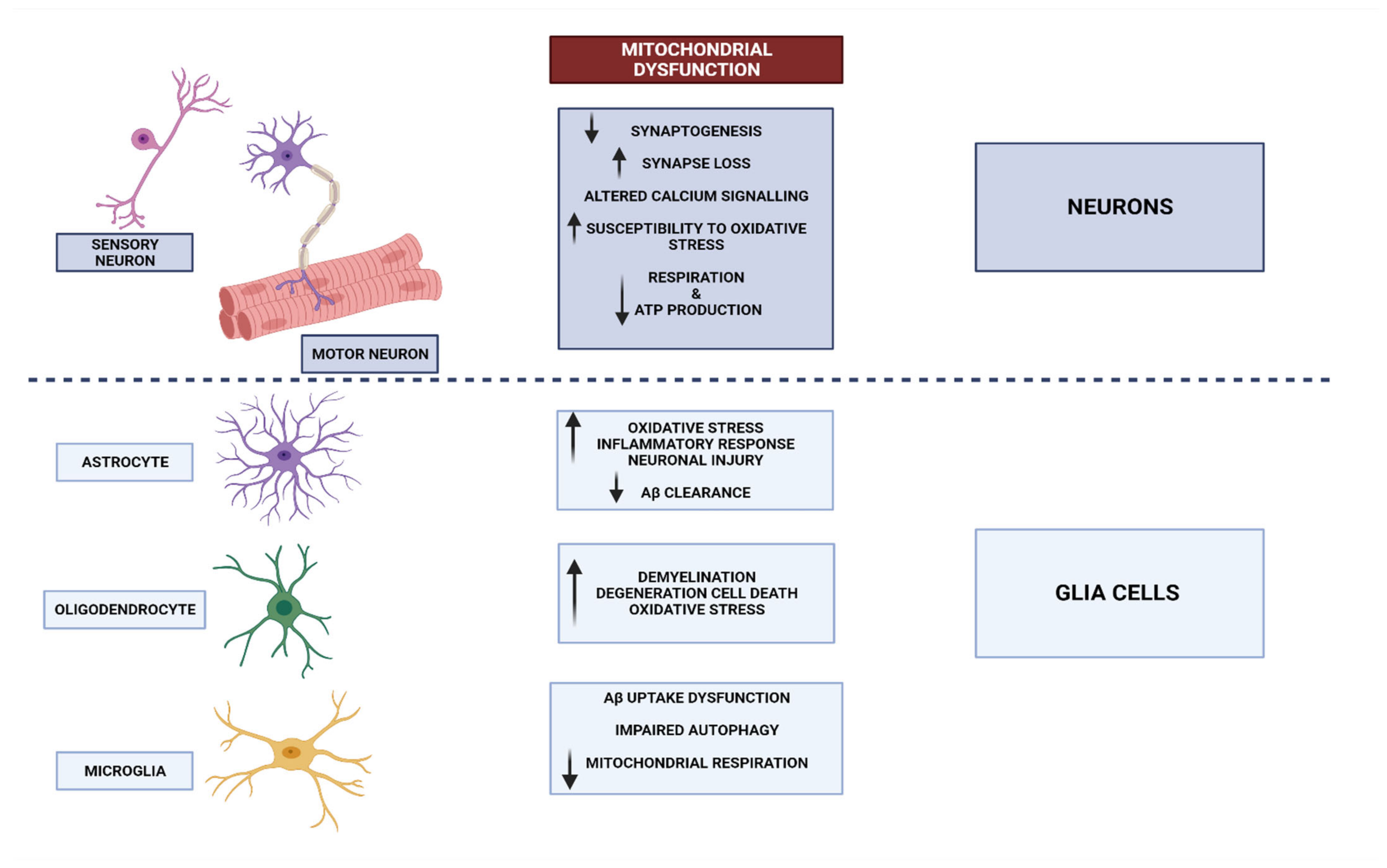
3. Neurodegenerative Disorders and Mitochondrial Dysfunction
3.1. Mitochondrial Dysfunctional Functioning in Neurodegenerative Disorders

3.1.1. Mitochondrial Dysfunction in Alzheimer’s Disease
3.1.2. Mitochondrial Dysfunction in Parkinson’s Disease
3.1.3. Mitochondrial Dysfunction in Other Neurodegenerative Disease
4. Mitochondrial DNA Mutations in Brain Disease
5. Impaired Oxidative Phosphorylation and Brain Disease
6. Mitochondrial Dynamics in Neuronal Health
7. Calcium Dysregulation and Its Impact on Mitochondria
8. Reactive Oxygen Species and Mitochondrial Dysfunction
9. Inflammation and Mitochondrial Impairment in Brain Disease
Inflammation in Metabolic Diseases: A Progress to Brain Diseases
10. Mitochondria and Neurotransmitter Systems in Psychiatric Illnesses
11. Mitochondrial Biomarkers in Brain Disease
11.1. Mitochondrial Biomarkers in Neurodegenerative Disorders
11.2. Mitochondrial Biomarkers in Psychiatric Illnesses
11.3. Challenges and Perspectives of Biomarkers in Neurodegenerative Disorders and Psychiatric Illnesses
11.3.1. Challenges
11.3.2. Future Perspectives
12. Therapeutic Strategies: Mitochondrial Protective Agents
13. Metabolic Modulators and Mitochondrial Function in Brain Diseases
14. Gene Therapy for Restoring Mitochondrial Function
15. Conclusions and Future Perspectives: Challenges and Opportunities
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- San-Millán, I. The Key Role of Mitochondrial Function in Health and Disease. Antioxidants 2023, 12, 782. [Google Scholar] [CrossRef]
- Lezi, E.; Swerdlow, R.H. Mitochondria in Neurodegeneration. Adv. Exp. Med. Biol. 2012, 942, 269–286. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Smith, H.J.; Yao, P.; Mair, W.B. Causal roles of mitochondrial dynamics in longevity and healthy aging. EMBO Rep. 2019, 20, e48395. [Google Scholar] [CrossRef] [PubMed]
- Solleiro-Villavicencio, H.; Rivas-Arancibia, S. Effect of Chronic Oxidative Stress on Neuroinflammatory Response Mediated by CD4(+)T Cells in Neurodegenerative Diseases. Front. Cell. Neurosci. 2018, 12, 114. [Google Scholar] [CrossRef]
- Missiroli, S.; Genovese, I.; Perrone, M.; Vezzani, B.; Vitto, V.A.M.; Giorgi, C. The Role of Mitochondria in Inflammation: From Cancer to Neurodegenerative Disorders. J. Clin. Med. 2020, 9, 740. [Google Scholar] [CrossRef] [PubMed]
- Teleanu, R.I.; Niculescu, A.-G.; Roza, E.; Vladâcenco, O.; Grumezescu, A.M.; Teleanu, D.M. Neurotransmitters—Key Factors in Neurological and Neurodegenerative Disorders of the Central Nervous System. Int. J. Mol. Sci. 2022, 23, 5954. [Google Scholar] [CrossRef] [PubMed]
- Daniels, T.E.; Olsen, E.M.; Tyrka, A.R. Stress and Psychiatric Disorders: The Role of Mitochondria. Annu. Rev. Clin. Psychol. 2020, 16, 165–186. [Google Scholar] [CrossRef]
- Dumont, M.; Lin, M.T.; Beal, M.F. Mitochondria and antioxidant targeted therapeutic strategies for Alzheimer’s disease. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S633–S643. [Google Scholar] [CrossRef]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best. Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef]
- Clemente-Suárez, V.J.; Bustamante-Sanchez, Á.; Tornero-Aguilera, J.F.; Ruisoto, P.; Mielgo-Ayuso, J. Inflammation in COVID-19 and the Effects of Non-Pharmacological Interventions during the Pandemic: A Review. Int. J. Mol. Sci. 2022, 23, 15584. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Moujalled, D.; Strasser, A.; Liddell, J.R. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021, 28, 2029–2044. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, K.; Eaton, S. Mitochondrial beta-oxidation. Eur. J. Biochem. 2004, 271, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef]
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 2004, 7, 254–261. [Google Scholar] [CrossRef]
- Barros, M.H.; McStay, G.P. Modular biogenesis of mitochondrial respiratory complexes. Mitochondrion 2019, 50, 94–114. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhai, Y.; Lou, J.; Liu, M.; Pang, X.; Sun, F. Thiabendazole inhibits ubiquinone reduction activity of mitochondrial respiratory complex II via a water molecule mediated binding feature. Protein Cell 2011, 2, 531–542. [Google Scholar] [CrossRef]
- Ramzan, R.; Kadenbach, B.; Vogt, S. Multiple Mechanisms Regulate Eukaryotic Cytochrome C Oxidase. Cells 2021, 10, 514. [Google Scholar] [CrossRef]
- Belhadj Slimen, I.; Najar, T.; Ghram, A.; Dabbebi, H.; Ben Mrad, M.; Abdrabbah, M. Reactive oxygen species, heat stress and oxidative-induced mitochondrial damage. A review. Int. J. Hyperth. 2014, 30, 513–523. [Google Scholar] [CrossRef]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef]
- Brand, M.D.; Affourtit, C.; Esteves, T.C.; Green, K.; Lambert, A.J.; Miwa, S.; Pakay, J.L.; Parker, N. Mitochondrial superoxide: Production, biological effects, and activation of uncoupling pro-teins. Free Radic. Biol. Med. 2004, 37, 755–767. [Google Scholar] [CrossRef]
- Tornero-Aguilera, J.F.; Jimenez-Morcillo, J.; Rubio-Zarapuz, A.; Clemente-Suárez, V.J. Central and Peripheral Fatigue in Physical Exercise Explained: A Narrative Review. Int. J. Environ. Res. Public Health 2022, 19, 3909. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Abramov, A.Y. Alpha-synuclein and Beta-Amyloid. Different Targets, Same Players: Calcium, Free Radicals and Mitochondria in the Mechanism of Neurodegeneration. Biochem. Biophys. Res. Commun. 2017, 483, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S. Calcium Signaling: From Basic to Bedside. Adv. Exp. Med. Biol. 2020, 1131, 1–6. [Google Scholar]
- Juárez, D.; González-Ravé, J.M.; Rubio-Arias, J.Á.; Clemente Suárez, V.J.; Valencia, M.; Abian Vicente, J. Isokinetic leg strength and power in elite handball players. J. Hum. Kinet. 2014, 41, 227–233. [Google Scholar]
- Bruni, F. Mitochondria: From Physiology to Pathology. Life 2021, 11, 991. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R. Apoptotic pathways: Paper wraps stone blunts scissors. Cell 2000, 102, 1–4. [Google Scholar] [CrossRef]
- Puthalakath, H.; Strasser, A. Keeping killers on a tight leash: Transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ. 2002, 9, 505–512. [Google Scholar] [CrossRef]
- Pellegrini, L.; Passer, B.J.; Tabaton, M.; Ganjei, J.K.; D’Adamio, L. Alternative, non-secretase processing of Alzheimer’s beta-amyloid precursor protein during apoptosis by caspase-6 and -8. J. Biol. Chem. 1999, 274, 21011–21016. [Google Scholar] [CrossRef]
- Vis, J.C.; Schipper, E.; de Boer-van Huizen, R.T.; Verbeek, M.M.; De Waal, R.M.W.; Wesseling, P.; ten Donkelaar, H.J.; Kremer, B. Expression pattern of apoptosis-related markers in Huntington’s disease. Acta Neuropathol. 2005, 109, 321–328. [Google Scholar] [CrossRef]
- Ofengeim, D.; Mazzitelli, S.; Ito, Y.; DeWitt, J.P.; Mifflin, L.; Zou, C.; Das, S.; Adiconis, X.; Chen, H.; Zhu, H.; et al. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E8788–E8797. [Google Scholar] [CrossRef]
- Yang, Y.; Klionsky, D.J. Autophagy and disease: Unanswered questions. Cell Death Differ. 2020, 27, 858–871. [Google Scholar] [CrossRef]
- Sasaki, S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2011, 70, 349–359. [Google Scholar] [CrossRef] [PubMed]
- van den Heuvel, M.P.; Sporns, O. Network hubs in the human brain. Trends Cogn. Sci. 2013, 17, 683–696. [Google Scholar] [CrossRef]
- Pino, A.; Fumagalli, G.; Bifari, F.; Decimo, I. New neurons in adult brain: Distribution, molecular mechanisms and therapies. Biochem. Pharmacol. 2017, 141, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of Mitochondrial Biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [PubMed]
- Kaminsky, N.; Bihari, O.; Kanner, S.; Barzilai, A. Connecting Malfunctioning Glial Cells and Brain Degenera-tive Disorders. Genom. Proteom. Bioinform. 2016, 14, 155–165. [Google Scholar]
- Vicente-Gutierrez, C.; Bonora, N.; Bobo-Jimenez, V.; Jimenez-Blasco, D.; Lopez-Fabuel, I.; Fernandez, E.; Josephine, C.; Bonvento, G.; Enriquez, J.A.; Almeida, A.; et al. Astrocytic Mitochondrial ROS Modulate Brain Metabolism and Mouse Behaviour. Nat. Metab. 2019, 1, 201–211. [Google Scholar] [CrossRef]
- Yang, S.; Qin, C.; Hu, Z.-W.; Zhou, L.-Q.; Yu, H.-H.; Chen, M.; Bosco, D.B.; Wang, W.; Wu, L.-J.; Tian, D.-S. Microglia Reprogram Metabolic Profiles for Phenotype and Function Changes in Central Nervous System. Neurobiol. Dis. 2021, 152, 105290. [Google Scholar] [CrossRef]
- Díaz-García, J.; Clemente-Suárez, V.J.; Fuentes-García, J.P.; Villafaina, S. Combining HIIT Plus Cognitive Task Increased Mental Fatigue but Not Physical Workload in Tennis Players. Appl. Sci. 2023, 13, 7046. [Google Scholar] [CrossRef]
- Lassmann, H. Classification of demyelinating diseases at the interface between etiology and pathogenesis. Curr. Opin. Neurol. 2001, 14, 253–258. [Google Scholar] [CrossRef]
- Ganat, Y.M.; Silbereis, J.; Cave, C.; Ngu, H.; Anderson, G.M.; Ohkubo, Y.; Ment, L.R.; Vaccarino, F.M. Early postnatal astroglial cells produce multilineage precursors and neural stem cells in vivo. J. Neurosci. 2006, 26, 8609–8621. [Google Scholar] [CrossRef]
- Fakhoury, M. Immune-mediated processes in neurodegeneration: Where do we stand? J. Neurol. 2016, 263, 1683–1701. [Google Scholar]
- Kovacs, G.G. Molecular pathology of neurodegenerative diseases: Principles and practice. J. Clin. Pathol. 2019, 72, 725–735. [Google Scholar] [CrossRef]
- Martin, J.B. Molecular Basis of the Neurodegenerative Disorders. N. Engl. J. Med. 1999, 340, 1970–1980. [Google Scholar] [CrossRef]
- Lamptey, R.N.L.; Chaulagain, B.; Trivedi, R.; Gothwal, A.; Layek, B.; Singh, J. A Review of the Common Neurodegenerative Disorders: Current Therapeutic Ap-proaches and the Potential Role of Nanotherapeutics. Int. J. Mol. Sci. 2022, 23, 1851. [Google Scholar]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Akbar, M.; Essa, M.M.; Daradkeh, G.; Abdelmegeed, M.A.; Choi, Y.; Mahmood, L.; Song, B.J. Mitochondrial dysfunction and cell death in neurodegenerative diseases through nitroxida-tive stress. Brain Res. 2016, 1637, 34–55. [Google Scholar]
- Johnson, J.; Mercado-Ayon, E.; Mercado-Ayon, Y.; Dong, Y.N.; Halawani, S.; Ngaba, L.; Lynch, D.R. Mitochondrial dysfunction in the development and progression of neurodegenerative diseases. Arch. Biochem. Biophys. 2021, 702, 108698. [Google Scholar]
- Clemente-Suárez, V.J.; Ramos-Campo, D.J.; Mielgo-Ayuso, J.; Dalamitros, A.A.; Nikolaidis, P.A.; Hormeño-Holgado, A.; Tornero-Aguilera, J.F. Nutrition in the Actual COVID-19 Pandemic. A Narrative Review. Nutrients 2021, 13, 1924. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coac-tivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Chew, K.; Zhao, L. Interactions of Mitochondrial Transcription Factor A with DNA Damage: Mechanistic Insights and Functional Implications. Genes 2021, 12, 1246. [Google Scholar] [CrossRef] [PubMed]
- Virbasius, J.V.; Scarpulla, R.C. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: A potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 1309–1313. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-D.; Yang, D.I.; Lin, T.K.; Shaw, F.Z.; Liou, C.W.; Chuang, Y.C. Roles of oxidative stress, apoptosis, PGC-1α and mitochondrial biogenesis in cerebral ische-mia. Int. J. Mol. Sci. 2011, 12, 7199–7215. [Google Scholar] [CrossRef]
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029. [Google Scholar] [CrossRef] [PubMed]
- Bouchez, C.; Devin, A. Mitochondrial Biogenesis and Mitochondrial Reactive Oxygen Species (ROS): A Complex Relationship Regulated by the cAMP/PKA Signaling Pathway. Cells 2019, 8, 287. [Google Scholar] [CrossRef]
- Meyer, J.N.; Leuthner, T.C.; Luz, A.L. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 2017, 391, 42–53. [Google Scholar]
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef]
- Clemente-Suárez, V.J.; Martín-Rodríguez, A.; Redondo-Flórez, L.; Ruisoto, P.; Navarro-Jiménez, E.; Ramos-Campo, D.J.; Tornero-Aguilera, J.F. Metabolic Health, Mitochondrial Fitness, Physical Activity, and Cancer. Cancers 2023, 15, 814. [Google Scholar] [CrossRef]
