Abstract
Non-alcoholic fatty liver disease (NAFLD) is a type of steatosis commonly associated with obesity, dyslipidemia, hypertension, and diabetes. Other diseases such as inherited alpha-1 antitrypsin deficiency (AATD) have also been related to the development of liver steatosis. The primary reasons leading to hepatic lipid deposits can be genetic and epigenetic, and the outcomes range from benign steatosis to liver failure, as well as to extrahepatic diseases. Progressive hepatocellular damage and dysregulated systemic immune responses can affect extrahepatic organs, specifically the heart and lungs. In this review, we discuss the similarities and differences between the molecular pathways of NAFLD and AATD, and the putative value of hepatic organoids as novel models to investigate the physio pathological mechanisms of liver steatosis.
1. Non-Alcoholic Fatty Liver Disease (NAFLD)
Non-alcoholic fatty liver disease (NAFLD) is a common liver condition characterized by an excess of lipid accumulation in hepatocytes (steatosis), which is present in about 25% of the adult population [1]. This term includes a range of liver diseases from benign steatosis to cirrhosis, passing through steatohepatitis (NASH) to hepatocellular carcinoma (HCC) [2]. There are different environmental or genetic risk factors that can lead to NAFLD [3], including insulin resistance and obesity. MAFLD (metabolic associated fatty liver disease) has been proposed as a new name that is expected to better mirror the heterogeneities and similarities between NAFLD and metabolic syndrome [4,5]; however, some controversies remain regarding this new name [6].
The pathology typically begins with an altered lipid homeostasis, the intracellular increment of fats followed by an uncontrolled inflammatory response, which can eventually lead to cirrhosis and/or to HCC [7]. Initially, most of the NAFLD patients are asymptomatic and blood markers typically do not reflect liver impairment [8]. The progression to NASH is associated with liver inflammation usually followed by fibrosis, whereas in some cases, the development of liver failure requires liver transplantation. However, cardiovascular diseases (CVD) are among the main causes of death among NAFLD patients [9].
It is widely accepted that free fatty acids act as primary triggers of NAFLD, although there are other factors implicated in disease progression such as dietary habits, obesity, insulin resistance, intestinal microbiota, or epigenetic factors [10]. Patients with NASH typically have high levels of blood endotoxins, suggesting that bacterial endotoxins play a role in NASH pathogenesis [11,12]. Among the intestinal microflora, Gram-negative bacilli seem to be the largest source of endotoxins, such as lipopolysaccharides (LPS). If intestinal enterobacteria invade the portal vein, they inflame the hepatic vasculature leading to persistent inflammation and progressive liver damage. In obese individuals, an increased expression of CD14, an endotoxin co-receptor in the liver, may result in leptin-induced endotoxin hyper responsiveness [13].
Steatosis is defined by the presence of lipid droplets (LDs) in the cytosol of more than 5% of hepatocytes, which is a consequence of altered lipid metabolism when fatty acid obtention exceeds fatty acid removal [14]. Lipid droplets are dynamic organelles composed of neutral lipids, mainly triglycerides and cholesterol esters [15], which act as energy storage but also as protectors against the deleterious effects of free fatty acids [16]. LDs are increasingly recognized as having important non-pathological roles in cell signalling and function. The properties of LDs are highly regulated by proteins coating the surface of LDs to control lipid trafficking and flux [17]. LDs also play roles in endoplasmic reticulum (ER) stress response, protein storage and degradation, and in infection and immunity [18]. Hence, although LDs formation, per se, is not a deleterious event, the accumulation of intrahepatic lipids is associated with increased circulating lipoproteins and increased risk of CVD [19], the main cause of death in NAFLD patients, as mentioned above.
