
Pharmacodynamic Model of the Dynamic Response of Pseudomonas aeruginosa Biofilms to Antibacterial Treatments

Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Dataset
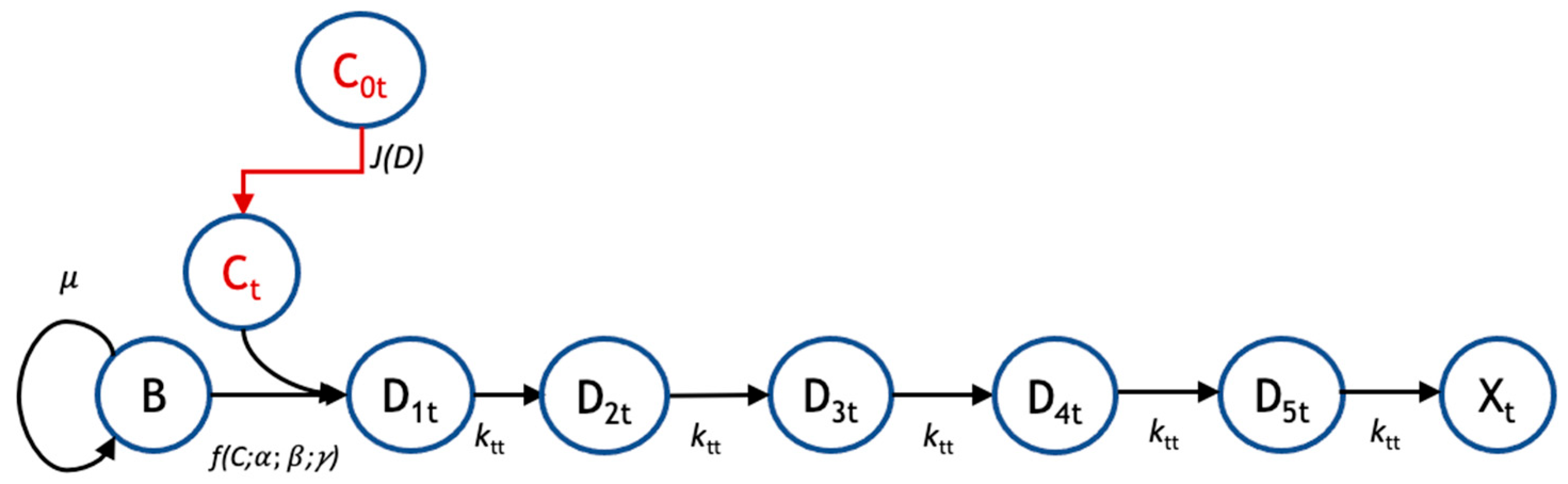
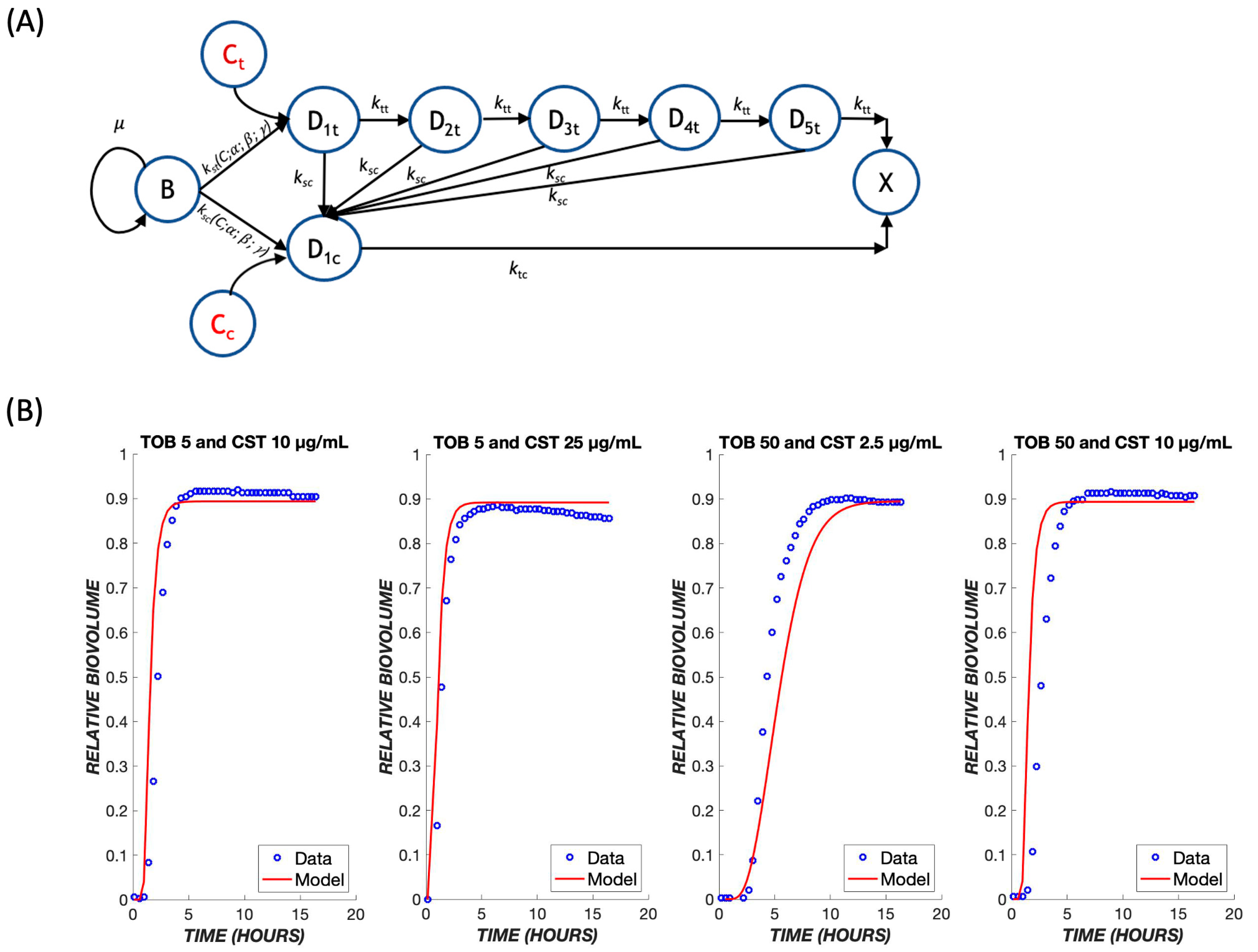
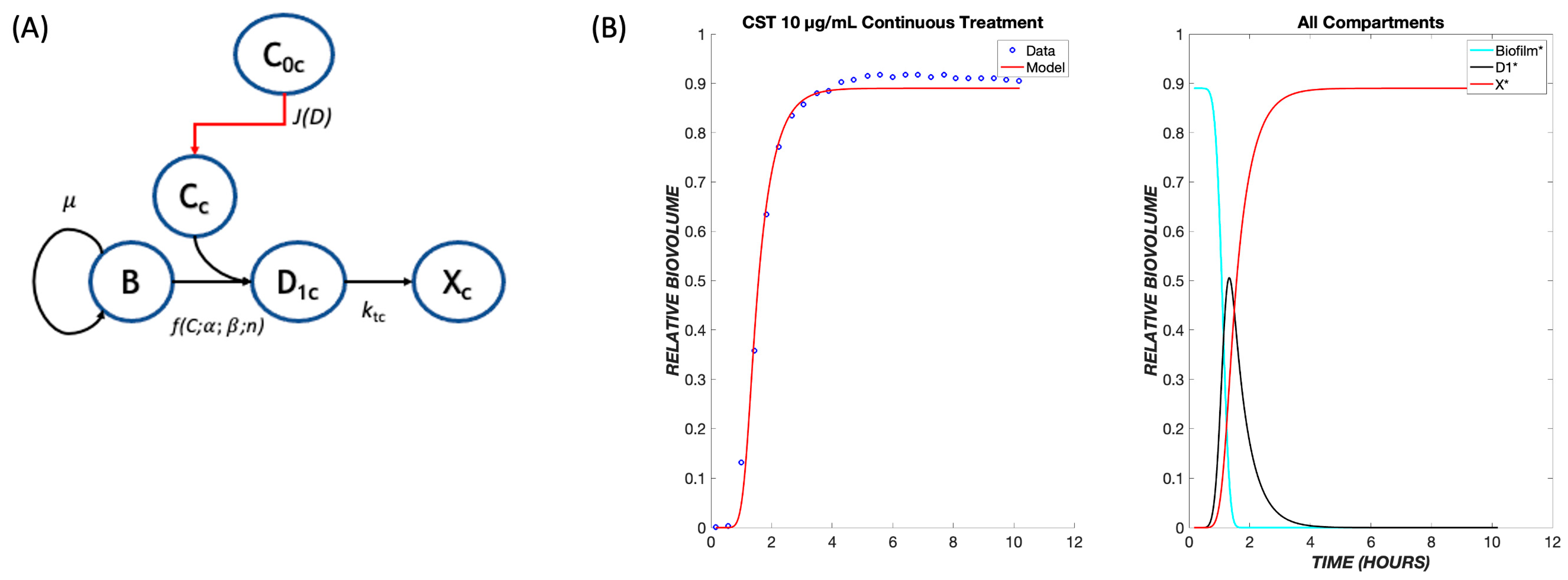
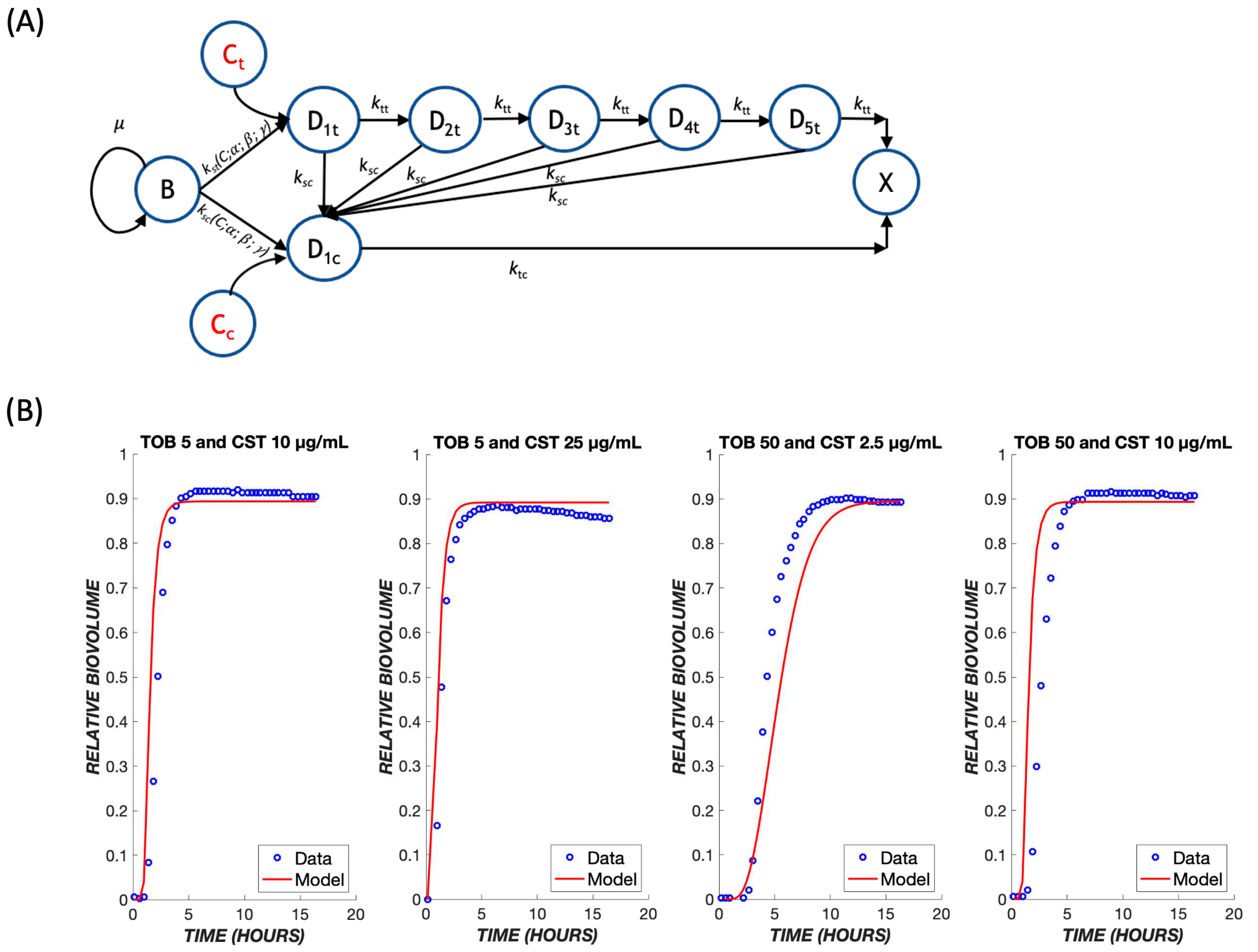
2.2. Mathematical Model
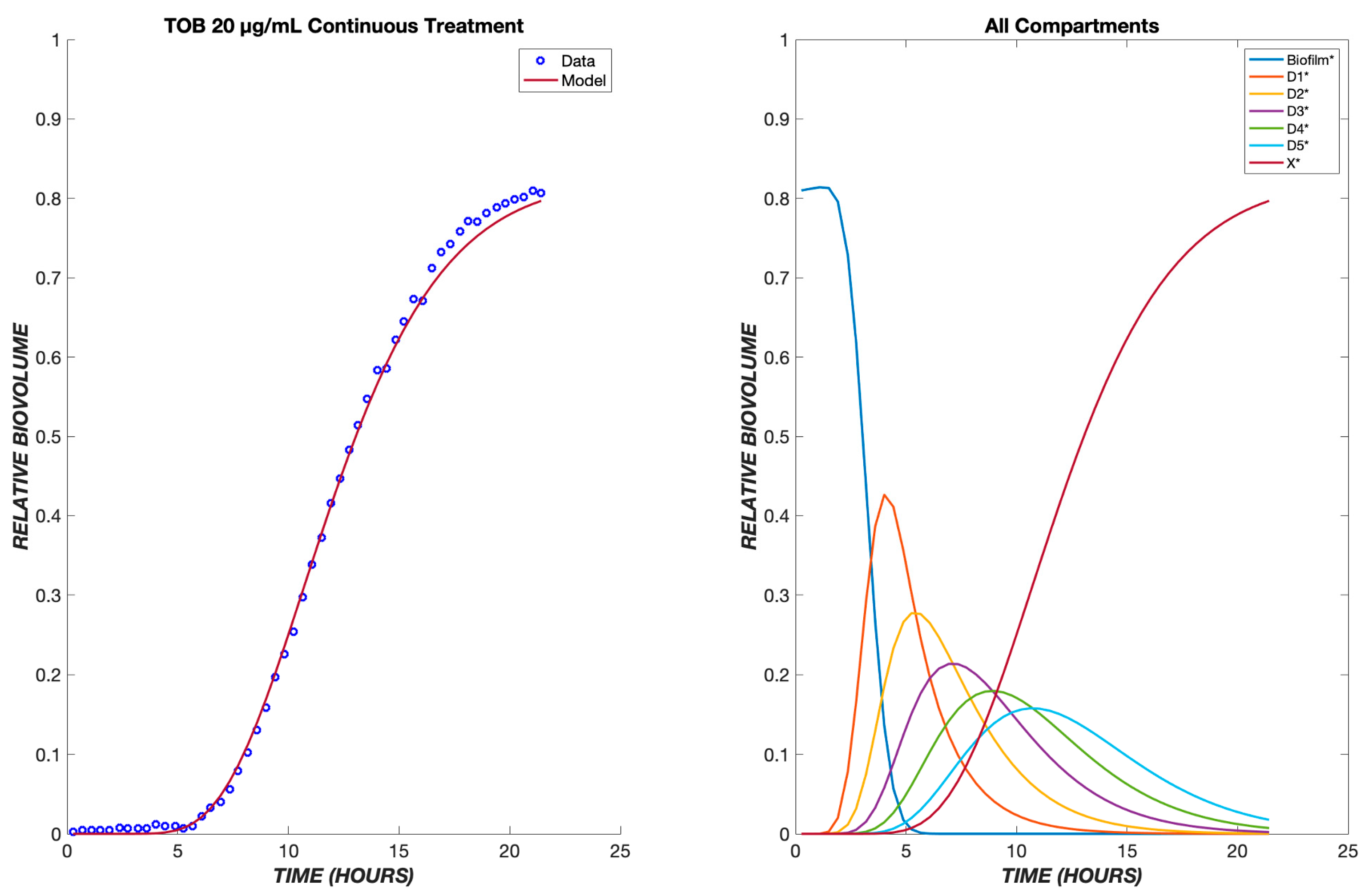
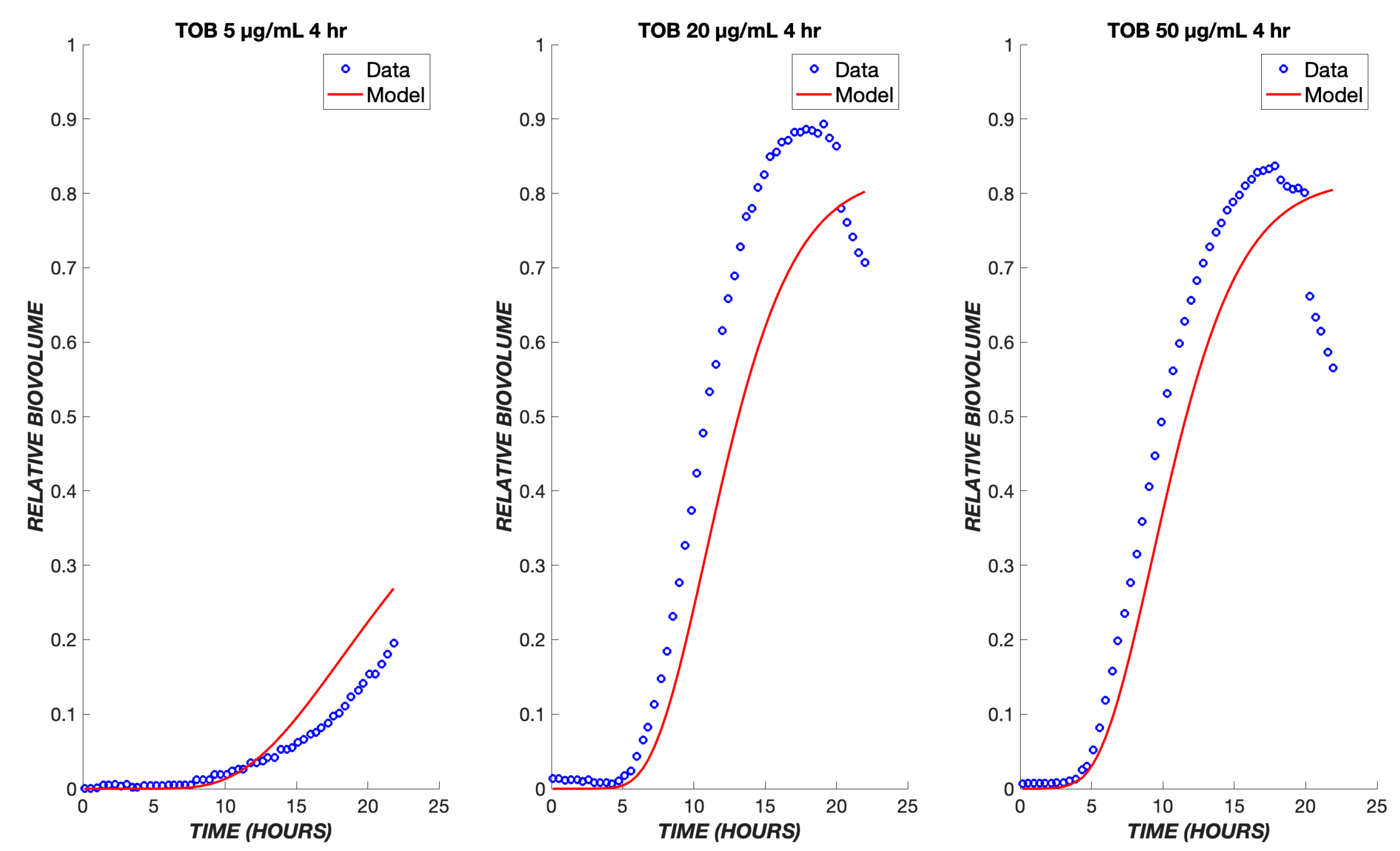
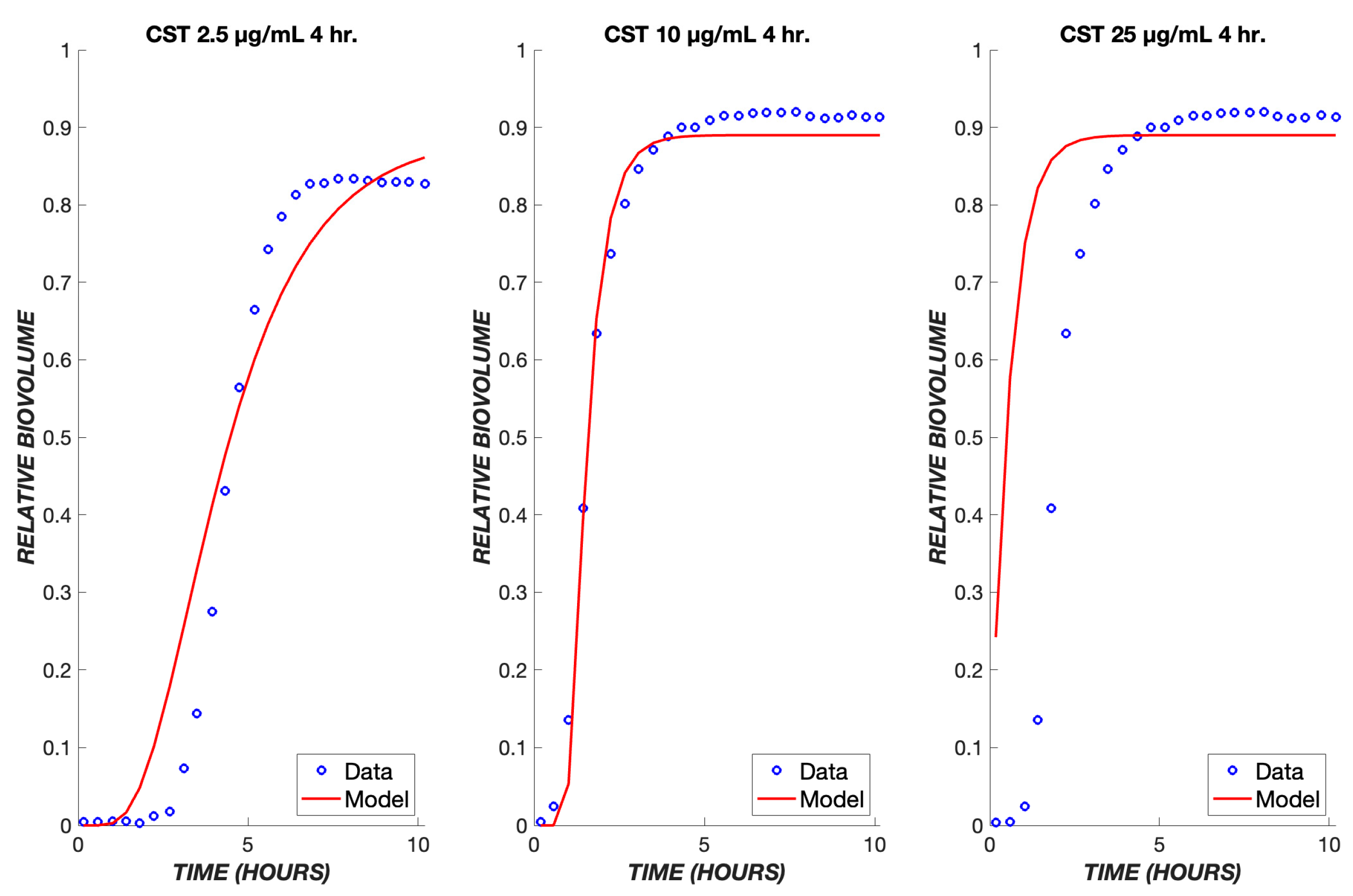
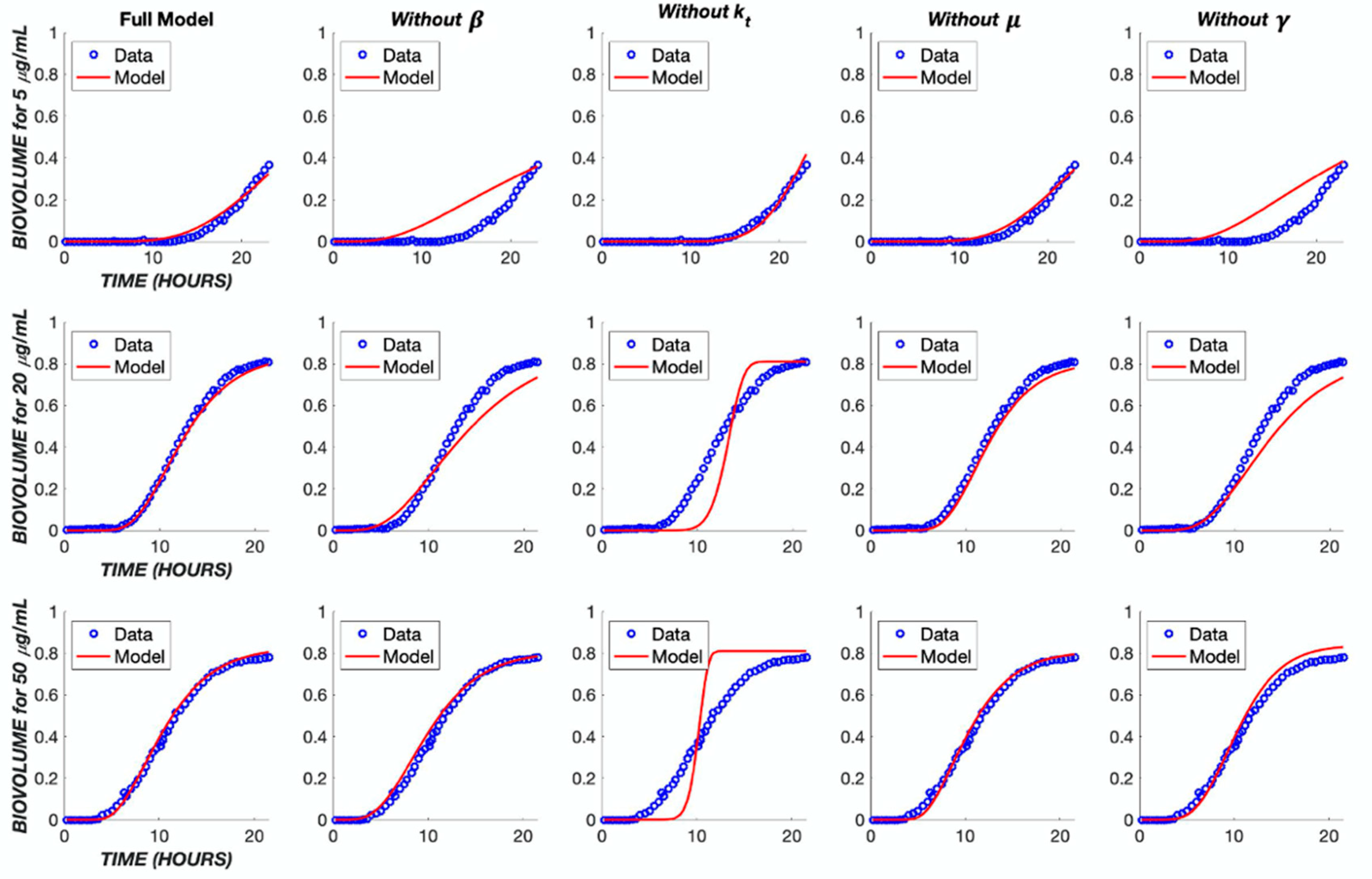
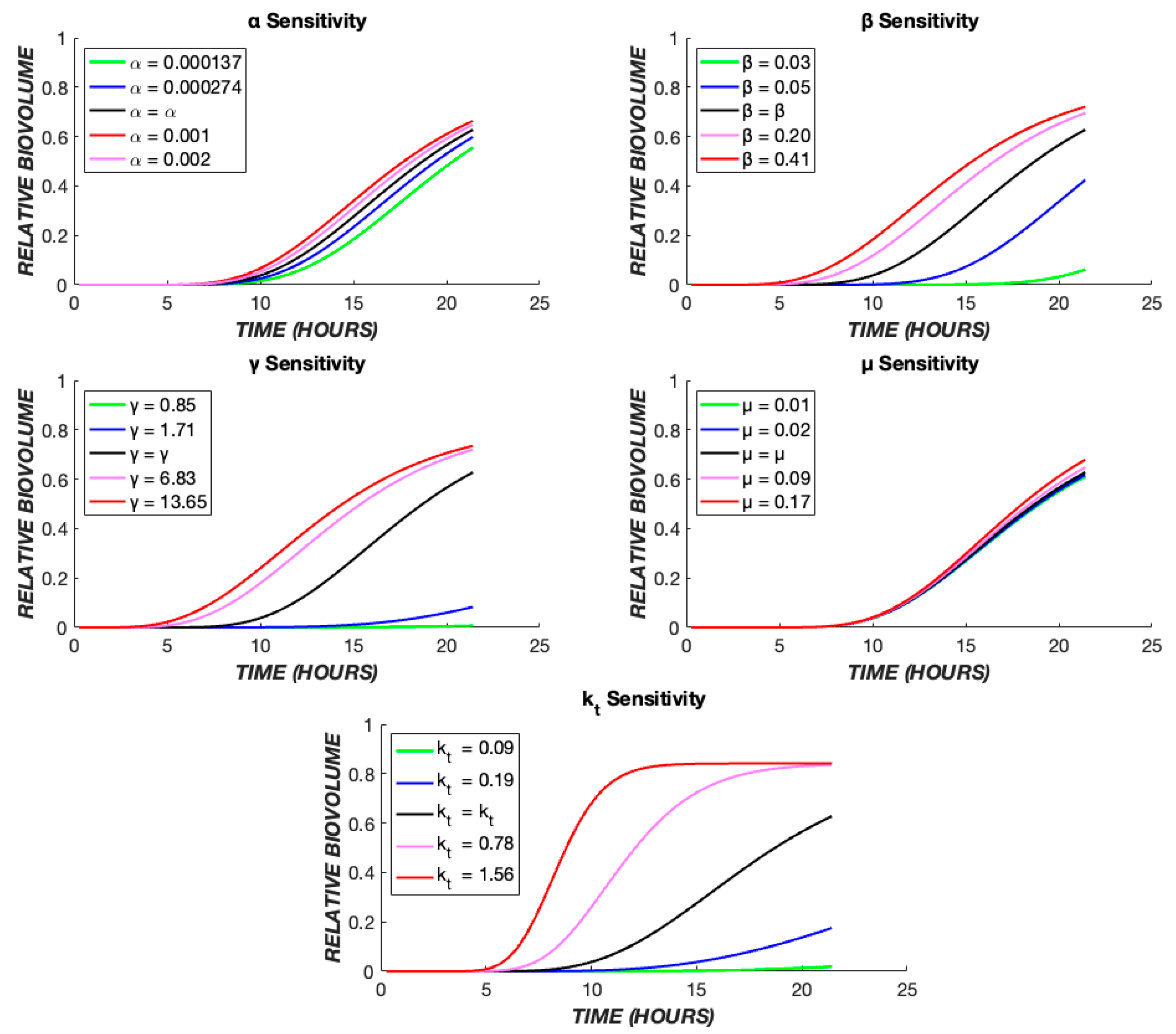
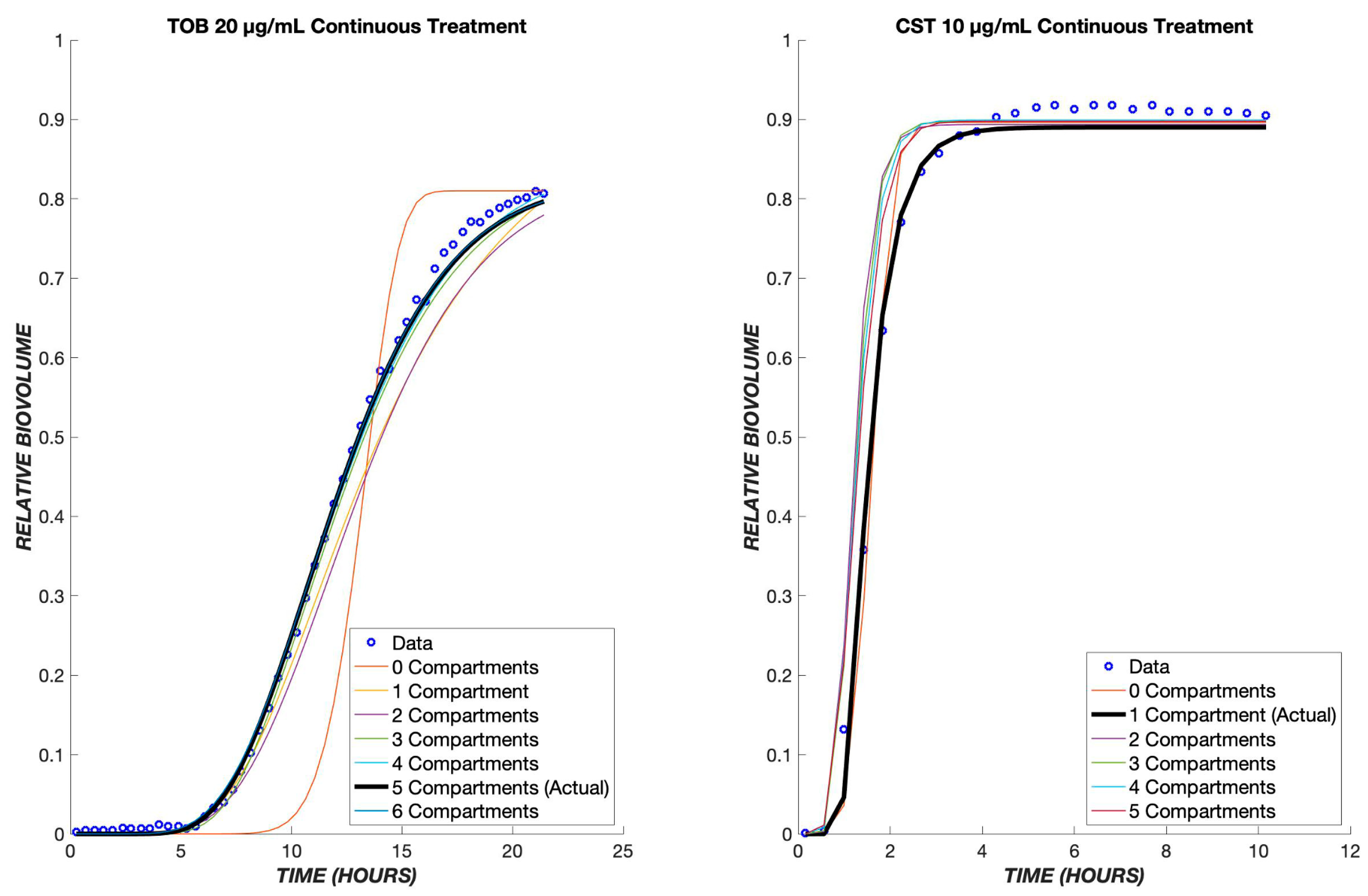
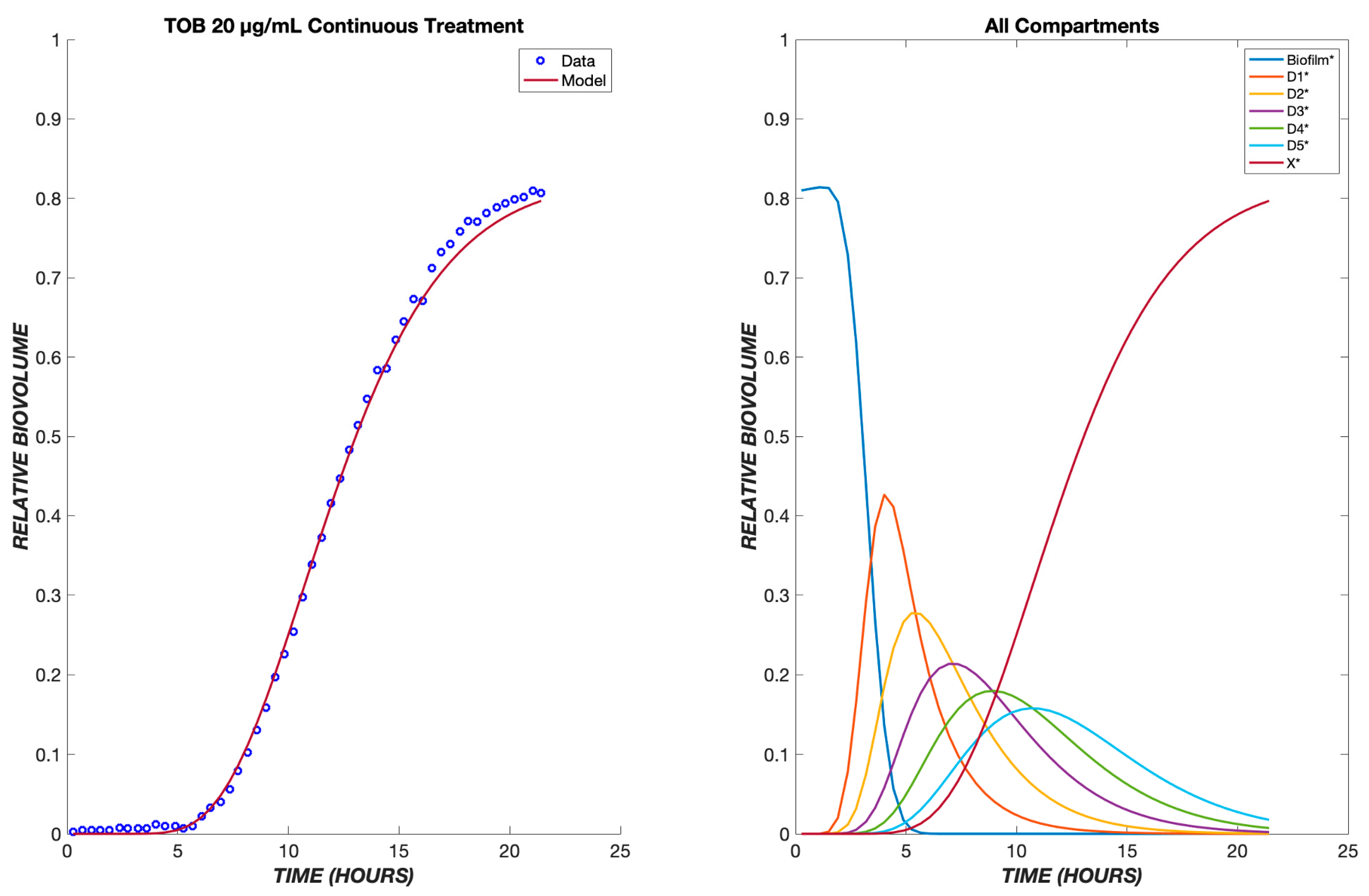
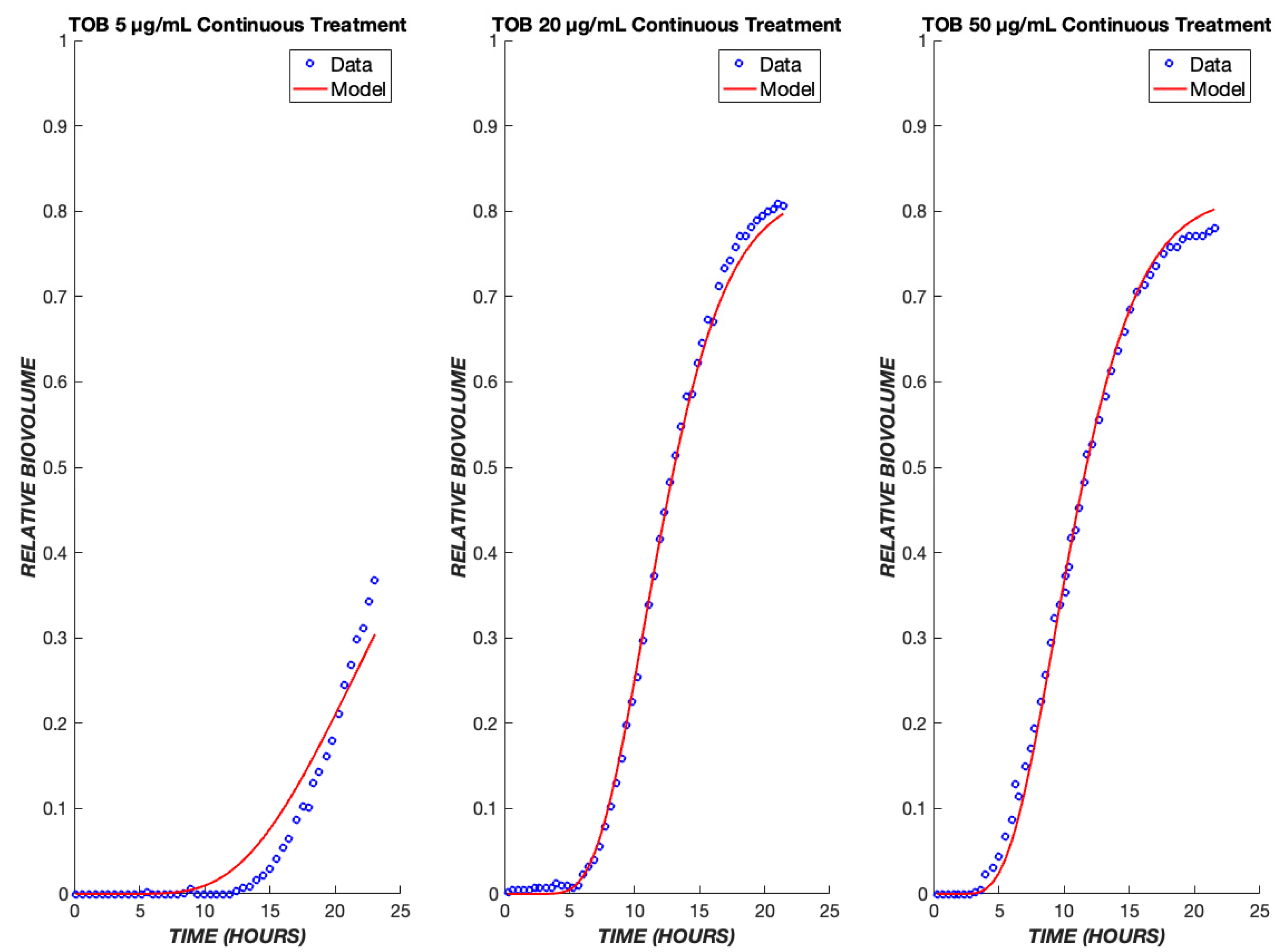
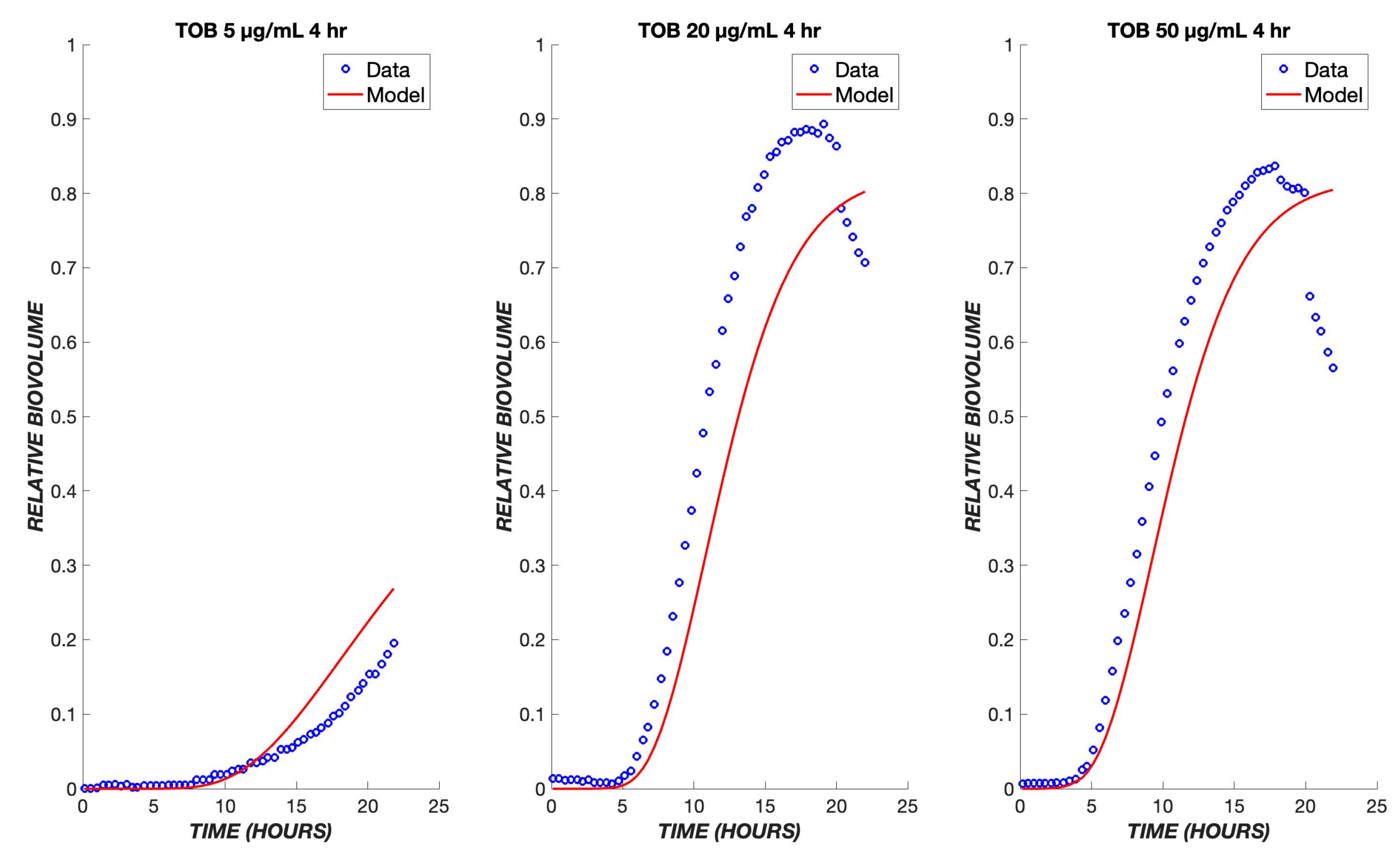
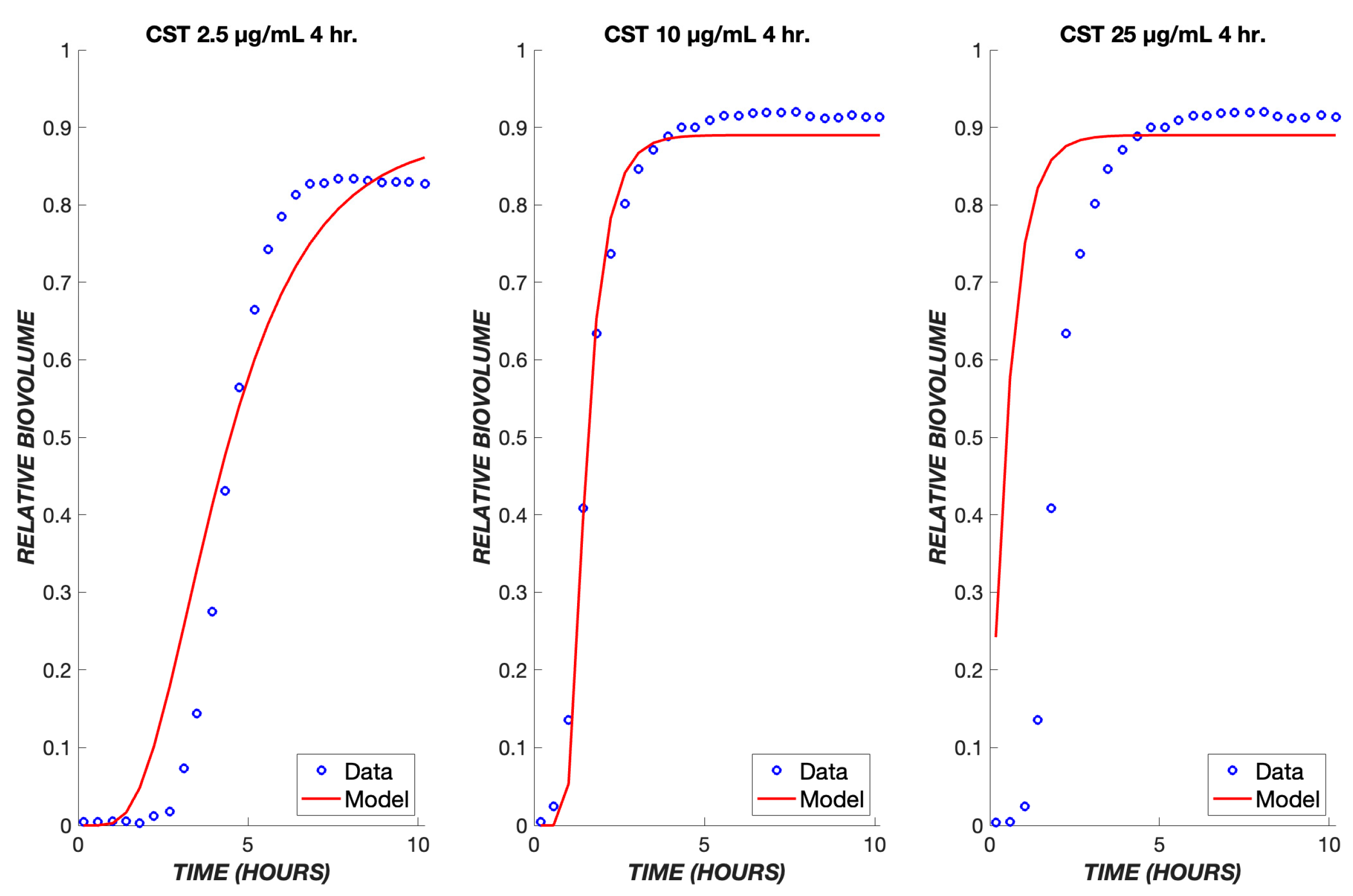
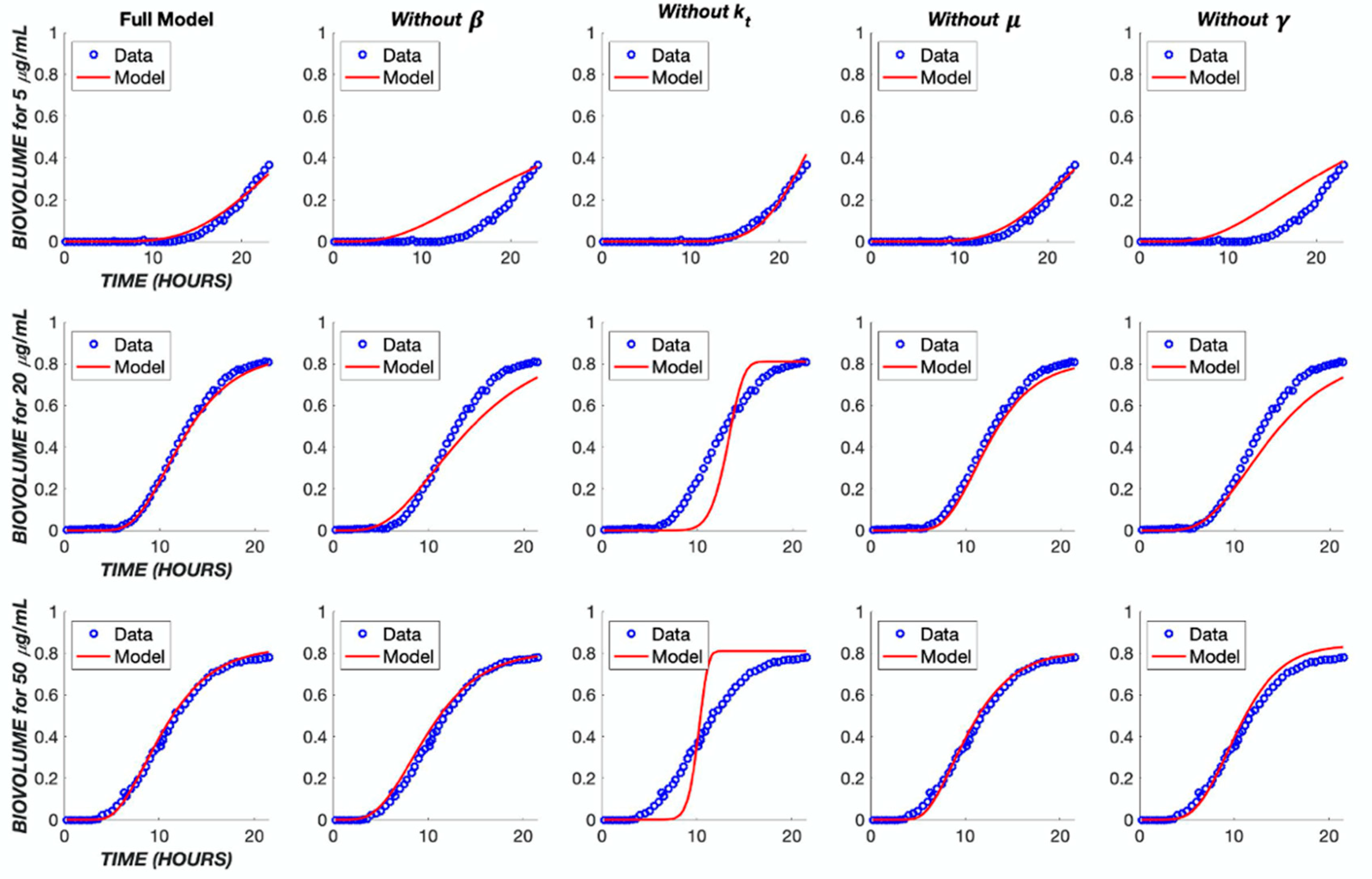
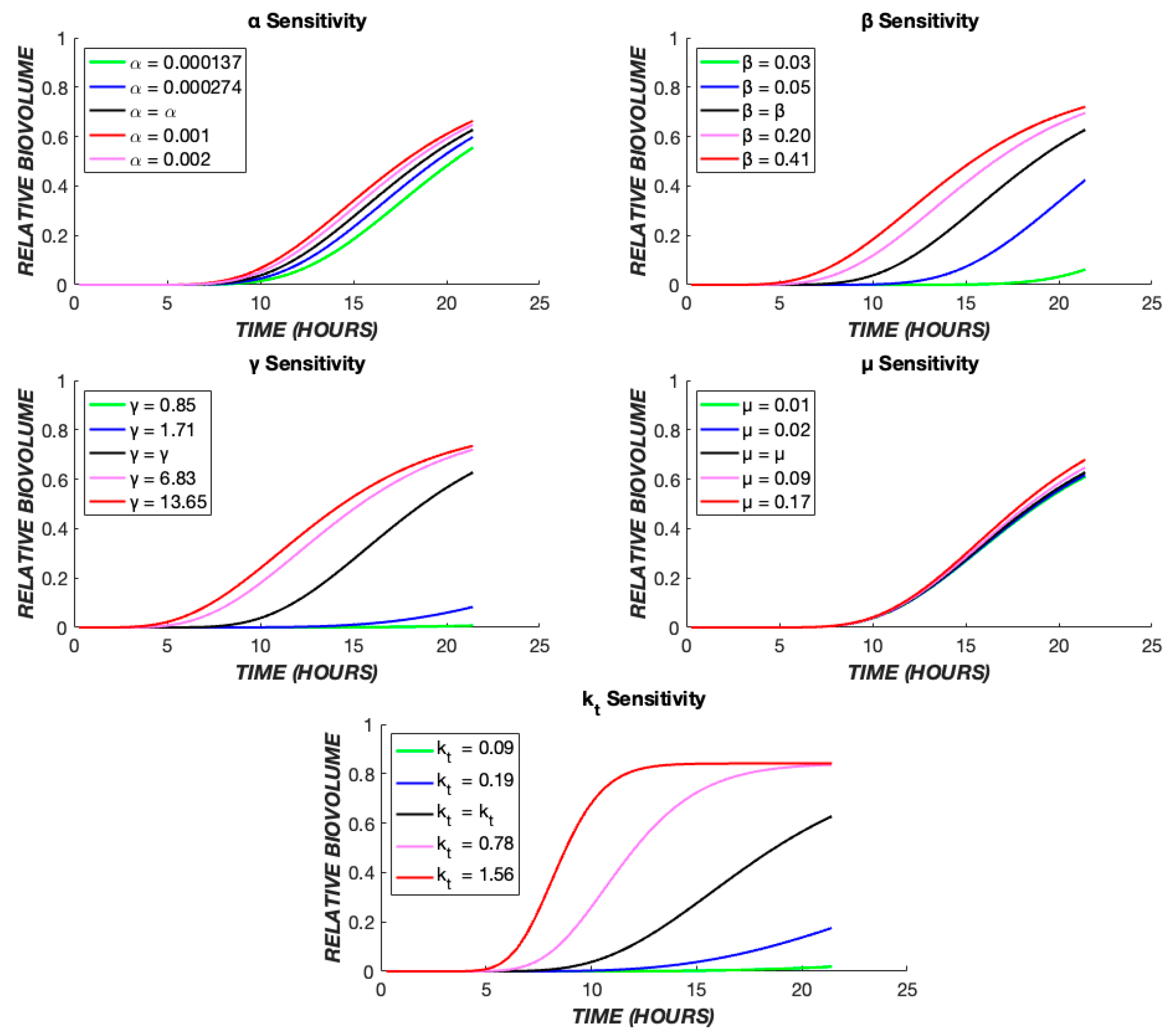
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Donlan, R.M. Biofilms: Microbial life on surfaces. Emerg. Infect. Dis. 2002, 8, 881–890. [Google Scholar] [CrossRef]
- Martinez-Melendez, A.; Morfin-Otero, R.; Villarreal-Trevino, L.; Baines, S.D.; Camacho-Ortiz, A.; Garza-Gonzalez, E. Analysis of biofilm production and expression of adhesion structures of circulating Clostridioides difficile strains from Mexico. Enfermedades Infecc. Microbiol. Clin. 2021, 40, 445–448. [Google Scholar] [CrossRef]