- Yapa, N.M.; Lisnyak, V.; Reljic, B.; Ryan, M.T. Mitochondrial dynamics in health and disease. FEBS Lett. 2021, 595, 1184–1204. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.; Brian, C.; Woods, J.; Pappa, A.; Panayiotidis, M.I.; Powers, R.; Franco, R. Mitochondrial dysfunction in glial cells: Implications for neuronal homeostasis and survival. Toxicology 2017, 391, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Pajarillo, E.; Nyarko-Danquah, I.; Aschner, M.; Lee, E. Role of Astrocytes in Parkinson’s Disease Asso-ciated with Genetic Mutations and Neurotoxicants. Cells 2023, 12, 622. [Google Scholar] [CrossRef]
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef]
- Branca, C.; Ferreira, E.; Nguyen, T.-V.; Doyle, K.; Caccamo, A.; Oddo, S. Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 4823–4835. [Google Scholar] [CrossRef]
- De Plano, L.M.; Calabrese, G.; Rizzo, M.G.; Oddo, S.; Caccamo, A. The Role of the Transcription Factor Nrf2 in Alzheimer’s Disease: Therapeutic Opportunities. Biomolecules 2023, 13, 549. [Google Scholar] [CrossRef]
- Pohland, M.; Pellowska, M.; Asseburg, H.; Hagl, S.; Reutzel, M.; Joppe, A.; Berressem, D.; Eckert, S.H.; Wurglics, M.; Schubert-Zsilavecz, M.; et al. MH84 improves mitochondrial dysfunction in a mouse model of early Alzheimer’s disease. Alzheimer’s Res. Ther. 2018, 10, 18. [Google Scholar] [CrossRef]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor form of brain-derived neurotrophic factor and ma-ture brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J. Neurochem. 2005, 93, 1412–1421. [Google Scholar] [CrossRef]
- Xia, D.-Y.; Huang, X.; Bi, C.F.; Mao, L.L.; Peng, L.J.; Qian, H.R. PGC-1α or FNDC5 Is Involved in Modulating the Effects of Aβ(1-42) Oligomers on Suppressing the Expression of BDNF, a Beneficial Factor for Inhibiting Neuronal Apoptosis, Aβ Deposition and Cognitive Decline of APP/PS1 Tg Mice. Front. Aging Neurosci. 2017, 9, 65. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef]
- Reddy, P.H. Mitochondrial oxidative damage in aging and Alzheimer’s disease: Implications for mitochon-drially targeted antioxidant therapeutics. J. Biomed. Biotechnol. 2006, 2006, 31372. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Parks, J.K.; Cassarino, D.S.; Maguire, D.J.; Maguire, R.S.; Bennett, J.P.; Davis, R.E.; Parker, W.D. Cybrids in Alzheimer’s disease: A cellular model of the disease? Neurology 1997, 49, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Lambert, T.J.; Storm, D.R.; Sullivan, J.M. MicroRNA132 modulates short-term synaptic plasticity but not basal release probability in hippocampal neurons. PLoS ONE 2010, 5, e15182. [Google Scholar] [CrossRef] [PubMed]
- Wingo, T.S.; Yang, J.; Fan, W.; Canon, S.M.; Gerasimov, E.S.; Lori, A.; Logsdon, B.; Yao, B.; Seyfried, N.T.; Lah, J.J.; et al. Brain microRNAs associated with late-life depressive symptoms are also associated with cognitive trajectory and dementia. NPJ Genom. Med. 2020, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Reddy, P.H. The role of synaptic microRNAs in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165937. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Reddy, P.H. A New Discovery of MicroRNA-455-3p in Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 72, S117–S130. [Google Scholar] [CrossRef] [PubMed]
- Gowda, P.; Reddy, P.H.; Kumar, S. Deregulated mitochondrial microRNAs in Alzheimer’s disease: Focus on synapse and mitochondria. Ageing Res. Rev. 2022, 73, 101529. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Chesselet, M.-F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef]
- Dexter, D.T.; Jenner, P. Parkinson disease: From pathology to molecular disease mechanisms. Free Radic. Biol. Med. 2013, 62, 132–144. [Google Scholar] [CrossRef]
- Schapira, A.H.V. Mitochondrial Pathology in parkinson’s disease. Mt. Sinai J. Med. 2011, 78, 872–881. [Google Scholar] [CrossRef]
- Heikkila, R.E.; Nicklas, W.J.; Vyas, I.; Duvoisin, R.C. Dopaminergic toxicity of rotenone and the 1-methyl-4-phenylpyridinium ion after their stereotaxic administration to rats: Implication for the mechanism of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity. Neurosci. Lett. 1985, 62, 389–394. [Google Scholar] [CrossRef] [PubMed]
- González-Rodríguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.; Reddy, S.; Zheng, K.; Betensky, R.A.; Simon, D.K. Association of PGC-1alpha polymorphisms with age of onset and risk of Parkinson’s disease. BMC Med. Genet. 2011, 12, 69. [Google Scholar] [CrossRef]
- Ritz, B.R.; Manthripragada, A.D.; Costello, S.; Lincoln, S.J.; Farrer, M.J.; Cockburn, M.; Bronstein, J. Dopamine transporter genetic variants and pesticides in Parkinson’s disease. Environ. Health Perspect. 2009, 117, 964–969. [Google Scholar] [CrossRef]
- Parker, W.D., Jr.; Parks, J.K.; Swerdlow, R.H. Complex I deficiency in Parkinson’s disease frontal cortex. Brain Res. 2008, 1189, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Kruse, S.E.; Watt, W.C.; Marcinek, D.J.; Kapur, R.P.; Schenkman, K.A.; Palmiter, R.D. Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab. 2008, 7, 312–320. [Google Scholar] [CrossRef]
- Cardoso, S.M.; Esteves, A.R.; Arduíno, D.M. Mitochondrial metabolic control of microtubule dynamics impairs the autophagic pathway in Parkinson’s disease. Neurodegener. Dis. 2012, 10, 38–40. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Andersen, J.K. Redox imbalance in Parkinson’s disease. Biochim. Biophys. Acta 2008, 1780, 1362–1367. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.-L.; Wang, T.T.; Luby-Phelps, K.; German, D.C. Mitochondria mass is low in mouse substantia nigra dopamine neurons: Implications for Parkinson’s disease. Exp. Neurol. 2007, 203, 370–380. [Google Scholar] [CrossRef] [PubMed]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2017, 25, 24–34. [Google Scholar] [CrossRef]
- MacDonald, M.E.; Barnes, G.; Srinidhi, J.; Duyao, M.P.; Ambrose, C.M.; Myers, R.H.; Gray, J.; Conneally, P.M.; Young, A.; Penney, J. Gametic but not somatic instability of CAG repeat length in Huntington’s disease. J. Med. Genet. 1993, 30, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Weydt, P.; Pineda, V.V.; Torrence, A.E.; Libby, R.T.; Satterfield, T.F.; Lazarowski, E.R.; Gilbert, M.L.; Morton, G.J.; Bammler, T.K.; Strand, A.D.; et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab. 2006, 4, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Johri, A.; Chandra, A.; Flint Beal, M. PGC-1α, mitochondrial dysfunction, and Huntington’s disease. Free Radic. Biol. Med. 2013, 62, 37–46. [Google Scholar] [CrossRef]
- Tsunemi, T.; Ashe, T.D.; Morrison, B.E.; Soriano, K.R.; Au, J.; Roque, R.A.V.; Lazarowski, E.R.; Damian, V.A.; Masliah, E.; La Spada, A.R. PGC-1α rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci. Transl. Med. 2012, 4, 142ra97. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, V.; Montermini, L.; Lutz, Y.; Cova, L.; Hindelang, C.; Jiralerspong, S.; Trottier, Y.; Kish, S.J.; Faucheux, B.; Trouillas, P.; et al. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum. Mol. Genet. 1997, 6, 1771–1780. [Google Scholar] [CrossRef]
- Coppola, G.; Marmolino, D.; Lu, D.; Wang, Q.; Cnop, M.; Rai, M.; Acquaviva, F.; Cocozza, S.; Pandolfo, M.; Geschwind, D.H.; et al. Functional genomic analysis of frataxin deficiency reveals tissue-specific alterations and identifies the PPARgamma pathway as a therapeutic target in Friedreich’s ataxia. Hum. Mol. Genet. 2009, 18, 2452–2461. [Google Scholar] [CrossRef]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Baker, N.; Patel, J.; Khacho, M. Linking mitochondrial dynamics, cristae remodeling and supercomplex formation: How mitochondrial structure can regulate bioenergetics. Mitochondrion 2019, 49, 259–268. [Google Scholar] [CrossRef]
- Annesley, S.J.; Fisher, P.R. Mitochondria in Health and Disease. Cells 2019, 8, 680. [Google Scholar] [CrossRef]
- Kummer, E.; Ban, N. Mechanisms and regulation of protein synthesis in mitochondria. Nat. Rev. Mol. Cell Biol. 2021, 22, 307–325. [Google Scholar] [CrossRef]
- Fernandez-Vizarra, E.; Zeviani, M. Mitochondrial disorders of the OXPHOS system. FEBS Lett. 2021, 595, 1062–1106. [Google Scholar] [CrossRef]
- Russell, O.M.; Gorman, G.S.; Lightowlers, R.N.; Turnbull, D.M. Mitochondrial Diseases: Hope for the Future. Cell 2020, 181, 168–188. [Google Scholar] [CrossRef]
- Stenton, S.L.; Prokisch, H. Genetics of mitochondrial diseases: Identifying mutations to help diagnosis. EBioMedicine 2020, 56, 102784. [Google Scholar] [CrossRef]
- Hahn, A.; Zuryn, S. The Cellular Mitochondrial Genome Landscape in Disease. Trends Cell Biol. 2019, 29, 227–240. [Google Scholar] [CrossRef]
- Borna, N.N.; Kishita, Y.; Kohda, M.; Lim, S.C.; Shimura, M.; Wu, Y.; Mogushi, K.; Yatsuka, Y.; Harashima, H.; Hisatomi, Y.; et al. Mitochondrial ribosomal protein PTCD3 mutations cause oxidative phosphorylation defects with Leigh syndrome. Neurogenetics 2019, 20, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.; Zuryn, S. Mitochondrial Genome (mtDNA) Mutations that Generate Reactive Oxygen Species. Antioxidants 2019, 8, 392. [Google Scholar] [CrossRef] [PubMed]
- Ganetzky, R.D.; Stendel, C.; McCormick, E.M.; Zolkipli-Cunningham, Z.; Goldstein, A.C.; Klopstock, T.; Falk, M.J. MT-ATP6mitochondrial disease variants: Phenotypic and biochemical features analysis in 218 published cases and cohort of 14 new cases. Hum. Mutat. 2019, 40, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wu, J.; Sun, R.; Tao, X.; Wang, X.; Kang, Q.; Wang, H.; Zhang, L.; Liu, P.; Zhang, J.; et al. SIRT5 deficiency suppresses mitochondrial ATP production and promotes AMPK activation in response to energy stress. PLoS ONE 2019, 14, e0211796. [Google Scholar] [CrossRef]
- Newman, N.J.; Carelli, V.; Taiel, M.; Yu-Wai-Man, P. Visual Outcomes in Leber Hereditary Optic Neuropathy Patients with the m.11778G>A (MTND4) Mitochondrial DNA Mutation. J. Neuro-Ophthalmol. 2020, 40, 547–557. [Google Scholar] [CrossRef]
- Monzio Compagnoni, G.; Di Fonzo, A.; Corti, S.; Comi, G.P.; Bresolin, N.; Masliah, E. The Role of Mitochondria in Neurodegenerative Diseases: The Lesson from Alzheimer’s Disease and Parkinson’s Disease. Mol. Neurobiol. 2020, 57, 2959–2980. [Google Scholar] [CrossRef] [PubMed]
- Norat, P.; Soldozy, S.; Sokolowski, J.D.; Gorick, C.M.; Kumar, J.S.; Chae, Y.; Yağmurlu, K.; Prada, F.; Walker, M.; Levitt, M.R.; et al. Mitochondrial dysfunction in neurological disorders: Exploring mitochondrial transplantation. NPJ Regen. Med. 2020, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Falabella, M.; Vernon, H.J.; Hanna, M.G.; Claypool, S.M.; Pitceathly, R.D.S. Cardiolipin, Mitochondria, and Neurological Disease. Trends Endocrinol. Metab. 2021, 32, 224–237. [Google Scholar] [CrossRef]
- Ricoy-Campo, J.R.; Cabello, A. [Mitochondriopathies]. Rev. Neurol. 2003, 37, 775–779. [Google Scholar] [PubMed]
- Finsterer, J. Mitochondriopathies. Eur. J. Neurol. 2004, 11, 163–186. [Google Scholar] [CrossRef]
- Lagler, F.B. Current and Emerging Therapies for Mitochondriopathies. Handb. Exp. Pharmacol. 2020, 261, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Hage, R.; Vignal-Clermont, C. Leber Hereditary Optic Neuropathy: Review of Treatment and Management. Front. Neurol. 2021, 12, 651639. [Google Scholar] [CrossRef]
- Newman, N.J.; Yu-Wai-Man, P.; Carelli, V.; Moster, M.L.; Biousse, V.; Vignal-Clermont, C.; Sergott, R.C.; Klopstock, T.; Sadun, A.A.; Barboni, P.; et al. Efficacy and Safety of Intravitreal Gene Therapy for Leber Hereditary Optic Neuropathy Treated within 6 Months of Disease Onset. Ophthalmology 2021, 128, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Stenton, S.L.; Sheremet, N.L.; Catarino, C.B.; Andreeva, N.A.; Assouline, Z.; Barboni, P.; Barel, O.; Berutti, R.; Bychkov, I.; Caporali, L.; et al. Impaired complex I repair causes recessive Leber’s hereditary optic neuropathy. J. Clin. Investig. 2021, 131, e138267. [Google Scholar] [CrossRef]