2. Alpha-1 Antitrypsin Deficiency (AATD)
Inherited alpha-1 antitrypsin deficiency (AATD) is a rare monogenic disorder (ORPHA 60) mainly related to lung and/or liver diseases, but also to neutrophilic panniculitis or systemic vasculitis [20]. AATD is characterized by low levels of circulating alpha-1 antitrypsin (AAT), an acute phase glycoprotein encoded by the SERPINA1 gene, in which more than 120 allelic variants have been described [21]. Some mutations in the SERPINA1 gene have no clinical relevance and are considered as normal variants or M alleles; however, deficient alleles, typically resulting from point mutations or small deletions, are related to low levels or functional activity of AAT, and mild to severe clinical manifestations. Among the deficient alleles, the most clinically relevant and best recognized is the Z allele (Glu342Lys), originating from a point mutation in exon 5 [22]. According to current data, the homozygosity in the Z allele is present in about 96% of AATD patients, whereas the remaining 4% are heterozygous carriers or contain other rare alleles [23].
AAT is primarily synthetized by hepatocytes (about 80%) and acts not only as a main inhibitor of neutrophil elastase and proteinase-3 [24,25], but also as a modulator of caspase activity and apoptosis, as an antioxidant, and/or as a broad immunomodulatory protein [26,27]. The complex tertiary structure of AAT makes it extremely vulnerable to conformational changes, as it happens in the Z allele where a change in just one amino acid triggers AAT polymerization. As mentioned above, AATD mainly affects the liver and lungs; hepatic manifestations are due to AAT intrahepatic polymer accumulation and cytotoxicity [28], whereas lung pathologies are due to low circulating levels, mostly polymeric forms of AAT resulting in an insufficient inhibition of neutrophil proteases [29]. On the other hand, among AATD carriers there is a great variability in clinical presentations: from asymptomatic to those who develop early onset emphysema [30] and/or liver steatosis, fibrosis, cirrhosis, or hepatocarcinoma [31]. This suggests that, in addition to the mutations in SERPINA1 gene, other genetic and/or environmental factors contribute to the clinical manifestations.
It has been demonstrated that AAT polymers accumulate in the ER by mechanisms that are not completely understood. Although polymer formation triggers the unfolded protein response [32] to be cleared out of the cell by autophagy or the ER-associated degradation (ERAD) pathways, aggregates can remain in hepatocytes, eliciting cellular stress and inflammation, which lead to liver damage [33,34,35].
The liver disease in AATD people with a homozygous Z allele has been associated with liver steatosis [36]. Concordantly, transgenic mice expressing the human Z allele displayed an altered lipid metabolism with increased levels of hepatic triglycerides and cholesterol [37], as well as high numbers of LDs [36]. Likewise, AATD patients with a homozygous Z allele seem to have lower serum levels of cholesterol and triglycerides than non-AATD patients [36], pointing to hampered lipoprotein secretion and a lower risk of CVD [38].
3. Meta-Inflammation in NAFLD and AATD
Meta-inflammation is defined as a low-grade chronic inflammation associated with metabolic syndrome [39]. Most scientists agree that meta-inflammation, as a component of immune system, links chronic inflammatory diseases and obesity [40]. In this scenario, adipose tissue macrophages can react to high concentrations of fatty acids and initiate signalling pathways promoting monocyte mobilization and differentiation into macrophages, which further contribute to the inflammatory response [41,42].
Macrophages derived from hematopoietic progenitors are involved in homeostatic and pathogenic processes. In adult tissues, the functions of macrophages are dependent on the microenvironment, and thus macrophages can acquire a proinflammatory (M1) or an anti-inflammatory (anti-fibrotic) (M2) phenotype [43,44]. Bone-marrow monocyte-derived macrophages can also acquire a pro-inflammatory phenotype and contribute to inflammation [45]. Because of lipid accumulation in NAFLD, not only is macrophage polarization altered in favour of the M1 phenotype, but macrophages also undergo metabolic reprogramming leading to increased fatty acid intake and worsen steatosis [46]. Activated liver Kupffer cells release pro-inflammatory cytokines, which in turn activate hepatic stellate cells, hepatocytes, or endothelial cells [47,48], promoting monocyte infiltration and boosting macrophage population. Furthermore, fat accumulation in Kupffer cells leads to oxidative stress and structural changes in the plasmatic and mitochondrial membranes, while in the context of AATD, due to AAT protein accumulation in the ER, this also leads to activation of the unfolded protein response [14]. An increase in free fatty acids intensifies lipid oxidation, mainly in the mitochondria and peroxisomes, as well as free-radical production, which can lead to mitochondrial damage and fragmentation [49,50]. On the other hand, ER stress induced by misfolded proteins triggering the unfolded protein response elicits p53 expression, mitochondrial cytochrome c release, and apoptosis [51]. Hence, liver Kupffer cells can contribute not only to the sustained meta-inflammation, but also to the progression of NAFLD (Figure 1).
