

The Complex Role of Chaperone-Mediated Autophagy in Cancer Diseases


Abstract
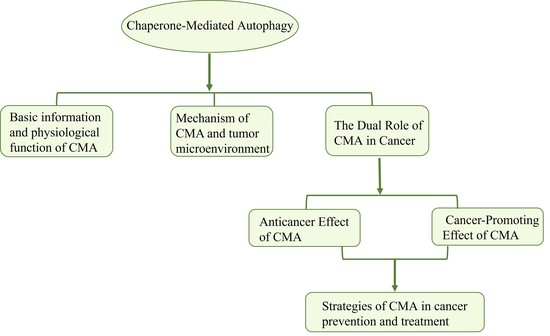
1. Introduction
2. Chaperone-Mediated Autophagy
2.1. CMA Components
2.1.1. Substrate Proteins
2.1.2. Main Companion
2.1.3. CMA Receptors
2.2. CMA-Related Physiological Functions
2.3. Regulation of CMA
CMA and Tumor Microenvironment
3. The Dual Role of CMA in Cancer
3.1. Anticancer Effect of CMA
3.2. Cancer-Promoting Effect of CMA
4. Prevention and Treatment of CMA in Cancer
4.1. Prevention
4.2. Treatment
5. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Knecht, E.; Aguado, C.; Carcel, J.; Esteban, I.; Esteve, J.M.; Ghislat, G.; Moruno, J.F.; Vidal, J.M.; Saez, R. Intracellular protein degradation in mammalian cells: Recent developments. Cell. Mol. Life Sci. 2009, 66, 2427–2443. [Google Scholar] [CrossRef]
- Ciechanover, A. Intracellular protein degradation: From a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Best Pract. Res. Clin. Haematol. 2017, 30, 341–355. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Assaye, M.A.; Gizaw, S.T. Chaperone-Mediated Autophagy and Its Implications for Neurodegeneration and Cancer. Int. J. Gen. Med. 2022, 15, 5635–5649. [Google Scholar] [CrossRef]
- Gomez-Sintes, R.; Arias, E. Chaperone-mediated autophagy and disease: Implications for cancer and neurodegeneration. Mol. Asp. Med. 2021, 82, 101025. [Google Scholar] [CrossRef]
- Kon, M.; Kiffin, R.; Koga, H.; Chapochnick, J.; Macian, F.; Varticovski, L.; Cuervo, A.M. Chaperone-mediated autophagy is required for tumor growth. Sci. Transl. Med. 2011, 3, 109ra117. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Shan, D.; Yan, W.; Zhang, Z.; Song, Q.; Jiang, Y.; Zhang, X.; Zhang, Z.; Wang, Z.; Wang, Y.; et al. Chaperone-mediated autophagy affects tumor cell proliferation and cisplatin resistance in esophageal squamous cell carcinoma. Thorac. Cancer 2021, 12, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, A.; Fujita, Y.; Hosaka, Y.; Kadota, T.; Ito, A.; Yagishita, S.; Watanabe, N.; Fujimoto, S.; Kawamoto, H.; Saito, N.; et al. Chaperone-mediated autophagy receptor modulates tumor growth and chemoresistance in non-small cell lung cancer. Cancer Sci. 2020, 111, 4154–4165. [Google Scholar] [CrossRef] [PubMed]
- Dice, J.F. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem. Sci. 1990, 15, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, P.; Bourdenx, M.; Madrigal-Matute, J.; Tiano, S.; Diaz, A.; Bartholdy, B.A.; Will, B.; Cuervo, A.M. Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Biol. 2019, 17, e3000301. [Google Scholar] [CrossRef]
- Rios, J.; Sequeida, A.; Albornoz, A.; Budini, M. Chaperone Mediated Autophagy Substrates and Components in Cancer. Front. Oncol. 2020, 10, 614677. [Google Scholar] [CrossRef]
- Alfaro, I.E.; Albornoz, A.; Molina, A.; Moreno, J.; Cordero, K.; Criollo, A.; Budini, M. Chaperone Mediated Autophagy in the Crosstalk of Neurodegenerative Diseases and Metabolic Disorders. Front. Endocrinol. 2018, 9, 778. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef]