- Rabin, N.; Zheng, Y.; Opoku-Temeng, C.; Du, Y.; Bonsu, E.; Sintim, H.O. Biofilm formation mechanisms and targets for developing antibiofilm agents. Future Med. Chem. 2015, 7, 493–512. [Google Scholar] [CrossRef] [PubMed]
- Ehre, C.; Ridley, C.; Thornton, D.J. Cystic fibrosis: An inherited disease affecting mucin-producing organs. Int. J. Biochem. Cell Biol. 2014, 52, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Khatoon, Z.; McTiernan, C.D.; Suuronen, E.J.; Mah, T.F.; Alarcon, E.I. Bacterial biofilm formation on implantable devices and approaches to its treatment and prevention. Heliyon 2018, 4, e01067. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, J.; Bode-Boger, S.M.; Troger, U.; Martens-Lobenhoffer, J.; Mulrooney, T.; Mittelstadt, H.; Russlies, M.; Kirchner, R.; Knobloch, J.K. Successful treatment of extensively drug-resistant Pseudomonas aeruginosa osteomyelitis using a colistin- and tobramycin-impregnated PMMA spacer. Int. J. Antimicrob. Agents 2014, 44, 363–366. [Google Scholar] [CrossRef]
- Kaiser, P.; Wachter, J.; Windbergs, M. Therapy of infected wounds: Overcoming clinical challenges by advanced drug delivery systems. Drug Deliv. Transl. Res. 2021, 11, 1545–1567. [Google Scholar] [CrossRef] [PubMed]
- Mouton, J.W.; Jacobs, N.; Tiddens, H.; Horrevorts, A.M. Pharmacodynamics of tobramycin in patients with cystic fibrosis. Diagn. Microbiol. Infect. Dis. 2005, 52, 123–127. [Google Scholar] [CrossRef]