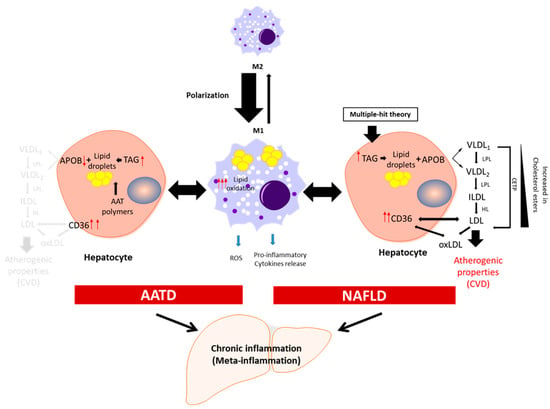
Figure 1.
Schematic presentation of the development of chronic inflammation in NAFLD and AATD. Activated liver macrophages promote inflammation characterized by cytokine and free radical (ROS) production and increased lipid oxidation. In this scenario, to diminish the net increment of lipids, hepatocytes fuse triglycerides (TAG) stored in lipid droplets into APOB-containing lipoproteins and increase the expression of the CD36 receptor to export the lipids out of the cells. In NAFLD patients, this increases the plasma lipoprotein levels with the concomitant risk of cardiovascular disease (CVD). In AATD patients, despite the increased expression of CD36, the accumulation of Z-AAT protein impairs lipids secretion and lowers the risk of CVD. LPL: lipoprotein lipase; HL: hepatic lipase; CETP: cholesteryl ester transfer protein; oxLDL: oxidized LDL.
In this scenario, a member of the class B scavenger receptor, CD36, plays a central role. CD36 binds oxidized low-density lipoproteins, long-chain fatty acids, phospholipids, and collagen [52,53]. Its high expression on macrophages, adipocytes, cardiomyocytes, and hepatic cells is important for fatty acid uptake and lipid metabolism. In fact, CD36 expression is much lower in normal hepatocytes than in hepatic steatosis and NAFLD [54]. An increased hepatic CD36 expression can enhance fatty acid uptake and triglyceride accumulation, although the precise role of CD36 in the pathogenesis of fatty liver remains unclear.
In addition to hepatocytes, monocytes and macrophages also express the AAT protein [55,56,57]. A study based on transgenic Z-AAT mice, reproducing most of the liver characteristics of AATD, showed high numbers of liver macrophages [58]. The characterization of these AATD-related macrophages revealed an altered immunophenotype with a population expressing F4/80hi and TIM4neg, known as a contributor in NAFLD progression [59]. As mentioned above, AAT possesses a broad spectrum of anti-inflammatory properties [60], whereas polymers of Z-AAT are pro-inflammatory, and their accumulation in monocytes and macrophages may trigger NLRP3 inflammasome activation [61]. Kupffer cells also express and accumulate the Z-AAT protein [62], but the effect of Z-AAT accumulation on AATD progression remains to be clarified [59].
4. Features of Lipid Metabolism in NAFLD and AATD
Lipids are key cellular components involved in maintaining the integrity of cellular membranes and energy homeostasis, although they also contribute to pathologies [63]. Lipid homeostasis in the liver depends on the equilibrated balance between lipid acquisition (de novo formation and uptake), storage, and removal [64]. Neutral lipids (sterol esters and triglycerides) are stored in LDs, and in a healthy liver, these lipids do not exceed 5% [65]. Fatty acids stored as sterol esters and triglycerides are used during liver homeostasis to generate energy via fatty acid oxidation or are transported to other organs in very-low-density lipoprotein (VLDL) [66,67] (Figure 1).