- Martinez, R.A.S.; Pinky, P.D.; Harlan, B.A.; Brewer, G.J. GTP energy dependence of endocytosis and autophagy in the aging brain and Alzheimer’s disease. Geroscience 2023, 45, 757–780. [Google Scholar] [CrossRef]
- Kiffin, R.; Kaushik, S.; Zeng, M.; Bandyopadhyay, U.; Zhang, C.; Massey, A.C.; Martinez-Vicente, M.; Cuervo, A.M. Altered dynamics of the lysosomal receptor for chaperone-mediated autophagy with age. J. Cell Sci. 2007, 120, 782–791. [Google Scholar] [CrossRef]
- Li, W.; Yang, Q.; Mao, Z. Chaperone-mediated autophagy: Machinery, regulation and biological consequences. Cell Mol. Life Sci. 2011, 68, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, Y.; Araya, J.; Fujita, Y.; Kuwano, K. Role of chaperone-mediated autophagy in the pathophysiology including pulmonary disorders. Inflamm. Regen. 2021, 41, 29. [Google Scholar] [CrossRef]
- Zhu, L.; He, S.; Huang, L.; Ren, D.; Nie, T.; Tao, K.; Xia, L.; Lu, F.; Mao, Z.; Yang, Q. Chaperone-mediated autophagy degrades Keap1 and promotes Nrf2-mediated antioxidative response. Aging Cell 2022, 21, e13616. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.; Pabla, N.; Dong, Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell Mol. Life Sci. 2013, 70, 4009–4021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Lai, W.; Liu, Y.; Wan, R.; Shen, Y. Chaperone-mediated autophagy attenuates H(2) O(2) -induced cardiomyocyte apoptosis by targeting poly (ADP-ribose) polymerase 1 (PARP1) for lysosomal degradation. Cell Biol. Int. 2022, 46, 1915–1926. [Google Scholar] [CrossRef]
- Valdor, R.; Mocholi, E.; Botbol, Y.; Guerrero-Ros, I.; Chandra, D.; Koga, H.; Gravekamp, C.; Cuervo, A.M.; Macian, F. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat. Immunol. 2014, 15, 1046–1054. [Google Scholar] [CrossRef]
- Perez, L.; McLetchie, S.; Gardiner, G.J.; Deffit, S.N.; Zhou, D.; Blum, J.S. LAMP-2C Inhibits MHC Class II Presentation of Cytoplasmic Antigens by Disrupting Chaperone-Mediated Autophagy. J. Immunol. 2016, 196, 2457–2465. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.B.; Fu, X.T.; Shi, Y.H.; Zhou, J.; Peng, Y.F.; Liu, W.R.; Shi, G.M.; Gao, Q.; Wang, X.Y.; Song, K.; et al. Lamp2a is required for tumor growth and promotes tumor recurrence of hepatocellular carcinoma. Int. J. Oncol. 2016, 49, 2367–2376. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhu, J.; Dou, J.; She, H.; Tao, K.; Xu, H.; Yang, Q.; Mao, Z. Phosphorylation of LAMP2A by p38 MAPK couples ER stress to chaperone-mediated autophagy. Nat. Commun. 2017, 8, 1763. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yang, Q.; Mao, Z. Signaling and induction of chaperone-mediated autophagy by the endoplasmic reticulum under stress conditions. Autophagy 2018, 14, 1094–1096. [Google Scholar] [CrossRef]
- Bandyopadhyay, U.; Sridhar, S.; Kaushik, S.; Kiffin, R.; Cuervo, A.M. Identification of regulators of chaperone-mediated autophagy. Mol. Cell 2010, 39, 535–547. [Google Scholar] [CrossRef]
- Anguiano, J.; Garner, T.P.; Mahalingam, M.; Das, B.C.; Gavathiotis, E.; Cuervo, A.M. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat. Chem. Biol. 2013, 9, 374–382. [Google Scholar] [CrossRef]