- Musken, M.; Pawar, V.; Schwebs, T.; Bahre, H.; Felgner, S.; Weiss, S.; Haussler, S. Breaking the Vicious Cycle of Antibiotic Killing and Regrowth of Biofilm-Residing Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2018, 62, e01635-18. [Google Scholar] [CrossRef]
- Lawrence, J.R.; Neu, T.R. Confocal laser scanning microscopy for analysis of microbial biofilms. Methods Enzymol. 1999, 310, 131–144. [Google Scholar] [CrossRef]
- Wood, S.R.; Kirkham, J.; Marsh, P.D.; Shore, R.C.; Nattress, B.; Robinson, C. Architecture of intact natural human plaque biofilms studied by confocal laser scanning microscopy. J. Dent. Res. 2000, 79, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, D.A.; Wu, A.W.; Zamora, D.; Daly, S.M.; Sturge, C.R.; Pybus, C.; Geller, B.L.; Goldberg, J.B.; Greenberg, D.E. Peptide-Conjugated Phosphorodiamidate Morpholino Oligomers Retain Activity against Multidrug-Resistant Pseudomonas aeruginosa In Vitro and In Vivo. mBio 2021, 12, e02411-20. [Google Scholar] [CrossRef] [PubMed]
- Chamoun, S.; Welander, J.; Martis-Thiele, M.M.; Ntzouni, M.; Claesson, C.; Vikstrom, E.; Turkina, M.V. Colistin Dependence in Extensively Drug-Resistant Acinetobacter baumannii Strain Is Associated with ISAjo2 and ISAba13 Insertions and Multiple Cellular Responses. Int. J. Mol. Sci. 2021, 22, 576. [Google Scholar] [CrossRef] [PubMed]