A composite route required for VLDL assembly is the lipidation of APOB100, a main and highly hydrophobic apolipoprotein. Initially, VLDLs are pre-assembled in ER lumen by the microsomal triglyceride transfer protein [68] and are subsequently moved to the secretory pathway as VLDL2 particles (TAG poor). These particles are secreted out of the hepatocytes or undergo additional lipidation through LD fusion and become VLDL1 particles (TAG enriched) [69]. A failure in APOB lipidation triggers its degradation because the protein is unable to fold correctly [70]. Therefore, VLDL secretion regulates the fat amount in the liver, and VLDL production and secretion are considered as contributors to CVDs [71]. An imbalanced lipid metabolism in NAFLD patients is related to higher levels of VLDL production (and consequently VLDL secretion), which links NAFLD with CVDs.
NAFLD is a multifactorial disorder, in which genetic alterations play a role [72]. For example, genes such as PNPLA3 [73] and TM6SF2 [74] are linked to a high risk of NAFLD. The patatin-like phospholipase domain-containing 3 gene (PNPLA3) encodes a membrane-associated lipase that mediates triacylglycerol hydrolysis to manage the increasing amount of lipids after a meal intake. The nonsynonymous transversion from cytosine to guanine (rs738409) renders an amino acid change at codon 148 (isoleucine to methionine) that results in an imbalance between the liver triglyceride content and VLDL secretion [75]. The results found by the authors point to a reduced mobilization of triacylglycerols from lipid droplets, even though VLDL assembly itself is not damaged or diminished, which could justify why this variant is not associated with a risk of CVD [75].
The transmembrane 6 superfamily member 2 (TM6SF2) gene, mainly expressed by hepatocytes, enterocytes, and renal cells, encodes for a protein located either in the ER membrane or in the ER−Golgi intermediate compartment. This protein participates in triglyceride secretion and LD formation, and thus regulates the liver fat content. A variant of the TM6SF2 gene (glutamic acid 167 to lysine) is implicated in reduced VLDL secretion [74]. Despite contradictory results in a mouse model using protein overexpression or silencing, lipid accumulation in humans has proven that this variant of the TM6SF2 gene is responsible of the reduced hepatic secretion of VLDL1, which is generated by the combination of VLDL2 and LDs. One putative explanation for this finding is the inability of the variant protein to stabilize APOB [76]. Hence, similarly to the above-described variant of the PNPLA3 gene, despite fat accumulation in the liver, there is no additional risk for CVD. These latter gene variants, to some extent, resemble the situation observed in AATD patients with liver disease, in which diminished hepatic lipid secretion and reduced risk of ischemic heart disease have been reported [36,38].
The lipid components of low-density lipoproteins (LDL) are involved in oxidation reactions, generating a variety of oxidized-lipid-derived products found in atheroma plaques [77]. The intracellular uptake of these oxidized products is mediated by the CD36 [78,79,80], a fatty acid translocase and signalling molecule acting as a receptor for lipoproteins; its derivatives [81]; and free fatty acids [82]. The expression of CD36 is upregulated in NAFLD [83,84,85]. Some studies based on mice models suggest that CD36 is a negative regulator of the autophagy and lipophagy induction. In line with this, CD36 knockdown in HepG2 cells increases lipophagy and β-oxidation, which contribute to lipid accumulation [86].
An increase in CD36 expression has also been linked with AATD [87]. Moreover, a functional relationship between AAT and CD36 has been described, where AAT prevented inflammasome activation in monocytes/macrophages through a signalling cascade involving CD36 [88].
5. Relationships between NAFLD, AATD, and Chronic Obstructive Pulmonary Disease (COPD)
NAFLD is a progressive liver disease evolving via NASH and fibrosis to cirrhosis, and eventually to hepatocellular carcinoma [89]. Investigations demonstrate that NAFLD, NASH, and liver fibrosis are prevalent in patients with COPD (by 41%, 37%, and 61%, respectively) [90]. Patients with COPD and NASH seem to have elevated TNF-α and leptin levels, unlike patients with COPD without liver damage [91]. It is thought that the chronic inflammatory synergy between NAFLD/NASH and COPD can trigger further injury and the progression of both diseases [91,92,93]. Additional studies are warranted to answer questions whether NAFLD and COPD develop together or separately.