- Sinnecker, T.; Andelova, M.; Mayr, M.; Rüegg, S.; Sinnreich, M.; Hench, J.; Frank, S.; Schaller, A.; Stippich, C.; Wuerfel, J.; et al. Diagnosis of adult-onset MELAS syndrome in a 63-year-old patient with suspected recurrent strokes—A case report. BMC Neurol. 2019, 19, 91. [Google Scholar] [CrossRef]
- Huang, G.; Wang, Y.; Yao, D. Myoclonic epilepsy with ragged red fibers syndrome associated with mitochondrial 3302A>G mutation in the MT-TL1 gene: A case report. Exp. Ther. Med. 2023, 25, 87. [Google Scholar] [CrossRef]
- Hamblet, N.S.; Castora, F.J. Elevated levels of the Kearns–Sayre syndrome mitochondrial DNA deletion in temporal cortex of Alzheimer’s patients. Mutat. Res. 1997, 379, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, A.; Martin, M.V.; Rollins, B.; Moon, E.A.; Bunney, W.E.; Macciardi, F.; Lupoli, S.; Smith, E.N.; Kelsoe, J.; Magnan, C.N.; et al. Mitochondrial mutations and polymorphisms in psychiatric disorders. Front. Genet. 2012, 3, 103. [Google Scholar] [CrossRef]
- Valiente-Pallejà, A.; Tortajada, J.; Bulduk, B.K.; Vilella, E.; Garrabou, G.; Muntané, G.; Martorell, L. Comprehensive summary of mitochondrial DNA alterations in the postmortem human brain: A systematic review. EBioMedicine 2022, 76, 103815. [Google Scholar] [CrossRef] [PubMed]
- Gu, G.; Reyes, P.F.; Golden, G.T.; Woltjer, R.L.; Hulette, C.; Montine, T.J.; Zhang, J. Mitochondrial DNA deletions/rearrangements in parkinson disease and related neurodegenerative disorders. J. Neuropathol. Exp. Neurol. 2002, 61, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Schon, K.R.; Ratnaike, T.; van den Ameele, J.; Horvath, R.; Chinnery, P.F. Mitochondrial Diseases: A Diagnostic Revolution. Trends Genet. 2020, 36, 702–717. [Google Scholar] [CrossRef]
- Schlieben, L.D.; Prokisch, H. The Dimensions of Primary Mitochondrial Disorders. Front. Cell Dev. Biol. 2020, 8, 600079. [Google Scholar] [CrossRef]
- Arthur, C.R.; Morton, S.L.; Dunham, L.D.; Keeney, P.M.; Bennett, J.P., Jr. Parkinson’s disease brain mitochondria have impaired respirasome assembly, age-related increases in distribution of oxidative damage to mtDNA and no differences in heteroplasmic mtDNA mutation abundance. Mol. Neurodegener. 2009, 4, 37. [Google Scholar] [CrossRef]
- Dölle, C.; Flønes, I.; Nido, G.S.; Miletic, H.; Osuagwu, N.; Kristoffersen, S.; Lilleng, P.K.; Larsen, J.P.; Tysnes, O.-B.; Haugarvoll, K.; et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat. Commun. 2016, 7, 13548. [Google Scholar] [CrossRef]
- Coxhead, J.; Kurzawa-Akanbi, M.; Hussain, R.; Pyle, A.; Chinnery, P.; Hudson, G. Somatic mtDNA variation is an important component of Parkinson’s disease. Neurobiol. Aging 2016, 38, 217.e1–217.e6. [Google Scholar] [CrossRef]
- Rubino, F.; Piredda, R.; Calabrese, F.M.; Simone, D.; Lang, M.; Calabrese, C.; Petruzzella, V.; Tommaseo-Ponzetta, M.; Gasparre, G.; Attimonelli, M. HmtDB, a genomic resource for mitochondrion-based human variability studies. Nucleic Acids Res. 2012, 40, D1150–D1159. [Google Scholar] [CrossRef]
- Marchbanks, R.; Ryan, M.; Day, I.; Owen, M.; McGuffin, P.; Whatley, S. A mitochondrial DNA sequence variant associated with schizophrenia and oxidative stress. Schizophr. Res. 2003, 65, 33–38. [Google Scholar] [CrossRef]
- Rollins, B.; Martin, M.V.; Sequeira, P.A.; Moon, E.A.; Morgan, L.Z.; Watson, S.J.; Schatzberg, A.; Akil, H.; Myers, R.M.; Jones, E.G.; et al. Mitochondrial variants in schizophrenia, bipolar disorder, and major depressive disorder. PLoS ONE 2009, 4, e4913. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, T.; Arai, M.; Miyashita, M.; Arai, M.; Obata, N.; Nohara, I.; Oshima, K.; Niizato, K.; Okazaki, Y.; Doi, N.; et al. Schizophrenia: Maternal inheritance and heteroplasmy of mtDNA mutations. Mol. Genet. Metab. 2012, 105, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Munakata, K.; Iwamoto, K.; Bundo, M.; Kato, T. Mitochondrial DNA 3243A>G mutation and increased expression of LARS2 gene in the brains of patients with bipolar disorder and schizophrenia. Biol. Psychiatry 2005, 57, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Bodenstein, D.F.; Kim, H.K.; Brown, N.C.; Navaid, B.; Young, L.T.; Andreazza, A.C. Mitochondrial DNA content and oxidation in bipolar disorder and its role across brain regions. NPJ Schizophr. 2019, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.-S.; Huang, Y.-W.; Ho, C.-S.; Hung, P.-L.; Hsu, M.-H.; Wang, T.-J.; Wu, T.-Y.; Lee, T.-H.; Huang, Z.-D.; Chang, P.-C.; et al. Oxidative Insults and Mitochondrial DNA Mutation Promote Enhanced Autophagy and Mitophagy Compromising Cell Viability in Pluripotent Cell Model of Mitochondrial Disease. Cells 2019, 8, 65. [Google Scholar] [CrossRef]
- Li, H.; Slone, J.; Fei, L.; Huang, T. Mitochondrial DNA Variants and Common Diseases: A Mathematical Model for the Diversity of Age-Related mtDNA Mutations. Cells 2019, 8, 608. [Google Scholar] [CrossRef] [PubMed]
- Nissanka, N.; Minczuk, M.; Moraes, C.T. Mechanisms of Mitochondrial DNA Deletion Formation. Trends Genet. 2019, 35, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Lu, P.; Beeraka, N.M.; Sukocheva, O.A.; Madhunapantula, S.V.; Liu, J.; Sinelnikov, M.Y.; Nikolenko, V.N.; Bulygin, K.V.; Mikhaleva, L.M.; et al. Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced responses in breast cancers. Semin. Cancer Biol. 2022, 83, 556–569. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Amato, P.; Sparman, M.; Gutierrez, N.M.; Tippner-Hedges, R.; Ma, H.; Kang, E.; Fulati, A.; Lee, H.-S.; Sritanaudomchai, H.; et al. Human embryonic stem cells derived by somatic cell nuclear transfer. Cell 2013, 153, 1228–1238. [Google Scholar] [CrossRef]
- Artika, I.M. Allotopic expression of mitochondrial genes: Basic strategy and progress. Genes Dis. 2020, 7, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Nuevo-Tapioles, C.; Santacatterina, F.; Sánchez-Garrido, B.; de Arenas, C.N.; Robledo-Bérgamo, A.; Martínez-Valero, P.; Cantarero, L.; Pardo, B.; Hoenicka, J.; Murphy, M.P.; et al. Effective therapeutic strategies in a preclinical mouse model of Charcot-Marie-Tooth disease. Hum. Mol. Genet. 2021, 30, 2441–2455. [Google Scholar] [CrossRef] [PubMed]
- Goikoetxea-Usandizaga, N.; Serrano-Maciá, M.; Delgado, T.C.; Simón, J.; Ramos, D.F.; Barriales, D.; Cornide, M.E.; Jiménez, M.; Pérez-Redondo, M.; Lachiondo-Ortega, S.; et al. Mitochondrial bioenergetics boost macrophage activation, promoting liver regeneration in metabolically compromised animals. Hepatology 2022, 75, 550–566. [Google Scholar] [CrossRef]
- Miret-Casals, L.; Sebastián, D.; Brea, J.; Rico-Leo, E.M.; Palacín, M.; Fernández-Salguero, P.M.; Loza, M.I.; Albericio, F.; Zorzano, A. Identification of New Activators of Mitochondrial Fusion Reveals a Link between Mitochondrial Morphology and Pyrimidine Metabolism. Cell Chem. Biol. 2018, 25, 268–278.e4. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Ding, M.; Tang, D.; Gao, E.; Li, C.; Wang, K.; Qi, B.; Qiu, J.; Zhao, H.; Chang, P.; et al. Targeting mitochondrial dynamics by regulating Mfn2 for therapeutic intervention in diabetic cardiomyopathy. Theranostics 2019, 9, 3687–3706. [Google Scholar] [CrossRef]
- Supinski, G.S.; Schroder, E.A.; Callahan, L.A. Mitochondria and Critical Illness. Chest 2020, 157, 310–322. [Google Scholar] [CrossRef]
- Nelson, M.A.; McLaughlin, K.L.; Hagen, J.T.; Coalson, H.S.; Schmidt, C.; Kassai, M.; Kew, K.A.; McClung, J.M.; Neufer, P.D.; Brophy, P.; et al. Intrinsic OXPHOS limitations underlie cellular bioenergetics in leukemia. eLife 2021, 10, e63104. [Google Scholar] [CrossRef]
- Garrido-Pérez, N.; Vela-Sebastián, A.; López-Gallardo, E.; Emperador, S.; Iglesias, E.; Meade, P.; Jiménez-Mallebrera, C.; Montoya, J.; Bayona-Bafaluy, M.P.; Ruiz-Pesini, E. Oxidative Phosphorylation Dysfunction Modifies the Cell Secretome. Int. J. Mol. Sci. 2020, 21, 3374. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S.; Trombetti, F.; Pagliarani, A.; Ventrella, V.; Algieri, C.; Tioli, G.; Lenaz, G. Molecular and Supramolecular Structure of the Mitochondrial Oxidative Phosphorylation System: Implications for Pathology. Life 2021, 11, 242. [Google Scholar] [CrossRef]
- Sarker, A.H.; Cooper, P.K.; Hazra, T.K. DNA glycosylase NEIL2 functions in multiple cellular processes. Prog. Biophys. Mol. Biol. 2021, 164, 72–80. [Google Scholar] [CrossRef]
- Wang, C.-H.; Wei, Y.-H. Roles of Mitochondrial Sirtuins in Mitochondrial Function, Redox Homeostasis, Insulin Resistance and Type 2 Diabetes. Int. J. Mol. Sci. 2020, 21, 5266. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD(+) metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- He, L.; Liu, F.; Li, J. Mitochondrial Sirtuins and Doxorubicin-induced Cardiotoxicity. Cardiovasc. Toxicol. 2021, 21, 179–191. [Google Scholar] [CrossRef]
- Shahgaldi, S.; Kahmini, F.R. A comprehensive review of Sirtuins: With a major focus on redox homeostasis and metabolism. Life Sci. 2021, 282, 119803. [Google Scholar] [CrossRef]
- Palmeira, C.M.; Teodoro, J.S.; Amorim, J.A.; Steegborn, C.; Sinclair, D.A.; Rolo, A.P. Mitohormesis and metabolic health: The interplay between ROS, cAMP and sirtuins. Free Radic. Biol. Med. 2019, 141, 483–491. [Google Scholar] [CrossRef]
- Yuan, X.; Liu, Y.; Bijonowski, B.M.; Tsai, A.-C.; Fu, Q.; Logan, T.M.; Ma, T.; Li, Y. NAD(+)/NADH redox alterations reconfigure metabolism and rejuvenate senescent human mesenchymal stem cells in vitro. Commun. Biol. 2020, 3, 774. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, J.; Li, X.; Zhou, X.; Hu, Y.; Chu, S.; Peng, Y.; Chen, N. Research progress on adenosine in central nervous system diseases. CNS Neurosci. Ther. 2019, 25, 899–910. [Google Scholar] [CrossRef]
- Zhao, L.; Cao, J.; Hu, K.; He, X.; Yun, D.; Tong, T.; Han, L. Sirtuins and their Biological Relevance in Aging and Age-Related Diseases. Aging Dis. 2020, 11, 927–945. [Google Scholar] [CrossRef]
- Clemente-Suárez, V.J.; Robles-Pérez, J.J. Psycho-physiological response of soldiers in urban combat. An. Psicol. 2013, 29, 598–603. [Google Scholar]
- Van der Velpen, V.; Teav, T.; Gallart-Ayala, H.; Mehl, F.; Konz, I.; Clark, C.; Oikonomidi, A.; Peyratout, G.; Henry, H.; Delorenzi, M.; et al. Systemic and central nervous system metabolic alterations in Alzheimer’s disease. Alzheimers Res. Ther. 2019, 11, 93. [Google Scholar] [CrossRef]
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef] [PubMed]
- Rekatsina, M.; Paladini, A.; Piroli, A.; Zis, P.; Pergolizzi, J.V.; Varrassi, G. Pathophysiology and Therapeutic Perspectives of Oxidative Stress and Neurodegenerative Diseases: A Narrative Review. Adv. Ther. 2020, 37, 113–139. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.-R.; Hu, L.-S.; Li, G.-Y. SH-SY5Y human neuroblastoma cell line: In vitro cell model of dopaminergic neurons in Parkinson’s disease. Chin. Med. J. 2010, 123, 1086–1092. [Google Scholar] [PubMed]
- Agholme, L.; Lindström, T.; Kågedal, K.; Marcusson, J.; Hallbeck, M. An in vitro model for neuroscience: Differentiation of SH-SY5Y cells into cells with morphological and biochemical characteristics of mature neurons. J. Alzheimers Dis. 2010, 20, 1069–1082. [Google Scholar] [CrossRef]
- He, L.; Wang, J.; Yang, Y.; Li, J.; Tu, H. Mitochondrial Sirtuins in Parkinson’s Disease. Neurochem. Res. 2022, 47, 1491–1502. [Google Scholar] [CrossRef]
- Angelova, P.R.; Esteras, N.; Abramov, A.Y. Mitochondria and lipid peroxidation in the mechanism of neurodegeneration: Finding ways for prevention. Med. Res. Rev. 2021, 41, 770–784. [Google Scholar] [CrossRef]
- Taso, O.V.; Philippou, A.; Moustogiannis, A.; Zevolis, E.; Koutsilieris, M. Lipid peroxidation products and their role in neurodegenerative diseases. Ann. Res. Hosp. 2019, 3, 2. [Google Scholar] [CrossRef]
- Segura-Aguilar, J. On the Role of Aminochrome in Mitochondrial Dysfunction and Endoplasmic Reticulum Stress in Parkinson’s Disease. Front. Neurosci. 2019, 13, 271. [Google Scholar] [CrossRef]
- Segura-Aguilar, J.; Mannervik, B.; Inzunza, J.; Varshney, M.; Nalvarte, I.; Muñoz, P. Astrocytes protect dopaminergic neurons against aminochrome neurotoxicity. Neural Regen. Res. 2022, 17, 1861–1866. [Google Scholar] [CrossRef]