- Angeles-Martinez, L.; Hatzimanikatis, V. Spatio-temporal modeling of the crowding conditions and metabolic variability in microbial communities. PLoS Comput. Biol. 2021, 17, e1009140. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Low, W.L.; Gupta, A.; Amin, M.C.; Radecka, I.; Britland, S.T.; Raj, P.; Kenward, K.M. Strategies for antimicrobial drug delivery to biofilm. Curr. Pharm. Des. 2015, 21, 43–66. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.J.; Park, J.H.; Goo, S.; Chae, J.W.; Kim, J.; Shin, S.; Yun, H.Y. Dose Optimization of Vancomycin Using a Mechanism-based Exposure-Response Model in Pediatric Infectious Disease Patients. Clin. Ther. 2021, 43, 185–194 e116. [Google Scholar] [CrossRef]
- Wen, X.; Gehring, R.; Stallbaumer, A.; Riviere, J.E.; Volkova, V.V. Limitations of MIC as sole metric of pharmacodynamic response across the range of antimicrobial susceptibilities within a single bacterial species. Sci. Rep. 2016, 6, 37907. [Google Scholar] [CrossRef]
- Holford, N. Pharmacodynamic principles and the time course of immediate drug effects. Transl. Clin. Pharmacol. 2017, 25, 157–161. [Google Scholar] [CrossRef]
- Danhof, M.; de Lange, E.C.; Della Pasqua, O.E.; Ploeger, B.A.; Voskuyl, R.A. Mechanism-based pharmacokinetic-pharmacodynamic (PK-PD) modeling in translational drug research. Trends Pharmacol. Sci. 2008, 29, 186–191. [Google Scholar] [CrossRef]
- Rajman, I. PK/PD modelling and simulations: Utility in drug development. Drug Discov. Today 2008, 13, 341–346. [Google Scholar] [CrossRef]