AATD is the most common genetic cause of emphysema, and, as a result, the lack of normal levels of AAT do not protect the lungs from damage, leading to an increased risk for developing COPD. Subjects with homozygous ZZ and heterozygous MZ AATD genotypes seem to also have a higher risk of developing NAFLD than non-deficient subjects [94]. For decades, intravenous therapy with human-plasma-purified AAT has been used to treat patients with AATD-related emphysema. The lung-protective effects of AAT are attributed to the inhibition of proteases involved in lung matrix fragmentation, macrophage activation, and endothelial cell apoptosis [95]. Therapy with AAT can rescue epithelial cells from free-heme-mediated pro-inflammatory activation, cell death, and dysfunctional autophagy [96], and prevent acute lung rejection in a mouse orthotopic lung transplantation model [97]. We have also demonstrated hepatoprotective effects of AAT in three different mouse models of acute liver failure (ALF) via the inhibition of caspase-3 and TNF-α, which was also confirmed in a model of alcoholic steatohepatitis [98,99]. Moreover, AAT has been reported to inhibit the process of renal fibrosis through the suppression of TGF-β/Smad3 signalling [100], and to lower the liver stiffness, liver fibrosis, and lower levels of liver enzymes [36]. In INS-1E cells or a primary rat pancreatic islet model, we previously demonstrated that AAT increases insulin secretion in a glucose-dependent manner and protects INS-1E cells from cytokine-induced apoptosis [101]. Patients with NAFLD are associated with hepatic and adipose tissue insulin resistance, and typically have higher levels of serum TNF-α and IL-6, soluble TNFR1 (sTNFR1), and soluble IL-6 receptor (sIL-6R) than patients with a simple steatosis without signs of inflammation, ballooning cells, or fibrosis [102]. Persistent inflammation and altered lipid homeostasis may lead to the evolution of both NAFLD and COPD. In this context, therapy with the AAT protein is of great interest because of its broad tissue-protective effects, which might be beneficial for both NAFLD and COPD patients.
6. Organoids to Model Liver Disease
In vitro two-dimensional (2D) cell models are widely used to reproduce the physiopathology and molecular mechanisms of various diseases. Traditionally used human cell lines are relatively cheap, easy to handle, and can be genetically modified. Nevertheless, the use of cell lines to address questions related to specific human diseases is not always straightforward, and they also lack tissue organization and grow without a physiological context. Therefore, the more recently developed three-dimensional (3D) cell cultures have become better experimental tools [103]. The 3D cultures known as organoids can be generated from adult stem cells, embryonic stem cells (ESCs), or induced pluripotent stem cells (iPSCs). Organoids derived from human progenitor cells can be long-term expanded; they can recapitulate organ architecture with remarkable fidelity, with the presence of multiple cell types of the specific organ [104,105]; and they assume at least some functions of the organ. All these reasons make them a valuable tool for testing new therapeutic drugs and for disease modelling to investigate human diseases including neural disorders [106,107], cancer [108,109], lung diseases [110,111,112], liver diseases [113,114], and others.
In 2001, for the first time, Michalopoulos and colleagues described 3D liver organoids derived from rat hepatocytes [115] that had short-term survival in the culture. A long-term maintenance of cultured organoids was achieved in 2013 by Huch and colleagues from adult murine tissue by using matrigel, hepatocyte growth factor (HGF), epidermal growth factor (EGF), and factors induced under liver damage such as fibroblast growth factor and R-spondin [116]. In parallel, Takebe and collaborators described an alternative method to obtain liver organoids by combining human iPSC-derived hepatocytes, mesenchymal stem cells, and umbilical cord cells [117]. Subsequently, these modified protocols became useful for the modelling of different diseases [118]. Furthermore, patient-derived organoids are an excellent tool to study personalized presentations of pathophysiological conditions influenced by genetic and/or environmental factors.