- Chava, S.; Lee, C.; Aydin, Y.; Chandra, P.K.; Dash, A.; Chedid, M.; Thung, S.N.; Moroz, K.; Wu, T.; Nayak, N.C.; et al. Chaperone-mediated autophagy compensates for impaired macroautophagy in the cirrhotic liver to promote hepatocellular carcinoma. Oncotarget 2017, 8, 40019–40036. [Google Scholar] [CrossRef]
- Rovira, M.; Sereda, R.; Pladevall-Morera, D.; Ramponi, V.; Marin, I.; Maus, M.; Madrigal-Matute, J.; Diaz, A.; Garcia, F.; Munoz, J.; et al. The lysosomal proteome of senescent cells contributes to the senescence secretome. Aging Cell 2022, 21, e13707. [Google Scholar] [CrossRef]
- Wang, R.; Liu, Y.; Liu, L.; Chen, M.; Wang, X.; Yang, J.; Gong, Y.; Ding, B.S.; Wei, Y.; Wei, X. Tumor cells induce LAMP2a expression in tumor-associated macrophage for cancer progression. EBioMedicine 2019, 40, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Salinas, M.D.; Valdor, R. Chaperone-Mediated Autophagy in Pericytes: A Key Target for the Development of New Treatments against Glioblastoma Progression. Int. J. Mol. Sci. 2022, 23, 8886. [Google Scholar] [CrossRef] [PubMed]
- Valdor, R.; Garcia-Bernal, D.; Riquelme, D.; Martinez, C.M.; Moraleda, J.M.; Cuervo, A.M.; Macian, F.; Martinez, S. Glioblastoma ablates pericytes antitumor immune function through aberrant up-regulation of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2019, 116, 20655–20665. [Google Scholar] [CrossRef]
- Guo, B.; Li, L.; Guo, J.; Liu, A.; Wu, J.; Wang, H.; Shi, J.; Pang, D.; Cao, Q. M2 tumor-associated macrophages produce interleukin-17 to suppress oxaliplatin-induced apoptosis in hepatocellular carcinoma. Oncotarget 2017, 8, 44465–44476. [Google Scholar] [CrossRef]
- Andrade-Tomaz, M.; de Souza, I.; Rocha, C.R.R.; Gomes, L.R. The Role of Chaperone-Mediated Autophagy in Cell Cycle Control and Its Implications in Cancer. Cells 2020, 9, 2140. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Evaluation of HIF-1 inhibitors as anticancer agents. Drug Discov. Today 2007, 12, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.K.; Lee, S.H.; Moon, E.J.; Park, M.H.; Jang, H.; Weitzel, D.H.; Kim, H.H.; Basnet, N.; Kwon, D.Y.; Lee, C.T.; et al. Manassantin A inhibits tumour growth under hypoxia through the activation of chaperone-mediated autophagy by modulating Hsp90 activity. Br. J. Cancer 2023, 128, 1491–1502. [Google Scholar] [CrossRef]
- Hubert, V.; Weiss, S.; Rees, A.J.; Kain, R. Modulating Chaperone-Mediated Autophagy and Its Clinical Applications in Cancer. Cells 2022, 11, 2562. [Google Scholar] [CrossRef]
- Moreno-Blas, D.; Gorostieta-Salas, E.; Castro-Obregón, S. Connecting chaperone-mediated autophagy dysfunction to cellular senescence. Ageing Res. Rev. 2018, 41, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Bonhoure, A.; Vallentin, A.; Martin, M.; Senff-Ribeiro, A.; Amson, R.; Telerman, A.; Vidal, M. Acetylation of translationally controlled tumor protein promotes its degradation through chaperone-mediated autophagy. Eur. J. Cell Biol. 2017, 96, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.L.; Huang, G.J.; Wang, H.J.; Chen, J.L.; Hsu, H.P.; Lu, T.J. Hispolon promotes MDM2 downregulation through chaperone-mediated autophagy. Biochem. Biophys. Res. Commun. 