- Haagensen, J.; Verotta, D.; Huang, L.; Engel, J.; Spormann, A.M.; Yang, K. Spatiotemporal pharmacodynamics of meropenem- and tobramycin-treated Pseudomonas aeruginosa biofilms. J. Antimicrob. Chemother. 2017, 72, 3357–3365. [Google Scholar] [CrossRef]
- Stewart, P.S. Biofilm accumulation model that predicts antibiotic resistance of Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 1994, 38, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.S.; White, B.; Boegli, L.; Hamerly, T.; Williamson, K.S.; Franklin, M.J.; Bothner, B.; James, G.A.; Fisher, S.; Vital-Lopez, F.G.; et al. Conceptual Model of Biofilm Antibiotic Tolerance That Integrates Phenomena of Diffusion, Metabolism, Gene Expression, and Physiology. J. Bacteriol. 2019, 201, e00307-19. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.S.; Zhang, T.; Xu, R.; Pitts, B.; Walters, M.C.; Roe, F.; Kikhney, J.; Moter, A. Reaction-diffusion theory explains hypoxia and heterogeneous growth within microbial biofilms associated with chronic infections. NPJ Biofilms Microbiomes 2016, 2, 16012. [Google Scholar] [CrossRef] [PubMed]
- Sheraton, M.V.; Yam, J.K.H.; Tan, C.H.; Oh, H.S.; Mancini, E.; Yang, L.; Rice, S.A.; Sloot, P.M.A. Mesoscopic Energy Minimization Drives Pseudomonas aeruginosa Biofilm Morphologies and Consequent Stratification of Antibiotic Activity Based on Cell Metabolism. Antimicrob. Agents Chemother. 2018, 62, e02544-17. [Google Scholar] [CrossRef]