- Creus-Muncunill, J.; Badillos-Rodríguez, R.; Garcia-Forn, M.; Masana, M.; Barriga, G.G.-D.; Guisado-Corcoll, A.; Alberch, J.; Malagelada, C.; Delgado-García, J.M.; Gruart, A.; et al. Increased translation as a novel pathogenic mechanism in Huntington’s disease. Brain 2019, 142, 3158–3175. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Plaza-Zabala, A.; Montpeyo, M.; Sebastian, D.; Vila, M.; Martinez-Vicente, M. Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy 2021, 17, 672–689. [Google Scholar] [CrossRef]
- Pantiya, P.; Thonusin, C.; Chattipakorn, N.; Chattipakorn, S.C. Mitochondrial abnormalities in neurodegenerative models and possible interventions: Focus on Alzheimer’s disease, Parkinson’s disease, Huntington’s disease. Mitochondrion 2020, 55, 14–47. [Google Scholar] [CrossRef]
- Burtscher, J.; Di Pardo, A.; Maglione, V.; Schwarzer, C.; Squitieri, F. Mitochondrial Respiration Changes in R6/2 Huntington’s Disease Model Mice during Aging in a Brain Region Specific Manner. Int. J. Mol. Sci. 2020, 21, 5412. [Google Scholar] [CrossRef]
- Fjodorova, M.; Louessard, M.; Li, Z.; De La Fuente, D.C.; Dyke, E.; Brooks, S.P.; Perrier, A.L.; Li, M. CTIP2-Regulated Reduction in PKA-Dependent DARPP32 Phosphorylation in Human Medium Spiny Neurons: Implications for Huntington Disease. Stem Cell Rep. 2019, 13, 448–457. [Google Scholar] [CrossRef]
- Henkel, N.D.; Wu, X.; O’donovan, S.M.; Devine, E.A.; Jiron, J.M.; Rowland, L.M.; Sarnyai, Z.; Ramsey, A.J.; Wen, Z.; Hahn, M.K.; et al. Schizophrenia: A disorder of broken brain bioenergetics. Mol. Psychiatry 2022, 27, 2393–2404. [Google Scholar] [CrossRef]
- Shroitman, N.K.; Yitzhaky, A.; Ben Shachar, D.; Gurwitz, D.; Hertzberg, L. Meta-analysis of brain samples of individuals with schizophrenia detects down-regulation of multiple ATP synthase encoding genes in both females and males. J. Psychiatr. Res. 2023, 158, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Ni, P.; Chung, S. Mitochondrial Dysfunction in Schizophrenia. Bioessays 2020, 42, e1900202. [Google Scholar] [CrossRef]
- Lancaster, T.M.; Dimitriadis, S.L.; Tansey, K.E.; Perry, G.; Ihssen, N.; Jones, D.K.; Singh, K.D.; Holmans, P.; Pocklington, A.; Smith, G.D.; et al. Structural and Functional Neuroimaging of Polygenic Risk for Schizophrenia: A Recall-by-Genotype–Based Approach. Schizophr. Bull. 2019, 45, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Rao, W.; Zhang, Y.; Li, K.; Zhang, X.Y. Association between cognitive impairment and apolipoprotein A1 or apolipoprotein B levels is regulated by apolipoprotein E variant rs429358 in patients with chronic schizophrenia. Aging 2021, 13, 16353–16366. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.M.; Zhu, R.R.; Tian, Y.; Uludag, K.; Chen, J.J.; Zhou, H.X.; Wang, L.; Kosten, T.R.; Zhang, X.Y. Association between MnSOD Activity and Cognitive Impairment in Unmedicated First-Episode Schizophrenia: Regulated by MnSOD Ala-9Val Gene Polymorphism. Antioxidants 2022, 11, 1981. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhang, Q.; Su, Y.; Chen, X.; Li, X.; Du, B.; Deng, X.; Ji, F.; Li, J.; Dong, Q.; et al. Effect of schizophrenia risk gene polymorphisms on cognitive and neural plasticity. Schizophr. Res. 2022, 248, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, S.; Noda, Y.; Tarumi, R.; Mimura, Y.; Yoshida, K.; Iwata, Y.; Elsalhy, M.; Kuromiya, M.; Kurose, S.; Masuda, F.; et al. Glutathione levels and activities of glutathione metabolism enzymes in patients with schizophrenia: A systematic review and meta-analysis. J. Psychopharmacol. 2019, 33, 1199–1214. [Google Scholar] [CrossRef] [PubMed]
- Juchnowicz, D.; Dzikowski, M.; Rog, J.; Waszkiewicz, N.; Karakuła, K.H.; Zalewska, A.; Maciejczyk, M.; Karakula-Juchnowicz, H. Pro/Antioxidant State as a Potential Biomarker of Schizophrenia. J. Clin. Med. 2021, 10, 4156. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Ta, Q.T.H.; Nguyen, T.K.O.; Nguyen, T.T.D.; Giau, V. Van. Type 3 Diabetes and Its Role Implications in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 3165. [Google Scholar] [CrossRef]
- González, A.; Calfío, C.; Churruca, M.; Maccioni, R.B. Glucose metabolism and AD: Evidence for a potential diabetes type 3. Alzheimer’s Res. Ther. 2022, 14, 56. [Google Scholar] [CrossRef]
- Wescott, A.P.; Kao, J.P.Y.; Lederer, W.J.; Boyman, L. Voltage-energized calcium-sensitive ATP production by mitochondria. Nat. Metab. 2019, 1, 975–984. [Google Scholar] [CrossRef]
- Plotegher, N.; Perocheau, D.; Ferrazza, R.; Massaro, G.; Bhosale, G.; Zambon, F.; Rahim, A.A.; Guella, G.; Waddington, S.N.; Szabadkai, G.; et al. Impaired cellular bioenergetics caused by GBA1 depletion sensitizes neurons to calcium overload. Cell Death Differ. 2020, 27, 1588–1603. [Google Scholar] [CrossRef]
- Boyman, L.; Karbowski, M.; Lederer, W.J. Regulation of Mitochondrial ATP Production: Ca2+ Signaling and Quality Control. Trends Mol. Med. 2020, 26, 21–39. [Google Scholar] [CrossRef]
- Clemente-Suárez, V.J.; Navarro-Jiménez, E.; Jimenez, M.; Hormeño-Holgado, A.; Martinez-Gonzalez, M.B.; Benitez-Agudelo, J.C.; Perez-Palencia, N.; Laborde-Cárdenas, C.C.; Tornero-Aguilera, J.F. Impact of COVID-19 Pandemic in Public Mental Health: An Extensive Narrative Review. Sustainability 2021, 13, 3221. [Google Scholar] [CrossRef]
- Devine, M.J.; Birsa, N.; Kittler, J.T. Miro sculpts mitochondrial dynamics in neuronal health and disease. Neurobiol. Dis. 2016, 90, 27–34. [Google Scholar] [CrossRef]
- Sebastián, D.; Palacín, M.; Zorzano, A. Mitochondrial Dynamics: Coupling Mitochondrial Fitness with Healthy Aging. Trends Mol. Med. 2017, 23, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Avdoshina, V.; Fields, J.A.; Castellano, P.; Dedoni, S.; Palchik, G.; Trejo, M.; Adame, A.; Rockenstein, E.; Eugenin, E.; Masliah, E.; et al. The HIV Protein gp120 Alters Mitochondrial Dynamics in Neurons. Neurotox. Res. 2016, 29, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Ferré, C.A.; Thouard, A.; Bétourné, A.; Le Dorze, A.-L.; Belenguer, P.; Miquel, M.-C.; Peyrin, J.-M.; Gonzalez-Dunia, D.; Szelechowski, M. HSPA9/Mortalin mediates axo-protection and modulates mitochondrial dynamics in neurons. Sci. Rep. 2021, 11, 17705. [Google Scholar] [CrossRef] [PubMed]
- Priyanka; Seth, P. Insights Into the Role of Mortalin in Alzheimer’s Disease, Parkinson’s Disease, and HIV-1-Associated Neurocognitive Disorders. Front. Cell Dev. Biol. 2022, 10, 903031. [Google Scholar] [CrossRef] [PubMed]
- Van Laar, V.S.; Berman, S.B. The interplay of neuronal mitochondrial dynamics and bioenergetics: Implications for Parkinson’s disease. Neurobiol. Dis. 2013, 51, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Seager, R.; Lee, L.; Henley, J.M.; Wilkinson, K.A. Mechanisms and roles of mitochondrial localisation and dynamics in neuronal function. Neuronal Signal. 2020, 4, NS20200008. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.R.; Abramov, A.Y. Mitochondrial calcium imbalance in Parkinson’s disease. Neurosci. Lett. 2018, 663, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Crews, L.; Tsigelny, I.; Hashimoto, M.; Masliah, E. Role of synucleins in Alzheimer’s disease. Neurotoxicity Res. 2009, 16, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Cascella, R.; Cecchi, C. Calcium Dyshomeostasis in Alzheimer’s Disease Pathogenesis. Int. J. Mol. Sci. 2021, 22, 4914. [Google Scholar] [CrossRef] [PubMed]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Praticò, D.; Elrod, J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019, 10, 3885. [Google Scholar] [CrossRef]
- Mira, R.G.; Cerpa, W. Building a Bridge Between NMDAR-Mediated Excitotoxicity and Mitochondrial Dysfunction in Chronic and Acute Diseases. Cell. Mol. Neurobiol. 2021, 41, 1413–1430. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.C.; Ashkavand, Z.; Norman, K.R. The Role of Mitochondrial Calcium Homeostasis in Alzheimer’s and Related Diseases. Int. J. Mol. Sci. 2020, 21, 9153. [Google Scholar] [CrossRef] [PubMed]
- Manolaras, I.; Del Bondio, A.; Griso, O.; Reutenauer, L.; Eisenmann, A.; Habermann, B.H.; Puccio, H. Mitochondrial dysfunction and calcium dysregulation in COQ8A-ataxia Purkinje neurons are rescued by CoQ10 treatment. Brain 2023, 146, 3836–3850. [Google Scholar] [CrossRef] [PubMed]
- Lennicke, C.; Cochemé, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell 2021, 81, 3691–3707. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rigoulet, M.; Yoboue, E.D.; Devin, A. Mitochondrial ROS generation and its regulation: Mechanisms involved in H(2)O(2) signaling. Antioxid. Redox Signal 2011, 14, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Walton, P.A.; Pizzitelli, M. Effects of peroxisomal catalase inhibition on mitochondrial function. Front. Physiol. 2012, 3, 108. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Sarniak, A.; Lipińska, J.; Tytman, K.; Lipińska, S. Endogenous mechanisms of reactive oxygen species (ROS) generation. Postep. Hig. Med. Dosw. Online 2016, 70, 1150–1165. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, C.E.; Carroll, K.S. Cysteine-mediated redox signaling: Chemistry, biology, and tools for discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef] [PubMed]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Janke, R.; Dodson, A.E.; Rine, J. Metabolism and epigenetics. Annu. Rev. Cell Dev. Biol. 2015, 31, 473–496. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Zeke, A.; Misheva, M.; Reményi, A.; Bogoyevitch, M.A. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Biol. Rev. 2016, 80, 793–835. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zheng, J.; Nussinov, R.; Ma, B. Release of Cytochrome C from Bax Pores at the Mitochondrial Membrane. Sci. Rep. 2017, 7, 2635. [Google Scholar] [CrossRef] [PubMed]
- Estaquier, J.; Vallette, F.; Vayssiere, J.-L.; Mignotte, B. The mitochondrial pathways of apoptosis. Adv. Exp. Med. Biol. 2012, 942, 157–183. [Google Scholar] [PubMed]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Suárez, V.J.; Mielgo-Ayuso, J.; Quiles, J.L.; Varela-Lopez, A.; Aranda, P. Effect of α-tocopherol megadoses on hematologic parameters and antioxidant capacity of rats in an ultraendurance probe. Physiol. Int. 2017, 104, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Peeters, A.; Shinde, A.B.; Dirkx, R.; Smet, J.; De Bock, K.; Espeel, M.; Vanhorebeek, I.; Vanlander, A.; Van Coster, R.; Carmeliet, P.; et al. Mitochondria in peroxisome-deficient hepatocytes exhibit impaired respiration, depleted DNA, and PGC-1α independent proliferation. Biochim. Biophys. Acta 2015, 1853, 285–298. [Google Scholar] [CrossRef] [PubMed]
- García-Giménez, J.L.; Pallardó, F.V. Maintenance of glutathione levels and its importance in epigenetic regulation. Front. Pharmacol. 2014, 5, 88. [Google Scholar] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine; Oxford University Press: New York, NY, USA, 2015. [Google Scholar]
- Matsui, R.; Ferran, B.; Oh, A.; Croteau, D.; Shao, D.; Han, J.; Pimentel, D.R.; Bachschmid, M.M. Redox Regulation via Glutaredoxin-1 and Protein S-Glutathionylation. Antioxid. Redox Signal 2020, 32, 677–700. [Google Scholar] [CrossRef]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef]
- Song, T.; Song, X.; Zhu, C.; Patrick, R.; Skurla, M.; Santangelo, I.; Green, M.; Harper, D.; Ren, B.; Forester, B.P.; et al. Mitochondrial dysfunction, oxidative stress, neuroinflammation, and metabolic alterations in the progression of Alzheimer’s disease: A meta-analysis of in vivo magnetic resonance spectroscopy studies. Ageing Res. Rev. 2021, 72, 101503. [Google Scholar]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar]
- Tõugu, V.; Palumaa, P. Coordination of zinc ions to the key proteins of neurodegenerative diseases: Aβ, APP, α-synuclein and PrP. Coord. Chem. Rev. 2012, 256, 2219–2224. [Google Scholar]
- Faller, P.; Hureau, C.; Berthoumieu, O. Role of metal ions in the self-assembly of the Alzheimer’s amyloid-β peptide. Inorg. Chem. 2013, 52, 12193–12206. [Google Scholar]