2010, 398, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Auzmendi-Iriarte, J.; Otaegi-Ugartemendia, M.; Carrasco-Garcia, E.; Azkargorta, M.; Diaz, A.; Saenz-Antonanzas, A.; Andermatten, J.A.; Garcia-Puga, M.; Garcia, I.; Elua-Pinin, A.; et al. Chaperone-Mediated Autophagy Controls Proteomic and Transcriptomic Pathways to Maintain Glioma Stem Cell Activity. Cancer Res. 2022, 82, 1283–1297. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Fan, Y.; Dan, W.; Liu, B.; Wang, Z.; Zeng, J.; Li, L. Chaperone-mediated autophagy in cancer: Advances from bench to bedside. Histol. Histopathol. 2020, 35, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.R.; Menck, C.F.M.; Cuervo, A.M. Chaperone-mediated autophagy prevents cellular transformation by regulating MYC proteasomal degradation. Autophagy 2017, 13, 928–940. [Google Scholar] [CrossRef]
- Li, W.; Dou, J.; Yang, J.; Xu, H.; She, H. Targeting Chaperone-Mediated Autophagy for Disease Therapy. Curr. Pharm. Rep. 2018, 4, 261–275. [Google Scholar] [CrossRef]
- Welsch, T.; Younsi, A.; Disanza, A.; Rodriguez, J.A.; Cuervo, A.M.; Scita, G.; Schmidt, J. Eps8 is recruited to lysosomes and subjected to chaperone-mediated autophagy in cancer cells. Exp. Cell Res. 2010, 316, 1914–1924. [Google Scholar] [CrossRef]
- Schoenherr, C.; Serrels, B.; Proby, C.; Cunningham, D.L.; Findlay, J.E.; Baillie, G.S.; Heath, J.K.; Frame, M.C. Eps8 controls Src- and FAK-dependent phenotypes in squamous carcinoma cells. J. Cell Sci. 2014, 127, 5303–5316. [Google Scholar] [CrossRef]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef]
- Xia, H.-G.; Najafov, A.; Geng, J.; Galan-Acosta, L.; Han, X.; Guo, Y.; Shan, B.; Zhang, Y.; Norberg, E.; Zhang, T.; et al. Degradation of HK2 by chaperone-mediated autophagy promotes metabolic catastrophe and cell death. J. Cell Biol. 2015, 210, 705–716. [Google Scholar] [CrossRef]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.G.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013, 27, 1718–1730. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Suh, Y.; Cuervo, A.M. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat. Commun. 2015, 6, 6823. [Google Scholar] [CrossRef]
- Xuan, Y.; Zhao, S.; Xiao, X.; Xiang, L.; Zheng, H.C. Inhibition of chaperone-mediated autophagy reduces tumor growth and metastasis and promotes drug sensitivity in colorectal cancer. Mol. Med. Rep. 2021, 23, 360. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Y.Y.; Yao, C.B.; Li, J.T.; Zhao, X.N.; Yang, H.B.; Zhang, M.; Yin, M.; Chen, J.; Lei, Q.Y. Acetylation targets HSD17B4 for degradation via the CMA pathway in response to estrone. Autophagy 2017, 13, 538–553. [Google Scholar] [CrossRef]
- Saha, T. LAMP2A overexpression in breast tumors promotes cancer cell survival via chaperone-mediated autophagy. Autophagy 2012, 8, 1643–1656. [Google Scholar] [CrossRef]
- Peng, J.Q.; Han, S.M.; Chen, Z.H.; Yang, J.; Pei, Y.Q.; Bao, C.; Qiao, L.; Chen, W.Q.; Liu, B. Chaperone-mediated autophagy regulates apoptosis and the proliferation of colon carcinoma cells. Biochem. Biophys. Res. Commun. 2020, 522, 348–354. [Google Scholar] [CrossRef]