- Janakiraman, V.; Englert, D.; Jayaraman, A.; Baskaran, H. Modeling growth and quorum sensing in biofilms grown in microfluidic chambers. Ann. Biomed. Eng. 2009, 37, 1206–1216. [Google Scholar] [CrossRef]
- Cogan, N.G.; Szomolay, B.; Dindos, M. Effect of periodic disinfection on persisters in a one-dimensional biofilm model. Bull. Math. Biol. 2013, 75, 94–123. [Google Scholar] [CrossRef]
- Roberts, M.E.; Stewart, P.S. Modeling antibiotic tolerance in biofilms by accounting for nutrient limitation. Antimicrob. Agents Chemother. 2004, 48, 48–52. [Google Scholar] [CrossRef]
- Kindler, O.; Pulkkinen, O.; Cherstvy, A.G.; Metzler, R. Burst statistics in an early biofilm quorum sensing model: The role of spatial colony-growth heterogeneity. Sci. Rep. 2019, 9, 12077. [Google Scholar] [CrossRef]
- Mattei, M.R.; Frunzo, L.; D’Acunto, B.; Pechaud, Y.; Pirozzi, F.; Esposito, G. Continuum and discrete approach in modeling biofilm development and structure: A review. J. Math. Biol. 2018, 76, 945–1003. [Google Scholar] [CrossRef]
- Nichols, W.W.; Dorrington, S.M.; Slack, M.P.; Walmsley, H.L. Inhibition of tobramycin diffusion by binding to alginate. Antimicrob. Agents Chemother. 1988, 32, 518–523. [Google Scholar] [CrossRef]
- Stewart, P.S. Diffusion in biofilms. J. Bacteriol. 2003, 185, 1485–1491. [Google Scholar] [CrossRef]