- Kim, A.C.; Lim, S.; Kim, Y.K. Metal Ion Effects on Aβ and Tau Aggregation. Int. J. Mol. Sci. 2018, 19, 128. [Google Scholar] [CrossRef]
- Wang, P.; Wang, Z.-Y. Metal ions influx is a double edged sword for the pathogenesis of Alzheimer’s disease. Ageing Res. Rev. 2017, 35, 265–290. [Google Scholar] [CrossRef]
- Tiiman, A.; Palumaa, P.; Tõugu, V. The missing link in the amyloid cascade of Alzheimer’s disease—Metal ions. Neurochem. Int. 2013, 62, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Tramutola, A.; Lanzillotta, C.; Perluigi, M.; Butterfield, D.A. Oxidative stress, protein modification and Alzheimer disease. Brain Res. Bull. 2017, 133, 88–96. [Google Scholar] [CrossRef]
- Aluise, C.D.; Robinson, R.A.; Cai, J.; Pierce, W.M.; Markesbery, W.R.; Butterfield, D.A. Redox proteomics analysis of brains from subjects with amnestic mild cognitive impairment compared to brains from subjects with preclinical Alzheimer’s disease: Insights into memory loss in MCI. J. Alzheimer’s Dis. 2011, 23, 257–269. [Google Scholar] [CrossRef]
- Ito, S.; Ohtsuki, S.; Kamiie, J.; Nezu, Y.; Terasaki, T. Cerebral clearance of human amyloid-beta peptide (1–40) across the blood-brain barrier is reduced by self-aggregation and formation of low-density lipoprotein receptor-related protein-1 ligand complexes. J. Neurochem. 2007, 103, 2482–2490. [Google Scholar] [CrossRef] [PubMed]
- Sagare, A.; Deane, R.; Bell, R.D.; Johnson, B.; Hamm, K.; Pendu, R.; Marky, A.; Lenting, P.J.; Wu, Z.; Zarcone, T.; et al. Clearance of amyloid-beta by circulating lipoprotein receptors. Nat. Med. 2007, 13, 1029–1031. [Google Scholar] [CrossRef] [PubMed]
- Jeynes, B.; Provias, J. Evidence for Altered LRP/RAGE Expression in Alzheimer Lesion Pathogenesis. Curr. Alzheimer Res. 2008, 5, 432–437. [Google Scholar] [CrossRef]
- Liu, Q.; Smith, M.A.; Avilá, J.; DeBernardis, J.; Kansal, M.; Takeda, A.; Zhu, X.; Nunomura, A.; Honda, K.; Moreira, P.I.; et al. Alzheimer-specific epitopes of tau represent lipid peroxidation-induced conformations. Free Radic. Biol. Med. 2005, 38, 746–754. [Google Scholar] [CrossRef]
- Habib, L.K.; Lee, M.T.C.; Yang, J. Inhibitors of catalase-amyloid interactions protect cells from beta-amyloid-induced oxidative stress and toxicity. J. Biol. Chem. 2010, 285, 38933–38943. [Google Scholar]
- Ye, H.; Robak, L.A.; Yu, M.; Cykowski, M.; Shulman, J.M. Genetics and Pathogenesis of Parkinson’s Syndrome. Annu. Rev. Pathol. Mech. Dis. 2023, 18, 95–121. [Google Scholar] [CrossRef] [PubMed]
- Lotharius, J.; Brundin, P. Pathogenesis of Parkinson’s disease: Dopamine, vesicles and alpha-synuclein. Nat. Rev. Neurosci. 2002, 3, 932–942. [Google Scholar] [CrossRef]
- Graham, D.G. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol. Pharmacol. 1978, 14, 633–643. [Google Scholar] [PubMed]
- Sulzer, D.; Bogulavsky, J.; Larsen, K.E.; Behr, G.; Karatekin, E.; Kleinman, M.H.; Turro, N.; Krantz, D.; Edwards, R.H.; Greene, L.A.; et al. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc. Natl. Acad. Sci. USA 2000, 97, 11869–11874. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Chung, K.K.K. Alpha-synuclein impairs normal dynamics of mitochondria in cell and animal models of Parkinson’s disease. J. Neurochem. 2012, 122, 404–414. [Google Scholar] [CrossRef]
- Yakunin, E.; Kisos, H.; Kulik, W.; Grigoletto, J.; Wanders, R.J.; Sharon, R. The regulation of catalase activity by PPAR γ is affected by α-synuclein. Ann. Clin. Transl. Neurol. 2014, 1, 145–159. [Google Scholar] [CrossRef]
- Wang, Y.; Pan, Y.; Li, H. What is brain health and why is it important? BMJ 2020, 371, m3683. [Google Scholar] [CrossRef]
- Currais, A. Ageing and inflammation—A central role for mitochondria in brain health and disease. Ageing Res. Rev. 2015, 21, 30–42. [Google Scholar] [CrossRef]
- Liu, F.; Lu, J.; Manaenko, A.; Tang, J.; Hu, Q. Mitochondria in Ischemic Stroke: New Insight and Implications. Aging Dis. 2018, 9, 924–937. [Google Scholar] [CrossRef]
- Yan, F.; Tang, H.; Wang, L.; Huang, L.; Zhang, J. Editorial: Mitochondrial Dysfunction in Stroke. Front. Aging Neurosci. 2022, 14, 888952. [Google Scholar] [CrossRef]
- Monsour, M.; Gordon, J.; Lockard, G.; Alayli, A.; Borlongan, C.V. Mitochondria in Cell-Based Therapy for Stroke. Antioxidants 2023, 12, 178. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.O.; Ho, W.S. Mitochondrial Dysfunction, Macrophage, and Microglia in Brain Cancer. Front. Cell Dev. Biol. 2020, 8, 620788. [Google Scholar] [CrossRef] [PubMed]
- Widmann, C.N.; Heneka, M.T. Long-term cerebral consequences of sepsis. Lancet Neurol. 2014, 13, 630–636. [Google Scholar] [CrossRef]
- D’Avila, J.d.C.P.; Santiago, A.P.S.; Amâncio, R.T.; Galina, A.; Oliveira, M.F.; Bozza, F.A. Sepsis induces brain mitochondrial dysfunction. Crit. Care Med. 2008, 36, 1925–1932. [Google Scholar] [CrossRef]
- Joshi, A.U.; Saw, N.L.; Shamloo, M.; Mochly-Rosen, D. Drp1/Fis1 interaction mediates mitochondrial dysfunction, bioenergetic failure and cognitive decline in Alzheimer’s disease. Oncotarget 2018, 9, 6128–6143. [Google Scholar] [CrossRef]
- Haileselassie, B.; Joshi, A.U.; Minhas, P.S.; Mukherjee, R.; Andreasson, K.I.; Mochly-Rosen, D. Mitochondrial dysfunction mediated through dynamin-related protein 1 (Drp1) propagates impairment in blood brain barrier in septic encephalopathy. J. Neuroinflammation 2020, 17, 36. [Google Scholar] [CrossRef]
- Pei, L.; Wallace, D.C. Mitochondrial Etiology of Neuropsychiatric Disorders. Biol. Psychiatry 2018, 83, 722–730. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S. The neurobiology of stress: From serendipity to clinical relevance. Brain Res. 2000, 886, 172–189. [Google Scholar] [CrossRef]
- Morella, I.M.; Brambilla, R.; Morè, L. Emerging roles of brain metabolism in cognitive impairment and neuropsychiatric disorders. Neurosci. Biobehav. Rev. 2022, 142, 104892. [Google Scholar] [CrossRef]
- Van Horssen, J.; van Schaik, P.; Witte, M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci. Lett. 2019, 710, 132931. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; van Horssen, J. Oxidative stress and its impact on neurons and glia in multiple sclerosis lesions. Biochim. Biophys. Acta 2016, 1862, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Brummer, C.; Faerber, S.; Bruss, C.; Blank, C.; Lacroix, R.; Haferkamp, S.; Herr, W.; Kreutz, M.; Renner, K. Metabolic targeting synergizes with MAPK inhibition and delays drug resistance in melanoma. Cancer Lett. 2019, 442, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; van Horssen, J.; Mahad, D. Progressive multiple sclerosis: Pathology and pathogenesis. Nat. Rev. Neurol. 2012, 8, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Wong, S.; Giulivi, C. Evidence of reactive oxygen species-mediated damage to mitochondrial DNA in children with typical autism. Mol. Autism 2013, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Frye, R.E. Evidence linking oxidative stress, mitochondrial dysfunction, and inflammation in the brain of individuals with autism. Front. Physiol. 2014, 5, 150. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, A.; Venkatasubramanian, G.; Berk, M.; Debnath, M. Mitochondrial dysfunction in schizophrenia: Pathways, mechanisms and implications. Neurosci. Biobehav. Rev. 2015, 48, 10–21. [Google Scholar] [CrossRef]
- De Barcelos, I.P.; Troxell, R.M.; Graves, J.S. Mitochondrial Dysfunction and Multiple Sclerosis. Biology 2019, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Prasuhn, J.; Davis, R.L.; Kumar, K.R. Targeting Mitochondrial Impairment in Parkinson’s Disease: Challenges and Opportunities. Front. Cell Dev. Biol. 2020, 8, 615461. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Frye, R.E. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol. Psychiatry 2012, 17, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Bansal, Y.; Kuhad, A. Mitochondrial Dysfunction in Depression. Curr. Neuropharmacol. 2016, 14, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Palomo, A.; Dodd, S.; Anmella, G.; Carvalho, A.F.; Scaini, G.; Quevedo, J.; Pacchiarotti, I.; Vieta, E.; Berk, M. The Role of Mitochondria in Mood Disorders: From Physiology to Pathophysiology and to Treatment. Front. Psychiatry 2021, 12, 546801. [Google Scholar] [CrossRef] [PubMed]
- Myhill, S.; Booth, N.E.; McLaren-Howard, J. Chronic fatigue syndrome and mitochondrial dysfunction. Int. J. Clin. Exp. Med. 2009, 2, 1–16. [Google Scholar] [PubMed]
- Morris, G.; Berk, M. The many roads to mitochondrial dysfunction in neuroimmune and neuropsychiatric disorders. BMC Med. 2015, 13, 68. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Morris, J.K. New Therapeutics to Modulate Mitochondrial Function in Neurodegenerative Disorders. Curr. Pharm. Des. 2017, 23, 731–752. [Google Scholar] [CrossRef]
- Clemente-Suárez, V.J.; Martín-Rodríguez, A.; Yáñez-Sepúlveda, R.; Tornero-Aguilera, J.F. Mitochondrial Transfer as a Novel Therapeutic Approach in Disease Diagnosis and Treatment. Int. J. Mol. Sci. 2023, 24, 8848. [Google Scholar] [CrossRef] [PubMed]
- Harilal, S.; Jose, J.; Parambi, D.G.T.; Kumar, R.; Mathew, G.E.; Uddin, S.; Kim, H.; Mathew, B. Advancements in nanotherapeutics for Alzheimer’s disease: Current perspectives. J. Pharm. Pharmacol. 2019, 71, 1370–1383. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Favia, M.; Spera, I.; Campanella, A.; Lanza, M.; Castegna, A. Metabolic Features of Brain Function with Relevance to Clinical Features of Alzheimer and Parkinson Diseases. Molecules 2022, 27, 951. [Google Scholar] [CrossRef]
- Tanaka, H.; Gourley, D.D.; Dekhtyar, M.; Haley, A.P. Cognition, Brain Structure, and Brain Function in Individuals with Obesity and Related Disorders. Curr. Obes. Rep. 2020, 9, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.A.; Spencer, S.J. Obesity and neuroinflammation: A pathway to cognitive impairment. Brain Behav. Immun. 2014, 42, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Dye, L.; Boyle, N.B.; Champ, C.; Lawton, C. The relationship between obesity and cognitive health and decline. Proc. Nutr. Soc. 2017, 76, 443–454. [Google Scholar] [CrossRef]
- Merlo, S.; Spampinato, S.; Canonico, P.L.; Copani, A.; Sortino, M.A. Alzheimer’s disease: Brain expression of a metabolic disorder? Trends Endocrinol. Metab. 2010, 21, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Hussain, B.; Chang, J. Peripheral inflammation and blood–brain barrier disruption: Effects and mechanisms. CNS Neurosci. Ther. 2021, 27, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired Mitochondrial Activity in the Insulin-Resistant Offspring of Patients with Type 2 Diabetes. N. Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.K.; Hevener, A.L.; Barnard, R.J. Metabolic syndrome and insulin resistance: Underlying causes and modification by exercise training. Compr. Physiol. 2013, 3, 1–58. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E.; Daniele, G.; Galindo, C.; Abdul-Ghani, M.A.; DeFronzo, R.A.; Norton, L. Pioglitazone inhibits mitochondrial pyruvate metabolism and glucose production in hepatocytes. FEBS J. 2017, 284, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Zsurka, G.; Kunz, W.S. Mitochondrial involvement in neurodegenerative diseases. IUBMB Life 2013, 65, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.T.; McCarthy, M.J.; Vawter, M.P. Psychiatric drugs impact mitochondrial function in brain and other tissues. Schizophr. Res. 2020, 217, 136–147. [Google Scholar] [CrossRef]
- Spiers, J.G.; Chen, H.-J.C.; Bourgognon, J.-M.; Steinert, J.R. Dysregulation of stress systems and nitric oxide signaling underlies neuronal dysfunction in Alzheimer’s disease. Free. Radic. Biol. Med. 2019, 134, 468–483. [Google Scholar] [CrossRef] [PubMed]
- Foyet, S.H.; Balmus, I.-M.; Hervé, N.A.H.; Emmanuel, A.A.; Guenne, S.; Kiendrebéogo, M.; Ciobica, A. Ethnopharmacological approaches in mood and anxiety disorders. The relevance of the oxidative stress status. J. Complement. Integr. Med. 2017, 14. [Google Scholar] [CrossRef]