- Sohn, E.J.; Kim, J.H.; Oh, S.O.; Kim, J.Y. Regulation of self-renewal in ovarian cancer stem cells by fructose via chaperone-mediated autophagy. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1869, 166723. [Google Scholar] [CrossRef]
- Suzuki, J.; Nakajima, W.; Suzuki, H.; Asano, Y.; Tanaka, N. Chaperone-mediated autophagy promotes lung cancer cell survival through selective stabilization of the pro-survival protein, MCL1. Biochem. Biophys. Res. Commun. 2017, 482, 1334–1340. [Google Scholar] [CrossRef] [PubMed]
- Desideri, E.; Castelli, S.; Dorard, C.; Toifl, S.; Grazi, G.L.; Ciriolo, M.R.; Baccarini, M. Impaired degradation of YAP1 and IL6ST by chaperone-mediated autophagy promotes proliferation and migration of normal and hepatocellular carcinoma cells. Autophagy 2023, 19, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wang, X.W.; Liu, Z.H.; Sun, Y.M.; Tang, Y.X.; Zhou, D.H. Chaperone-mediated autophagy substrate proteins in cancer. Oncotarget 2017, 8, 51970–51985. [Google Scholar] [CrossRef]
- Ali, A.B.; Nin, D.S.; Tam, J.; Khan, M. Role of chaperone mediated autophagy (CMA) in the degradation of misfolded N-CoR protein in non-small cell lung cancer (NSCLC) cells. PLoS ONE 2011, 6, e25268. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, B.; Wang, J.; Wu, H.; Xu, S.; Zhang, J.; Wang, L. Discovery of LAMP-2A as potential biomarkers for glioblastoma development by modulating apoptosis through N-CoR degradation. Cell Commun. Signal 2021, 19, 40. [Google Scholar] [CrossRef] [PubMed]
- Sulzmaier, F.; Opoku-Ansah, J.; Ramos, J.W. Phosphorylation is the switch that turns PEA-15 from tumor suppressor to tumor promoter. Small GTPases 2012, 3, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Quintavalle, C.; Di Costanzo, S.; Zanca, C.; Tasset, I.; Fraldi, A.; Incoronato, M.; Mirabelli, P.; Monti, M.; Ballabio, A.; Pucci, P.; et al. Phosphorylation-regulated degradation of the tumor-suppressor form of PED by chaperone-mediated autophagy in lung cancer cells. J. Cell Physiol. 2014, 229, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Yang, K.; Chiao, L.; Deng, Y.; Zhou, X.; Zhang, Z.; Zeng, S.X.; Lu, H. Inhibition of tumor suppressor p73 by nerve growth factor receptor via chaperone-mediated autophagy. J. Mol. Cell Biol. 2020, 12, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yang, J.; Fan, X.; Hu, S.; Zhou, F.; Dong, J.; Zhang, S.; Shang, Y.; Jiang, X.; Guo, H.; et al. Chaperone-mediated autophagy regulates proliferation by targeting RND3 in gastric cancer. Autophagy 2016, 12, 515–528. [Google Scholar] [CrossRef]
- Zhang, S.; Hu, B.; You, Y.; Yang, Z.; Liu, L.; Tang, H.; Bao, W.; Guan, Y.; Shen, X. Sorting nexin 10 acts as a tumor suppressor in tumorigenesis and progression of colorectal cancer through regulating chaperone mediated autophagy degradation of p21(Cip1/WAF1). Cancer Lett. 2018, 419, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Bartrons, R.; Caro, J. Hypoxia, glucose metabolism and the Warburg’s effect. J. Bioenerg. Biomembr. 2007, 39, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Li, D.; Zhao, D.; Lin, R.; Chu, Y.; Zhang, H.; Zha, Z.; Liu, Y.; Li, Z.; Xu, Y.; et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol. Cell 2011, 42, 719–730. [Google Scholar] [CrossRef]