- Banat, I.M.; De Rienzo, M.A.; Quinn, G.A. Microbial biofilms: Biosurfactants as antibiofilm agents. Appl. Microbiol. Biotechnol. 2014, 98, 9915–9929. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.S. A model of biofilm detachment. Biotechnol. Bioeng. 1993, 41, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Olmos, A.; Garcia-Castillo, M.; Maiz, L.; Lamas, A.; Baquero, F.; Canton, R. In vitro prevention of Pseudomonas aeruginosa early biofilm formation with antibiotics used in cystic fibrosis patients. Int. J. Antimicrob. Agents 2012, 40, 173–176. [Google Scholar] [CrossRef]
- Moskowitz, S.M.; Foster, J.M.; Emerson, J.; Burns, J.L. Clinically feasible biofilm susceptibility assay for isolates of Pseudomonas aeruginosa from patients with cystic fibrosis. J. Clin. Microbiol. 2004, 42, 1915–1922. [Google Scholar] [CrossRef]
- Sou, T.; Kukavica-Ibrulj, I.; Soukarieh, F.; Halliday, N.; Levesque, R.C.; Williams, P.; Stocks, M.; Camara, M.; Friberg, L.E.; Bergstrom, C.A.S. Model-Based Drug Development in Pulmonary Delivery: Pharmacokinetic Analysis of Novel Drug Candidates for Treatment of Pseudomonas aeruginosa Lung Infection. J. Pharm. Sci. 2019, 108, 630–640. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Description | Units | TOB Value | CST Value |
|---|---|---|---|---|
| μB | Growth rate of biofilm population | h−1 | 0.0321 | 0.0001 |
| α | Rate constant for drug effect on biofilm | (μg/mL)γ−1 h−1 | 0.0002 | 0.0082 |
| β | Normalized drug diffusivity | h−1 | 0.2088 | 0.3986 |
| γ | Cooperativity in drug effect on biofilm | 3.5330 | 4.4313 | |