- Vaváková, M.; Ďuračková, Z.; Trebatická, J. Markers of Oxidative Stress and Neuroprogression in Depression Disorder. Oxid. Med. Cell. Longev. 2015, 2015, 898393. [Google Scholar] [CrossRef]
- Madireddy, S.; Madireddy, S. Therapeutic Interventions to Mitigate Mitochondrial Dysfunction and Oxidative Stress-Induced Damage in Patients with Bipolar Disorder. Int. J. Mol. Sci. 2022, 23, 1844. [Google Scholar] [CrossRef]
- Ermakov, E.A.; Dmitrieva, E.M.; Parshukova, D.A.; Kazantseva, D.V.; Vasilieva, A.R.; Smirnova, L.P. Oxidative Stress-Related Mechanisms in Schizophrenia Pathogenesis and New Treatment Perspectives. Oxidative Med. Cell. Longev. 2021, 2021, 8881770. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.A.; Yamauchi, Y.; Horikawa, H.; Monji, A.; Mizoguchi, Y.; Seki, Y.; Hayakawa, K.; Utsumi, H.; Kanba, K. Neurotransmitters, psychotropic drugs and microglia: Clinical implications for psychiatry. Curr. Med. Chem. 2013, 20, 331–344. [Google Scholar] [PubMed]
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in neuroplasticity and neurological disorders. Neuron 2008, 60, 748–766. [Google Scholar] [CrossRef]
- Ye, N.; Song, Z.; Zhang, A. Dual ligands targeting dopamine D2 and serotonin 5-HT1A receptors as new antipsychotical or anti-Parkinsonian agents. Curr. Med. Chem. 2013, 21, 437–457. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Suárez, V.J. Multidisciplinary intervention in the treatment of mixed anxiety and depression disorder. Physiol. Behav. 2020, 219, 112858. [Google Scholar] [CrossRef]
- Lopresti, A.L.; Hood, S.D.; Drummond, P.D. A review of lifestyle factors that contribute to important pathways associated with major depression: Diet, sleep and exercise. J. Affect. Disord. 2013, 148, 12–27. [Google Scholar] [CrossRef]
- Sacher, J.; Rekkas, P.V.; Wilson, A.A.; Houle, S.; Romano, L.; Hamidi, J.; Rusjan, P.; Fan, I.; Stewart, D.E.; Meyer, J.H. Relationship of monoamine oxidase—A distribution volume to postpartum depression and postpartum crying. Neuropsychopharmacology 2015, 40, 429–435. [Google Scholar] [CrossRef]
- Maes, M.; Galecki, P.; Chang, Y.S.; Berk, M. A review on the oxidative and nitrosative stress (O&NS) pathways in major depression and their possible contribution to the (neuro)degenerative processes in that illness. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 676–692. [Google Scholar] [CrossRef]
- Wichers, M.C.; Maes, M. The role of indoleamine 2,3-dioxygenase (IDO) in the pathophysiology of interferon-alpha-induced depression. J. Psychiatry Neurosci. 2004, 29, 11–17. [Google Scholar] [PubMed]
- Clemente-Suárez, V.J.; Bustamante-Sanchez, Á.; Mielgo-Ayuso, J.; Martínez-Guardado, I.; Martín-Rodríguez, A.; Tornero-Aguilera, J.F. Antioxidants and Sports Performance. Nutrients 2023, 15, 2371. [Google Scholar] [CrossRef] [PubMed]
- Spanos, M.; Gras-Najjar, J.; Letchworth, J.M.; Sanford, A.L.; Toups, J.V.; Sombers, L.A. Quantitation of hydrogen peroxide fluctuations and their modulation of dopamine dynamics in the rat dorsal striatum using fast-scan cyclic voltammetry. ACS Chem. Neurosci. 2013, 4, 782–789. [Google Scholar] [CrossRef]
- Kumar, H.; Lim, H.-W.; More, S.V.; Kim, B.-W.; Koppula, S.; Kim, I.S.; Choi, D.-K. The role of free radicals in the aging brain and Parkinson’s Disease: Convergence and parallelism. Int. J. Mol. Sci. 2012, 13, 10478–10504. [Google Scholar] [CrossRef] [PubMed]
- Marino, B.L.; de Souza, L.R.; Sousa, K.P.; Ferreira, J.V.; Padilha, E.C.; da Silva, C.H.; Taft, C.A.; Hage-Melim, L.I. Parkinson’s Disease: A Review from Pathophysiology to Treatment. Mini Rev. Med. Chem. 2020, 20, 754–767. [Google Scholar] [CrossRef]
- Zhang, Y.-F.; Cragg, S.J. Revisiting dopamine-acetylcholine imbalance in Parkinson’s disease: Glutamate co-transmission as an exciting partner in crime. Neuron 2021, 109, 1070–1071. [Google Scholar] [CrossRef]
- Kandimalla, R.; Reddy, P.H. Therapeutics of Neurotransmitters in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1049–1069. [Google Scholar] [CrossRef]
- Francis, P.T. Glutamatergic systems in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2003, 18, S15–S21. [Google Scholar] [CrossRef]
- Lauderback, C.M.; Hackett, J.M.; Huang, F.F.; Keller, J.N.; Szweda, L.I.; Markesbery, W.R.; Butterfield, D.A. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer’s disease brain: The role of Abeta1–42. J. Neurochem. 2001, 78, 413–416. [Google Scholar] [CrossRef]
- Soares, M.B.; Ramalho, J.B.; Izaguirry, A.P.; Pavin, N.F.; Spiazzi, C.C.; Schimidt, H.L.; Mello-Carpes, P.B.; Santos, F.W. Comparative effect of Camellia sinensis teas on object recognition test deficit and metabolic changes induced by cafeteria diet. Nutr. Neurosci. 2019, 22, 531–540. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Bemeur, C. Oxidative Stress in the Central Nervous System Complications of Chronic Liver Failure. In Studies on Hepatic Disorders; Springer: Berlin/Heidelberg, Germany, 2015; pp. 357–370. [Google Scholar] [CrossRef]
- Bota, D.A.; Davies, K.J.A. Mitochondrial Lon protease in human disease and aging: Including an etiologic classification of Lon-related diseases and disorders. Free Radic. Biol. Med. 2016, 100, 188–198. [Google Scholar] [CrossRef]
- Pomatto, L.C.D.; Carney, C.; Shen, B.; Wong, S.; Halaszynski, K.; Salomon, M.P.; Davies, K.J.; Tower, J. The Mitochondrial Lon Protease Is Required for Age-Specific and Sex-Specific Adaptation to Oxidative Stress. Curr. Biol. 2017, 27, 1–15. [Google Scholar] [CrossRef]
- Gragnoli, C. Proteasome modulator 9 gene SNPs, responsible for anti-depressant response, are in linkage with generalized anxiety disorder. J. Cell Physiol. 2014, 229, 1157–1159. [Google Scholar] [CrossRef] [PubMed]
- Gong, B.; Radulovic, M.; Figueiredo-Pereira, M.E.; Cardozo, C. The Ubiquitin-Proteasome System: Potential Therapeutic Targets for Alzheimer’s Disease and Spinal Cord Injury. Front. Mol. Neurosci. 2016, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 2005, 280, 17294–17300. [Google Scholar] [CrossRef]
- Kravtsova-Ivantsiv, Y.; Cohen, S.; Ciechanover, A. Modification by single ubiquitin moieties rather than polyubiquitination is sufficient for proteasomal processing of the p105 NF-kappaB precursor. Mol. Cell 2009, 33, 496–504. [Google Scholar] [CrossRef] [PubMed]
- González, A.N.B.; Pazos, M.I.L.; Calvo, D.J. Reactive Oxygen Species in the Regulation of the GABA Mediated Inhibitory Neurotransmission. Neuroscience 2020, 439, 137–145. [Google Scholar] [CrossRef]
- Obata, K. Synaptic inhibition and γ-aminobutyric acid in the mammalian central nervous system. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2013, 89, 139–156. [Google Scholar] [CrossRef]
- Calvo, D.J.; González, A.N.B. Dynamic Regulation of the GABAA Receptor Function by Redox Mechanisms. Mol. Pharmacol. 2016, 90, 326–333. [Google Scholar] [CrossRef]
- Tassone, A.; Meringolo, M.; Ponterio, G.; Bonsi, P.; Schirinzi, T.; Martella, G. Mitochondrial Bioenergy in Neurodegenerative Disease: Huntington and Parkinson. Int. J. Mol. Sci. 2023, 24, 7221. [Google Scholar] [CrossRef] [PubMed]
- Wallin, A.K.; Blennow, K.; Andreasen, N.; Minthon, L. CSF biomarkers for Alzheimer’s Disease: Levels of beta-amyloid, tau, phosphorylated tau relate to clinical symptoms and survival. Dement. Geriatr. Cogn. Disord 2006, 21, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Ikawa, M.; Okazawa, H.; Nakamoto, Y.; Yoneda, M. PET Imaging for Oxidative Stress in Neurodegenerative Disorders Associated with Mitochondrial Dysfunction. Antioxidants 2020, 9, 861. [Google Scholar] [CrossRef] [PubMed]
- Buneeva, O.; Fedchenko, V.; Kopylov, A.; Medvedev, A. Mitochondrial Dysfunction in Parkinson’s Disease: Focus on Mitochondrial DNA. Biomedicines 2020, 8, 591. [Google Scholar] [CrossRef] [PubMed]
- Belloli, S.; Morari, M.; Murtaj, V.; Valtorta, S.; Moresco, R.M.; Gilardi, M.C. Translation Imaging in Parkinson’s Disease: Focus on Neuroinflammation. Front. Aging Neurosci. 2020, 12, 152. [Google Scholar] [CrossRef]
- Siddiqui, A.; Rivera-Sánchez, S.; Castro, M.d.R.; Acevedo-Torres, K.; Rane, A.; Torres-Ramos, C.A.; Nicholls, D.G.; Andersen, J.K.; Ayala-Torres, S. Mitochondrial DNA damage Is associated with reduced mitochondrial bioenergetics in Huntington’s disease. Free Radic. Biol. Med. 2012, 53, 1478–1488. [Google Scholar] [CrossRef]
- Reddy, P.H. Role of mitochondria in neurodegenerative diseases: Mitochondria as a therapeutic target in Alzheimer’s disease. CNS Spectr. 2009, 14, 8–13. [Google Scholar] [CrossRef]
- Gaitsch, H.; Franklin, R.J.M.; Reich, D.S. Cell-free DNA-based liquid biopsies in neurology. Brain 2023, 146, 1758–1774. [Google Scholar] [CrossRef]
- Atlante, A.; Amadoro, G.; Latina, V.; Valenti, D. Therapeutic Potential of Targeting Mitochondria for Alzheimer’s Disease Treatment. J. Clin. Med. 2022, 11, 6742. [Google Scholar] [CrossRef]
- Blair, I.A.; Farmer, J.; Hersch, S.; Larkindale, J.; Lynch, D.R.; Napierala, J.; Napierala, M.; Payne, R.M.; Subramony, S.H. The current state of biomarker research for Friedreich’s ataxia: A report from the 2018 FARA biomarker meeting. Future Sci. OA 2019, 5, FSO398. [Google Scholar] [CrossRef]
- Mandal, P.K.; Gaur, S.; Roy, R.G.; Samkaria, A.; Ingole, R.; Goel, A. Schizophrenia, Bipolar and Major Depressive Disorders: Overview of Clinical Features, Neurotransmitter Alterations, Pharmacological Interventions, and Impact of Oxidative Stress in the Disease Process. ACS Chem. Neurosci. 2022, 13, 2784–2802. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.; Johansson, A.; Wibom, R.; Nennesmo, I.; von Döbeln, U.; Hagenfeldt, L.; Hällström, T. Alterations of mitochondrial function and correlations with personality traits in selected major depressive disorder patients. J. Affect. Disord. 2003, 76, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E.; Vassall, S.; Kaur, G.; Lewis, C.; Karim, M.; Rossignol, D. Emerging biomarkers in autism spectrum disorder: A systematic review. Ann. Transl. Med. 2019, 7, 792. [Google Scholar] [CrossRef] [PubMed]
- Prasuhn, J.; Kunert, L.; Brüggemann, N. Neuroimaging Methods to Map In Vivo Changes of OXPHOS and Oxidative Stress in Neurodegenerative Disorders. Int. J. Mol. Sci. 2022, 23, 7263. [Google Scholar] [CrossRef] [PubMed]
- Beversdorf, D.Q. Phenotyping, Etiological Factors, and Biomarkers: Toward Precision Medicine in Autism Spectrum Disorders. J. Dev. Behav. Pediatr. 2016, 37, 659–673. [Google Scholar] [CrossRef]
- Scaglia, F. The role of mitochondrial dysfunction in psychiatric disease. Dev. Disabil. Res. Rev. 2010, 16, 136–143. [Google Scholar] [CrossRef]
- Khadimallah, I.; Jenni, R.; Cabungcal, J.-H.; Cleusix, M.; Fournier, M.; Beard, E.; Klauser, P.; Knebel, J.-F.; Murray, M.M.; Retsa, C.; et al. Mitochondrial, exosomal miR137-COX6A2 and gamma synchrony as biomarkers of parvalbumin interneurons, psychopathology, and neurocognition in schizophrenia. Mol. Psychiatry 2022, 27, 1192–1204. [Google Scholar] [CrossRef]
- Flippo, K.H.; Strack, S. An emerging role for mitochondrial dynamics in schizophrenia. Schizophr. Res. 2017, 187, 26–32. [Google Scholar] [CrossRef]
- Kim, Y.; Vadodaria, K.C.; Lenkei, Z.; Kato, T.; Gage, F.H.; Marchetto, M.C.; Santos, R. Mitochondria, Metabolism, and Redox Mechanisms in Psychiatric Disorders. Antioxid. Redox Signal. 2019, 31, 275–317. [Google Scholar] [CrossRef] [PubMed]
- Emwas, A.-H.; Roy, R.; McKay, R.T.; Ryan, D.; Brennan, L.; Tenori, L.; Luchinat, C.; Gao, X.; Zeri, A.C.; Gowda, G.A.N.; et al. Recommendations and Standardization of Biomarker Quantification Using NMR-Based Metabolomics with Particular Focus on Urinary Analysis. J. Proteome Res. 2016, 15, 360–373. [Google Scholar] [CrossRef]
- Montagnana, M.; Lippi, G.; Salvagno, G.L.; Guidi, G.C. Reference ranges and diagnostic thresholds of laboratory markers of cardiac damage and dysfunction in a population of apparently healthy black Africans. Clin. Chem. Lab. Med. 2008, 46, 714–716. [Google Scholar] [CrossRef]
- Feczko, E.; Miranda-Dominguez, O.; Marr, M.; Graham, A.M.; Nigg, J.T.; Fair, D.A. The Heterogeneity Problem: Approaches to Identify Psychiatric Subtypes. Trends Cogn. Sci. 2019, 23, 584–601. [Google Scholar] [CrossRef]