- Xiao, S.; Xu, G.; Wang, Z.; Chong, T. Chaperon-mediated autophagy can promote proliferation and invasion of renal carcinoma cells and inhibit apoptosis through PKM2. Oncol. Rep. 2021, 46, 214. [Google Scholar] [CrossRef]
- Yang, T.; Ren, C.; Qiao, P.; Han, X.; Wang, L.; Lv, S.; Sun, Y.; Liu, Z.; Du, Y.; Yu, Z. PIM2-mediated phosphorylation of hexokinase 2 is critical for tumor growth and paclitaxel resistance in breast cancer. Oncogene 2018, 37, 5997–6009. [Google Scholar] [CrossRef]
- Han, Q.; Deng, Y.; Chen, S.; Chen, R.; Yang, M.; Zhang, Z.; Sun, X.; Wang, W.; He, Y.; Wang, F.; et al. Downregulation of ATG5-dependent macroautophagy by chaperone-mediated autophagy promotes breast cancer cell metastasis. Sci. Rep. 2017, 7, 4759. [Google Scholar] [CrossRef]
- Su, C.M.; Hsu, T.W.; Chen, H.A.; Wang, W.Y.; Huang, C.Y.; Hung, C.C.; Yeh, M.H.; Su, Y.H.; Huang, M.T.; Liao, P.H. Chaperone-mediated autophagy degrade Dicer to promote breast cancer metastasis. J. Cell Physiol. 2023, 238, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Xie, X.; Chen, Y.; Lin, Y.; Cai, Z.; Ding, L.; Wu, Y.; Peng, Y.; Tang, S.; Xu, H. Chaperone-mediated Autophagy Governs Progression of Papillary Thyroid Carcinoma via PPARgamma-SDF1/CXCR4 Signaling. J. Clin. Endocrinol. Metab. 2020, 105, 3308–3323. [Google Scholar] [CrossRef]
- Xue, N.; Lai, F.; Du, T.; Ji, M.; Liu, D.; Yan, C.; Zhang, S.; Yu, X.; Jin, J.; Chen, X. Chaperone-mediated autophagy degradation of IGF-1Rbeta induced by NVP-AUY922 in pancreatic cancer. Cell Mol. Life Sci. 2019, 76, 3433–3447. [Google Scholar] [CrossRef]
- Wu, J.H.; Guo, J.P.; Shi, J.; Wang, H.; Li, L.L.; Guo, B.; Liu, D.X.; Cao, Q.; Yuan, Z.Y. CMA down-regulates p53 expression through degradation of HMGB1 protein to inhibit irradiation-triggered apoptosis in hepatocellular carcinoma. World J. Gastroenterol. 2017, 23, 2308–2317. [Google Scholar] [CrossRef]
- Jin, Y.; Pan, Y.; Zheng, S.; Liu, Y.; Xu, J.; Peng, Y.; Zhang, Z.; Wang, Y.; Xiong, Y.; Xu, L.; et al. Inactivation of EGLN3 hydroxylase facilitates Erk3 degradation via autophagy and impedes lung cancer growth. Oncogene 2022, 41, 1752–1766. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Hou, T.; Gao, Y.; Dan, W.; Liu, T.; Liu, B.; Chen, Y.; Xie, H.; Yang, Z.; Chen, J.; et al. Acetylation-dependent regulation of TPD52 isoform 1 modulates chaperone-mediated autophagy in prostate cancer. Autophagy 2021, 17, 4386–4400. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Wang, B.; Liu, W.; Xu, Q.; Hou, L.; Song, J.; Guo, Q.; Li, N. Dysfunction of chaperone-mediated autophagy in human diseases. Mol. Cell Biochem. 2021, 476, 1439–1454. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cuervo, A.M. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat. Med. 2008, 14, 959–965. [Google Scholar] [CrossRef]
- Galan-Acosta, L.; Xia, H.; Yuan, J.; Vakifahmetoglu-Norberg, H. Activation of chaperone-mediated autophagy as a potential anticancer therapy. Autophagy 2015, 11, 2370–2371. [Google Scholar] [CrossRef]