| kt | Intercompartmental transit rate of drug | h−1 | 0.5424 | 1.8924 |
| Parameter | Full Model | Without μB | Without β | Without γ | Without kt |
|---|---|---|---|---|---|
| μB | 0.0321 | - | 0.5372 | 0.0289 | 0.0000 |
| α | 0.0002 | 0.0012 | 0.0072 | 0.0094 | 0.0351 |
| β | 0.2088 | 0.0815 | - | 0.2480 | 0.0200 |
| γ | 3.5330 | 3.2862 | 1.0705 | - | 3.4315 |
| kt | 0.5424 | 0.4370 | 0.4228 | 0.8053 | - |
| Error | 0.0561 | 0.0971 | 0.4337 | 0.7883 | 1.6449 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roychowdhury, S.; Roth, C.M. Pharmacodynamic Model of the Dynamic Response of Pseudomonas aeruginosa Biofilms to Antibacterial Treatments. Biomedicines 2023, 11, 2316. https://doi.org/10.3390/biomedicines11082316
Roychowdhury S, Roth CM. Pharmacodynamic Model of the Dynamic Response of Pseudomonas aeruginosa Biofilms to Antibacterial Treatments. Biomedicines. 2023; 11(8):2316. https://doi.org/10.3390/biomedicines11082316
Chicago/Turabian StyleRoychowdhury, Swarnima, and Charles M. Roth. 2023. "Pharmacodynamic Model of the Dynamic Response of Pseudomonas aeruginosa Biofilms to Antibacterial Treatments" Biomedicines 11, no. 8: 2316. https://doi.org/10.3390/biomedicines11082316
APA StyleRoychowdhury, S., & Roth, C. M. (2023). Pharmacodynamic Model of the Dynamic Response of Pseudomonas aeruginosa Biofilms to Antibacterial Treatments. Biomedicines, 11(8), 2316. https://doi.org/10.3390/biomedicines11082316