- Wörheide, M.A.; Krumsiek, J.; Kastenmüller, G.; Arnold, M. Multi-omics integration in biomedical research—A metabolomics-centric review. Anal. Chim. Acta 2021, 1141, 144–162. [Google Scholar] [CrossRef]
- Češková, E.; Šilhán, P. From Personalized Medicine to Precision Psychiatry? Neuropsychiatr. Dis. Treat. 2021, 17, 3663–3668. [Google Scholar] [CrossRef] [PubMed]
- Trigo, D.; Vitória, J.; da Cruz E Silva, O.A.B. Novel therapeutic strategies targeting mitochondria as a gateway in neurodegeneration. Neural Regen. Res. 2023, 18, 991–995. [Google Scholar] [CrossRef]
- Yen, C.; Lin, C.-L.; Chiang, M.-C. Exploring the Frontiers of Neuroimaging: A Review of Recent Advances in Understanding Brain Functioning and Disorders. Life 2023, 13, 1472. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhanunni, A.; Pretorius, T.B. Teacher Burnout in the Time of COVID-19: Antecedents and Psychological Consequences. Int. J. Environ. Res. Public. Health 2023, 20, 4204. [Google Scholar] [CrossRef]
- Carreira Míguez, M.; Clemente Suárez, V.J. Physical activity levels affect mental health and behavior in men. J. Men’s Health 2023, 1, 12. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Rolo, A.P.; Palmeira, C.M. Diabetes and mitochondrial function: Role of hyperglycemia and oxidative stress. Toxicol. Appl. Pharmacol. 2006, 212, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-Y.; Yang, Y.; Ming, M.; Liu, B. Mitochondrial ROS generation for regulation of autophagic pathways in cancer. Biochem. Biophys. Res. Commun. 2011, 414, 5–8. [Google Scholar] [CrossRef]
- Morán, M.; Moreno-Lastres, D.; Marín-Buera, L.; Arenas, J.; Martín, M.A.; Ugalde, C. Mitochondrial respiratory chain dysfunction: Implications in neurodegeneration. Free Radic. Biol. Med. 2012, 53, 595–609. [Google Scholar] [CrossRef]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef]
- Eckert, A.; Keil, U.; Marques, C.A.; Bonert, A.; Frey, C.; Schüssel, K.; Müller, W.E. Mitochondrial dysfunction, apoptotic cell death, and Alzheimer’s disease. Biochem. Pharmacol. 2003, 66, 1627–1634. [Google Scholar] [CrossRef] [PubMed]
- Carmo, C.; Naia, L.; Lopes, C.; Rego, A.C. Mitochondrial Dysfunction in Huntington’s Disease. Adv. Exp. Med. Biol. 2018, 1049, 59–83. [Google Scholar] [PubMed]
- Shi, P.; Gal, J.; Kwinter, D.M.; Liu, X.; Zhu, H. Mitochondrial dysfunction in amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2010, 1802, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Orsucci, D.; Mancuso, M.; Ienco, E.C.; LoGerfo, A.; Siciliano, G. Targeting mitochondrial dysfunction and neurodegeneration by means of coenzyme Q10 and its analogues. Curr. Med. Chem. 2011, 18, 4053–4064. [Google Scholar] [CrossRef]
- Melli, G.; Taiana, M.; Camozzi, F.; Triolo, D.; Podini, P.; Quattrini, A.; Taroni, F.; Lauria, G. Alpha-lipoic acid prevents mitochondrial damage and neurotoxicity in experimental chemotherapy neuropathy. Exp. Neurol. 2008, 214, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Acuna-Castroviejo, D.; Escames, G.; Rodriguez, M.I.; Lopez, L.C. Melatonin role in the mitochondrial function. Front. Biosci. 2007, 12, 947–963. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Liu, Y.-Y.; Liu, X.-Y.; Jia, S.-W.; Zhao, J.; Cui, D.; Wang, L. Resveratrol protects neurons and the myocardium by reducing oxidative stress and ameliorating mitochondria damage in a cerebral ischemia rat model. Cell. Physiol. Biochem. 2014, 34, 854–864. [Google Scholar] [CrossRef]
- Ferrari, R.; Ciampalini, G.; Agnoletti, G.; Cargnoni, A.; Ceconi, C.; Visioli, O. Effect of L-carnitine derivatives on heart mitochondrial damage induced by lipid peroxidation. Pharmacol. Res. Commun. 1988, 20, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, H.; Ghasemi, F.; Barreto, G.E.; Rafiee, R.; Sathyapalan, T.; Sahebkar, A. Effects of curcumin on mitochondria in neurodegenerative diseases. Biofactors 2020, 46, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Katila, N.; Bhurtel, S.; Park, P.-H.; Choi, D.-Y. Metformin attenuates rotenone-induced oxidative stress and mitochondrial damage via the AKT/Nrf2 pathway. Neurochem. Int. 2021, 148, 105120. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Dai, G.; Li, G.; Yang, E.S. Coenzyme Q10 reduces β-amyloid plaque in an APP/PS1 transgenic mouse model of Alzheimer’s disease. J. Mol. Neurosci. 2010, 41, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Fouad, G.I. Combination of Omega 3 and Coenzyme Q10 Exerts Neuroprotective Potential Against Hypercholesterolemia-Induced Alzheimer’s-Like Disease in Rats. Neurochem. Res. 2020, 45, 1142–1155. [Google Scholar] [CrossRef]
- Komaki, H.; Faraji, N.; Komaki, A.; Shahidi, S.; Etaee, F.; Raoufi, S.; Mirzaei, F. Investigation of protective effects of coenzyme Q10 on impaired synaptic plasticity in a male rat model of Alzheimer’s disease. Brain Res. Bull. 2019, 147, 14–21. [Google Scholar] [CrossRef]
- Bagheri, S.; Haddadi, R.; Saki, S.; Kourosh-Arami, M.; Rashno, M.; Mojaver, A.; Komaki, A. Neuroprotective effects of coenzyme Q10 on neurological diseases: A review article. Front. Neurosci. 2023, 17, 1188839. [Google Scholar] [CrossRef] [PubMed]
- Sanoobar, M.; Eghtesadi, S.; Azimi, A.; Khalili, M.; Jazayeri, S.; Gohari, M.R. Coenzyme Q10 supplementation reduces oxidative stress and increases antioxidant enzyme activity in patients with relapsing–remitting multiple sclerosis. Int. J. Neurosci. 2013, 123, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Weyer, G.; Babej-Dölle, R.M.; Hadler, D.; Hofmann, S.; Herrmann, W.M. A controlled study of 2 doses of idebenone in the treatment of Alzheimer’s disease. Neuropsychobiology 1997, 36, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Gutzmann, H.; Hadler, D. Sustained efficacy and safety of idebenone in the treatment of Alzheimer’s disease: Update on a 2-year double-blind multicentre study. J. Neural Transm. Suppl. 1998, 54, 301–310. [Google Scholar] [CrossRef]
- Storch, A. [Coenzyme Q10 in Parkinson’s disease. Symptomatic or neuroprotective effects?]. Nervenarzt 2007, 78, 1378–1382. [Google Scholar] [CrossRef]
- Thal, L.J.; Grundman, M.; Berg, J.; Ernstrom, K.; Margolin, R.; Pfeiffer, E.; Weiner, M.F.; Zamrini, E.; Thomas, R.G. Idebenone treatment fails to slow cognitive decline in Alzheimer’s disease. Neurology 2003, 61, 1498–1502. [Google Scholar] [CrossRef]
- De Araújo, D.P.; Lobato, R.D.F.G.; Cavalcanti, J.R.L.D.P.; Sampaio, L.R.L.; Araújo, P.V.P.; Silva, M.C.C.; Neves, K.R.T.; Fonteles, M.M.D.F.; De Sousa, F.C.F.; Vasconcelos, S.M.M. The contributions of antioxidant activity of lipoic acid in reducing neurogenerative progression of Parkinson’s disease: A review. Int. J. Neurosci. 2010, 121, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Maczurek, A.; Hager, K.; Kenklies, M.; Sharman, M.; Martins, R.; Engel, J.; Carlson, D.A.; Münch, G. Lipoic acid as an anti-inflammatory and neuroprotective treatment for Alzheimer’s disease. Adv. Drug Deliv. Rev. 2008, 60, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Jardim, F.R.; De Rossi, F.T.; Nascimento, M.X.; Barros, R.G.d.S.; Borges, P.A.; Prescilio, I.C.; De Oliveira, M.R. Resveratrol and Brain Mitochondria: A Review. Mol. Neurobiol. 2018, 55, 2085–2101. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Rosales-Corral, S.; Galano, A.; Zhou, X.J.; Xu, B. Mitochondria: Central Organelles for Melatonin’s Antioxidant and Anti-Aging Actions. Molecules 2018, 23, 509. [Google Scholar] [CrossRef]
- Gunata, M.; Parlakpinar, H.; Acet, H.A. Melatonin: A review of its potential functions and effects on neurological diseases. Rev. Neurol. 2019, 176, 148–165. [Google Scholar] [CrossRef] [PubMed]
- Alikiaii, B.; Bagherniya, M.; Askari, G.; Sathyapalan, T.; Sahebkar, A. Evaluation of the effect of curcumin on pneumonia: A systematic review of preclinical studies. Phytotherapy Res. 2021, 35, 1939–1952. [Google Scholar] [CrossRef]
- Bagherniya, M.; Nobili, V.; Blesso, C.N.; Sahebkar, A. Medicinal plants and bioactive natural compounds in the treatment of non-alcoholic fatty liver disease: A clinical review. Pharmacol. Res. 2018, 130, 213–240. [Google Scholar] [CrossRef]
- Ghandadi, M.; Sahebkar, A. Curcumin: An Effective Inhibitor of Interleukin-6. Curr. Pharm. Des. 2017, 23, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Mollazadeh, H.; Cicero, A.F.G.; Blesso, C.N.; Pirro, M.; Majeed, M.; Sahebkar, A. Immune modulation by curcumin: The role of interleukin-10. Crit. Rev. Food Sci. Nutr. 2019, 59, 89–101. [Google Scholar] [CrossRef]
- Panahi, Y.; Ahmadi, Y.; Teymouri, M.; Johnston, T.P.; Sahebkar, A. Curcumin as a potential candidate for treating hyperlipidemia: A review of cellular and metabolic mechanisms. J. Cell. Physiol. 2018, 233, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Teymouri, M.; Pirro, M.; Johnston, T.P.; Sahebkar, A. Curcumin as a multifaceted compound against human papilloma virus infection and cervical cancers: A review of chemistry, cellular, molecular, and preclinical features. Biofactors 2017, 43, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Mohseni, M.; Sahebkar, A.; Askari, G.; Johnston, T.P.; Alikiaii, B.; Bagherniya, M. The clinical use of curcumin on neurological disorders: An updated systematic review of clinical trials. Phytother. Res. 2021, 35, 6862–6882. [Google Scholar] [CrossRef]
- Cole, G.M.; Yang, F.; Lim, G.P.; Cummings, J.L.; Masterman, D.L.; Frautschy, S.A. A rationale for curcuminoids for the prevention or treatment of Alzheimer’s disease. Curr. Med. Chem. Immunol. Endocr. Metab. Agents 2003, 3, 15–25. [Google Scholar] [CrossRef]
- Virmani, M.A.; Cirulli, M. The Role of l-Carnitine in Mitochondria, Prevention of Metabolic Inflexibility and Disease Initiation. Int. J. Mol. Sci. 2022, 23, 2717. [Google Scholar] [CrossRef]
- Plioplys, A.V.; Plioplys, S. Serum levels of carnitine in chronic fatigue syndrome: Clinical correlates. Neuropsychobiology 1995, 32, 132–138. [Google Scholar] [CrossRef]
- Latham, L.E.; Wang, C.; Patterson, T.A.; Slikker, W.; Liu, F. Neuroprotective Effects of Carnitine and Its Potential Application to Ameliorate Neurotoxicity. Chem. Res. Toxicol. 2021, 34, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Casariego, A.; Burgos-Peláez, R.; Martínez-Faedo, C.; Calvo-Gracia, F.; Valero-Zanuy, M.; Luengo-Pérez, L.M.; Cuerda-Compés, C. Metabolic Effects of L-carnitine on Type 2 Diabetes Mellitus: Systematic Review and Meta-analysis. Exp. Clin. Endocrinol. Diabetes 2013, 121, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Förster, L.; Indra, D.; Rosenberger, K.; Zver, L.; Hofbauer, R. L-carnitine exerts a nutrigenomic effect via direct modulation of nuclear receptor signaling in adipocytes, hepatocytes and SKMC, demonstrating its nutritional impact. Nutr. Res. 2021, 85, 84–98. [Google Scholar] [CrossRef]
- Virmani, M.; Caso, V.; Spadoni, A.; Rossi, S.; Russo, F.; Gaetani, F. The action of acetyl-L-carnitine on the neurotoxicity evoked by amyloid fragments and peroxide on primary rat cortical neurones. Ann. N. Y. Acad. Sci. 2001, 939, 162–178. [Google Scholar] [CrossRef] [PubMed]
- Abdul, H.M.; Calabrese, V.; Calvani, M.; Butterfield, D.A. Acetyl-L-carnitine-induced up-regulation of heat shock proteins protects cortical neurons against amyloid-beta peptide 1–42-mediated oxidative stress and neurotoxicity: Implications for Alzheimer’s disease. J. Neurosci. Res. 2006, 84, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Dhitavat, S.; Ortiz, D.; Shea, T.B.; Rivera, E.R. Acetyl-L-carnitine protects against amyloid-beta neurotoxicity: Roles of oxidative buffering and ATP levels. Neurochem. Res. 2002, 27, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Rotermund, C.; Machetanz, G.; Fitzgerald, J.C. The Therapeutic Potential of Metformin in Neurodegenerative Diseases. Front. Endocrinol. 2018, 9, 400. [Google Scholar] [CrossRef]