- Liao, Y.; Yang, Y.; Pan, D.; Ding, Y.; Zhang, H.; Ye, Y.; Li, J.; Zhao, L. HSP90alpha Mediates Sorafenib Resistance in Human Hepatocellular Carcinoma by Necroptosis Inhibition under Hypoxia. Cancers 2021, 13, 243. [Google Scholar] [CrossRef]
- Liu, D.X.; Li, P.P.; Guo, J.P.; Li, L.L.; Guo, B.; Jiao, H.B.; Wu, J.H.; Chen, J.M. Exosomes derived from HBV-associated liver cancer promote chemoresistance by upregulating chaperone-mediated autophagy. Oncol. Lett. 2019, 17, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, Y.; Garcia-Canaveras, J.C.; Guo, L.; Kan, M.; Yu, S.; Blair, I.A.; Rabinowitz, J.D.; Yang, X. Chaperone-mediated autophagy regulates the pluripotency of embryonic stem cells. Science 2020, 369, 397–403. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Type of Cancer | CMA Action Factor and Its Activity | CMA Activity | Cellular Physiological Processes | Subjects | Cancer Development | Reference |
|---|---|---|---|---|---|---|
| Pancreatic carcinoma | IGF-IR, Down | Up | Cell signaling | The human PC Cell lines | Inhibition | [73] |
| Ovarian carcinoma | HK2, Down | Up | Glucose metabolism | Cell lines | Inhibition | [48] |
| Breast carcinoma | HSD17B4, Down | Up | Protein modification | Breast cancer tissues | Inhibition | [52] |
| ATG5, Down | Up | Gene transcription | Breast cancer Cell lines | Promotion | [70] | |
| Hepatocellular carcinoma | HMGB1, Down | Up | Gene expression | Cell lines | Promotion | [74] |
| TP53, Down | Up | Gene expression | Cell lines | Inhibition | [49] | |
| Colorectal carcinoma | P21, Down | Up | Gene expression | CRC tissues | Promotion | [65] |
| Gastric carcinoma | RND3, Down | Up | Cell growth | GC tissues | Promotion | [64] |
| Papillary thyroid carcinoma | PPAR γ, Down | Up | Cell signaling | PTC tissues | Promotion | [72] |
| Glioblastoma | N-CoR, Down | Up | Protein modification | Glioma cell line U87-MG and tissues | Promotion | [60] |
| Lung carcinoma | N-CoR, Down | Up | Protein folding | Lung cancer cell lines | Promotion | [59] |
| ErK3, Down | Up | Protein modification | Cell lines | Inhibition | [75] | |
| MCL1, Up | Down | Protein modification | NSCLC cell lines | Promotion | [56] | |
| Renal carcinoma | PKM2, Up | Down | Glucose metabolism | Cell lines | Promotion | [68] |
| Prostate carcinoma | TPD52, Up | Down | Protein modification | C57BL/6 mouse | Promotion | [76] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Wang, L.; He, H.; Liu, Y.; Jiang, Y.; Yang, J. The Complex Role of Chaperone-Mediated Autophagy in Cancer Diseases. Biomedicines 2023, 11, 2050. https://doi.org/10.3390/biomedicines11072050
Liu J, Wang L, He H, Liu Y, Jiang Y, Yang J. The Complex Role of Chaperone-Mediated Autophagy in Cancer Diseases. Biomedicines. 2023; 11(7):2050. https://doi.org/10.3390/biomedicines11072050
Chicago/Turabian StyleLiu, Jing, Lijuan Wang, Hua He, Yueying Liu, Yiqun Jiang, and Jinfeng Yang. 2023. "The Complex Role of Chaperone-Mediated Autophagy in Cancer Diseases" Biomedicines 11, no. 7: 2050. https://doi.org/10.3390/biomedicines11072050
APA StyleLiu, J., Wang, L., He, H., Liu, Y., Jiang, Y., & Yang, J. (2023). The Complex Role of Chaperone-Mediated Autophagy in Cancer Diseases. Biomedicines, 11(7), 2050. https://doi.org/10.3390/biomedicines11072050