- Kaufman, B.A.; Li, C.; Soleimanpour, S.A. Mitochondrial regulation of β-cell function: Maintaining the momentum for insulin release. Mol. Asp. Med. 2015, 42, 91–104. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009, 47, 333–343. [Google Scholar] [CrossRef]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef]
- Ihara, Y.; Nukina, N.; Miura, R.; Ogawara, M. Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer’s disease. J. Biochem. 1986, 99, 1807–1810. [Google Scholar] [CrossRef]
- Quntanilla, R.A.; Tapia-Monsalves, C. The Role of Mitochondrial Impairment in Alzheimer’s Disease Neurodegeneration: The Tau Connection. Curr. Neuropharmacol. 2020, 18, 1076–1091. [Google Scholar] [CrossRef]
- Meier-Ruge, W.; Bertoni-Freddari, C.; Iwangoff, P. Changes in brain glucose metabolism as a key to the pathogenesis of Alzheimer’s disease. Gerontology 1994, 40, 246–252. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef]
- Moon, H.E.; Paek, S.H. Mitochondrial Dysfunction in Parkinson’s Disease. Exp. Neurobiol. 2015, 24, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Tagle, D.; Valdes, J.; Elmer, L.; Allard, M.; Castilla, L.; Swaroop, M.; Blanchard, K.; Collins, F.S.; Snell, R.; Holloway, T.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar]
- Damiano, M.; Galvan, L.; Déglon, N.; Brouillet, E. Mitochondria in Huntington’s disease. Biochim. Biophys. Acta 2010, 1802, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Arduino, D.M.; Esteves, A.R.; Oliveira, C.R.; Cardoso, S.M. Mitochondrial metabolism modulation: A new therapeutic approach for Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2010, 9, 105–119. [Google Scholar] [CrossRef] [PubMed]
- de Sousa, C.N.S.; da Silva Leite, C.M.G.; da Silva Medeiros, I.; Vasconcelos, L.C.; Cabral, L.M.; Patrocínio, C.F.V.; Patrocínio, M.L.V.; Mouaffak, F.; Kebir, O.; Macedo, D.; et al. Alpha-lipoic acid in the treatment of psychiatric and neurological disorders: A systematic review. Metab. Brain Dis. 2019, 34, 39–52. [Google Scholar] [CrossRef]
- Spindler, M.; Beal, M.F.; Henchcliffe, C. Coenzyme Q10 effects in neurodegenerative disease. Neuropsychiatr. Dis. Treat. 2009, 5, 597–610. [Google Scholar] [CrossRef]
- Hikosaka, K.; Yaku, K.; Okabe, K.; Nakagawa, T. Implications of NAD metabolism in pathophysiology and therapeutics for neurodegenerative diseases. Nutr. Neurosci. 2021, 24, 371–383. [Google Scholar] [CrossRef]
- Ormazabal, A.; Casado, M.; Molero-Luis, M.; Montoya, J.; Rahman, S.; Aylett, S.-B.; Hargreaves, I.; Heales, S.; Artuch, R. Can folic acid have a role in mitochondrial disorders? Drug Discov. Today 2015, 20, 1349–1354. [Google Scholar] [CrossRef]
- Adhihetty, P.J.; Beal, M.F. Creatine and its potential therapeutic value for targeting cellular energy impairment in neurodegenerative diseases. NeuroMolecular Med. 2008, 10, 275–290. [Google Scholar] [CrossRef]
- Sun, A.Y.; Wang, Q.; Simonyi, A.; Sun, G.Y. Resveratrol as a therapeutic agent for neurodegenerative diseases. Mol. Neurobiol. 2010, 41, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Hagen, T.M.; Ingersoll, R.T.; Lykkesfeldt, J.; Liu, J.; Wehr, C.M.; Vinarsky, V.; Bartholomew, J.C.; Ames, B.N. (R)-alpha-lipoic acid-supplemented old rats have improved mitochondrial function, decreased oxidative damage, and increased metabolic rate. FASEB J. 1999, 13, 411–418. [Google Scholar] [CrossRef]
- Tibullo, D.; Li Volti, G.; Giallongo, C.; Grasso, S.; Tomassoni, D.; Anfuso, C.D.; Lupo, G.; Amenta, F.; Avola, R.; Bramanti, V. Biochemical and clinical relevance of alpha lipoic acid: Antioxidant and anti-inflammatory activity, molecular pathways and therapeutic potential. Inflamm. Res. 2017, 66, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Tai, S.; Zheng, Q.; Zhai, S.; Cai, T.; Xu, L.; Yang, L.; Jiao, L.; Zhang, C. Alpha-lipoic acid mediates clearance of iron accumulation by regulating iron metabolism in a Parkinson’s disease model induced by 6-OHDA. Front. Neurosci. 2020, 14, 612. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yang, S.; Wang, H. α-Lipoic acid alleviates ferroptosis in the MPP(+)-induced PC12 cells via activating the PI3K/Akt/Nrf2 pathway. Cell Biol. Int. 2021, 45, 422–431. [Google Scholar] [CrossRef]
- Shinto, L.; Quinn, J.; Montine, T.; Dodge, H.H.; Woodward, W.; Baldauf-Wagner, S.; Waichunas, D.; Bumgarner, L.; Bourdette, D.; Silbert, L.; et al. A randomized placebo-controlled pilot trial of omega-3 fatty acids and alpha lipoic acid in Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 38, 111–120. [Google Scholar] [CrossRef]
- Hager, K.; Kenklies, M.; McAfoose, J.; Engel, J.; Münch, G. Alpha-lipoic acid as a new treatment option for Alzheimer’s disease—A 48 months follow-up analysis. J. Neural Transm. Suppl. 2007, 72, 189–193. [Google Scholar] [CrossRef]
- Fava, A.; Pirritano, D.; Plastino, M.; Cristiano, D.; Puccio, G.; Colica, C.; Ermio, C.; De Bartolo, M.; Mauro, G.; Bosco, D. The effect of lipoic acid therapy on cognitive functioning in patients with Alzheimer’s disease. J. Neurodegener. Dis. 2013, 2013, 1–7. [Google Scholar] [CrossRef]
- Horvath, T.L.; Diano, S.; Leranth, C.; Garcia-Segura, L.M.; Cowley, M.A.; Shanabrough, M.; Elsworth, J.D.; Sotonyi, P.; Roth, R.H.; Dietrich, E.H.; et al. Coenzyme Q induces nigral mitochondrial uncoupling and prevents dopamine cell loss in a primate model of Parkinson’s disease. Endocrinology 2003, 144, 2757–2760. [Google Scholar] [CrossRef]
- Yoritaka, A.; Kawajiri, S.; Yamamoto, Y.; Nakahara, T.; Ando, M.; Hashimoto, K.; Nagase, M.; Saito, Y.; Hattori, N. Randomized, double-blind, placebo-controlled pilot trial of reduced coenzyme Q10 for Parkinson’s disease. Park. Relat. Disord. 2015, 21, 911–916. [Google Scholar] [CrossRef]
- Zhu, Z.-G.; Sun, M.-X.; Zhang, W.-L.; Wang, W.-W.; Jin, Y.-M.; Xie, C.-L. The efficacy and safety of coenzyme Q10 in Parkinson’s disease: A meta-analysis of randomized controlled trials. Neurol. Sci. 2017, 38, 215–224. [Google Scholar] [CrossRef]
- Gong, B.; Pan, Y.; Vempati, P.; Zhao, W.; Knable, L.; Ho, L.; Wang, J.; Sastre, M.; Ono, K.; Sauve, A.A.; et al. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-γ coactivator 1α regulated β-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol. Aging 2013, 34, 1581–1588. [Google Scholar] [CrossRef]
- Hou, Y.; Lautrup, S.; Cordonnier, S.; Wang, Y.; Croteau, D.L.; Zavala, E.; Zhang, Y.; Moritoh, K.; O’connell, J.F.; Baptiste, B.A.; et al. NAD(+) supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, E1876–E1885. [Google Scholar] [CrossRef]
- Helman, T.; Braidy, N. Importance of NAD+ Anabolism in Metabolic, Cardiovascular and Neurodegenerative Disorders. Drugs Aging 2023, 40, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Roschel, H.; Gualano, B.; Ostojic, S.M.; Rawson, E.S. Creatine supplementation and brain health. Nutrients 2021, 13, 586. [Google Scholar] [CrossRef]
- Harmon, K.K.; Stout, J.R.; Fukuda, D.H.; Pabian, P.S.; Rawson, E.S.; Stock, M.S. The Application of Creatine Supplementation in Medical Rehabilitation. Nutrients 2021, 13, 1825. [Google Scholar] [CrossRef]
- Li, Z.; Wang, P.; Yu, Z.; Cong, Y.; Sun, H.; Zhang, J.; Zhang, J.; Sun, C.; Zhang, Y.; Ju, X. The effect of creatine and coenzyme q10 combination therapy on mild cognitive impairment in Parkinson’s disease. Eur. Neurol. 2015, 73, 205–211. [Google Scholar] [CrossRef]
- Kieburtz, K.; Tilley, B.C.; Elm, J.J.; Babcock, D.; Hauser, R.; Ross, G.W.; Augustine, A.H.; Augustine, E.U.; Aminoff, M.J.; Bodis-Wollner, I.G.; et al. Effect of creatine monohydrate on clinical progression in patients with Parkinson disease: A randomized clinical trial. JAMA 2015, 313, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Wang, C.; Jin, Y.; Meng, Q.; Liu, Q.; Wu, J.; Sun, H. CoenzymeQ10-Induced Activation of AMPK-YAP-OPA1 Pathway Alleviates Atherosclerosis by Improving Mitochondrial Function, Inhibiting Oxidative Stress and Promoting Energy Metabolism. Front. Pharmacol. 2020, 11, 1034. [Google Scholar] [CrossRef]
- Passaquin, A.-C.; Renard, M.; Kay, L.; Challet, C.; Mokhtarian, A.; Wallimann, T.; Ruegg, U.T. Creatine supplementation reduces skeletal muscle degeneration and enhances mitochondrial function in mdx mice. Neuromuscul. Disord. 2002, 12, 174–182. [Google Scholar] [CrossRef]
- Smith, R.A.; Hartley, R.C.; Cochemé, H.M.; Murphy, M.P. Mitochondrial pharmacology. Trends Pharmacol. Sci. 2012, 33, 341–352. [Google Scholar] [CrossRef]
- Mounsey, R.B.; Teismann, P. Mitochondrial dysfunction in Parkinson’s disease: Pathogenesis and neuroprotection. Park. Dis. 2011, 2011, 617472. [Google Scholar] [CrossRef]
- Reddy, P.H.; Tripathi, R.; Troung, Q.; Tirumala, K.; Reddy, T.P.; Anekonda, V.; Shirendeb, U.P.; Calkins, M.J.; Reddy, A.P.; Mao, P.; et al. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: Implications to mitochondria-targeted antioxidant therapeutics. Biochim. Biophys. Acta 2012, 1822, 639–649. [Google Scholar] [CrossRef]
- Shen, Y.; Wu, Q.; Shi, J.; Zhou, S. Regulation of SIRT3 on mitochondrial functions and oxidative stress in Parkinson’s disease. Biomed. Pharmacother. 2020, 132, 110928. [Google Scholar] [CrossRef]
- Cheng, A.; Wan, R.; Yang, J.-L.; Kamimura, N.; Son, T.G.; Ouyang, X.; Luo, Y.; Okun, E.; Mattson, M.P. Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nat. Commun. 2012, 3, 1250. [Google Scholar] [CrossRef]
- Silva-Pinheiro, P.; Minczuk, M. The potential of mitochondrial genome engineering. Nat. Rev. Genet. 2022, 23, 199–214. [Google Scholar] [CrossRef]
- Gammage, P.A.; Moraes, C.T.; Minczuk, M. Mitochondrial Genome Engineering: The Revolution May Not Be CRISPR-Ized. Trends Genet. 2018, 34, 101–110. [Google Scholar] [CrossRef]
- Sharma, G.; Sharma, A.R.; Bhattacharya, M.; Lee, S.-S.; Chakraborty, C. CRISPR-Cas9: A Preclinical and Clinical Perspective for the Treatment of Human Diseases. Mol. Ther. 2021, 29, 571–586. [Google Scholar] [CrossRef]
- Zhu, Y. Advances in CRISPR/Cas9. BioMed Res. Int. 2022, 2022, 9978571. [Google Scholar] [CrossRef]
- Tang, J.-X.; Pyle, A.; Taylor, R.W.; Oláhová, M. Interrogating Mitochondrial Biology and Disease Using CRISPR/Cas9 Gene Editing. Genes 2021, 12, 1604. [Google Scholar] [CrossRef]
- Ma, H.; Marti-Gutierrez, N.; Park, S.-W.; Wu, J.; Lee, Y.; Suzuki, K.; Koski, A.; Ji, D.; Hayama, T.; Ahmed, R.; et al. Correction of a pathogenic gene mutation in human embryos. Nature 2017, 548, 413–419. [Google Scholar] [CrossRef] [PubMed]
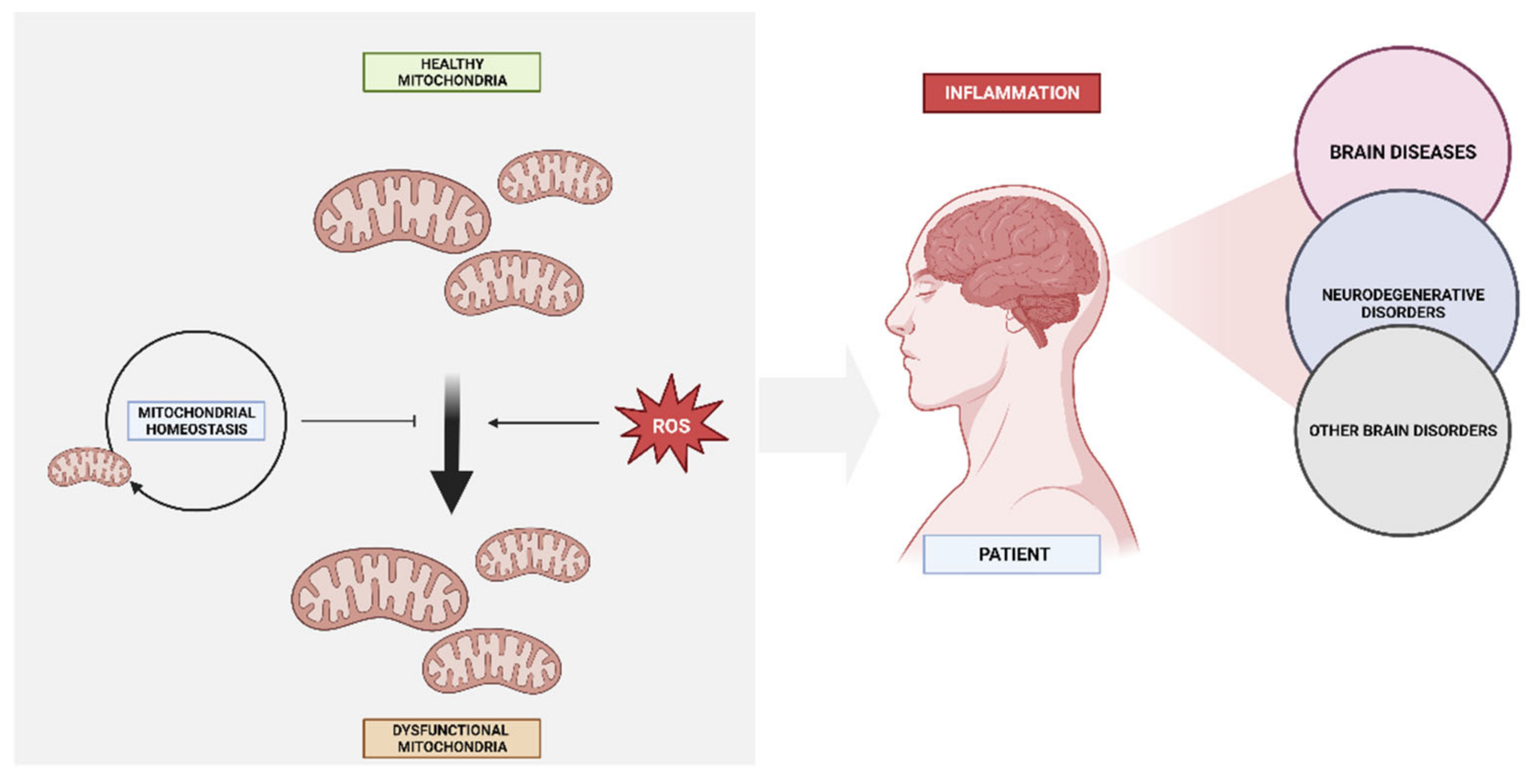
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clemente-Suárez, V.J.; Redondo-Flórez, L.; Beltrán-Velasco, A.I.; Ramos-Campo, D.J.; Belinchón-deMiguel, P.; Martinez-Guardado, I.; Dalamitros, A.A.; Yáñez-Sepúlveda, R.; Martín-Rodríguez, A.; Tornero-Aguilera, J.F. Mitochondria and Brain Disease: A Comprehensive Review of Pathological Mechanisms and Therapeutic Opportunities. Biomedicines 2023, 11, 2488. https://doi.org/10.3390/biomedicines11092488
Clemente-Suárez VJ, Redondo-Flórez L, Beltrán-Velasco AI, Ramos-Campo DJ, Belinchón-deMiguel P, Martinez-Guardado I, Dalamitros AA, Yáñez-Sepúlveda R, Martín-Rodríguez A, Tornero-Aguilera JF. Mitochondria and Brain Disease: A Comprehensive Review of Pathological Mechanisms and Therapeutic Opportunities. Biomedicines. 2023; 11(9):2488. https://doi.org/10.3390/biomedicines11092488
Chicago/Turabian StyleClemente-Suárez, Vicente Javier, Laura Redondo-Flórez, Ana Isabel Beltrán-Velasco, Domingo Jesús Ramos-Campo, Pedro Belinchón-deMiguel, Ismael Martinez-Guardado, Athanasios A. Dalamitros, Rodrigo Yáñez-Sepúlveda, Alexandra Martín-Rodríguez, and José Francisco Tornero-Aguilera. 2023. "Mitochondria and Brain Disease: A Comprehensive Review of Pathological Mechanisms and Therapeutic Opportunities" Biomedicines 11, no. 9: 2488. https://doi.org/10.3390/biomedicines11092488
APA StyleClemente-Suárez, V. J., Redondo-Flórez, L., Beltrán-Velasco, A. I., Ramos-Campo, D. J., Belinchón-deMiguel, P., Martinez-Guardado, I., Dalamitros, A. A., Yáñez-Sepúlveda, R., Martín-Rodríguez, A., & Tornero-Aguilera, J. F. (2023). Mitochondria and Brain Disease: A Comprehensive Review of Pathological Mechanisms and Therapeutic Opportunities. Biomedicines, 11(9), 2488. https://doi.org/10.3390/biomedicines11092488