Hepatic organoids generated from AATD patients have been proven as a new tool to study the pathophysiological characteristics of the liver [119,120]. These organoids have typical intrahepatic retention of Z-AAT polymers and show a positive diastase-resistant (PAS) staining [119,120]. Additionally, when AATD hepatic organoids were differentiated into hepatocytes, Huch and collaborators showed that these cells presented high ER stress and apoptosis [120]. Gomez-Mariano and colleagues further confirmed that differentiated liver organoids express albumin (ALB) and apolipoprotein B (APOB) genes, two specific hepatocyte markers [119].
Hepatocytes or hepatic parenchymal cells comprise approximately 60% of all liver cells, which form a 3D lattice filled by hepatic sinusoids. This latter provides nourishment for the parenchymal cells of the 3D structure, inter-luminal Kupffer, sinusoidal endothelial cells, and perisinusoidal stellate cells [121]. Different 3D cell strategies have been used for NAFLD modelling because the progression of this disease depends on the interactions among multiple liver cell types [122,123]. For example, Shen and collaborators treated hepatic stellate cells (HSC) with conditioned media from patient-derived liver organoids and proved that hepatocyte-derived VEGFA induces HSC activation and hepatocarcinoma progression, even in the absence of increased lipid accumulation [124]. Another NAFLD 3D model in vitro was based on the co-cultures of HepG2 (hepatocyte cell line) and LX-2 (HSC), forming hepatic spheroids and accumulating intracellular lipids after exposure to free fatty acid (FFA) [125]. To reproduce the NASH condition, spheroids were also generated using co-cultures of hepatocytes, HSC, and macrophages (ratio 4:1:1) in a culture medium containing high glucose and palmitate. In this model, treatment with the anti-CD47 antibody, a new therapeutic in obesity, did not improve steatosis, but reduced fibrosis and liver inflammation by inhibiting neutrophil and macrophage activation [126]. Likewise, the combination of hepatocytes (80%), Kupffer cells (10%), HSC (5%), and endothelial cells (5%) have been used to test a protective role of miR-122 in NAFLD. It has been suggested that miR-122 affects steatosis, fibrosis, and altered lipid metabolism because it changes the expression of lipases and fatty acid binding proteins implicated in intrahepatic lipid accumulation [127].
Other approaches to generate liver organoids are based on the differentiation of iPSCs [128]. To reproduce NAFLD, iPSCs-derived liver organoids were exposed to different free fatty acid treatments and analysed for the enlargement of hepatocytes (ballooning), as well as for organoids stiffness by atomic force microscopy [128]. More sophisticated approaches were also applied to deepen our knowledge regarding NAFLD development. For example, sirtuin-1 has been implicated in the progression of NAFLD due to its effects on de novo lipogenesis and beta-oxidation [129,130]. Different types of 3D models for NAFLD have been reviewed by Park and colleagues [131] and by Wang and collaborators [122].
Liver organoids generated from AATD patient-related liver disease seem to recapitulate hepatocyte alterations [119]. Like mature hepatocytes, differentiated hepatic organoids express albumin and apolipoprotein B, although it was found that Z-AATD-derived organoids show a lower expression of both genes, ALB and APOB [119]. A transcriptomic analysis carried out by our group confirmed the reduced APOB expression in Z-AATD organoids, which could at least partially explain why ZZ patients, despite their increased level of hepatic steatosis, show reduced levels of serum triglycerides and VLDL lipoproteins [36]. In addition to this downregulation of APOB transcription levels, other factors may contribute to the final amount of APOB [132]. Among differentially expressed genes, we also found CD36 as one of the upregulated genes in Z-AATD organoids when compared with the controls, somehow mimicking the behaviour displayed in NAFLD. The AATD-related steatosis is likely favoured by the increased expression in the CD36 receptor, although some work is still needed to unravel the relationship between them. On the other hand, the reduced APOB protein (Figure 1) might contribute to the diminished circulating levels of lipoproteins [36,133] and reduced risk of CVDs [38]. Yet, the association between AATD and CVDs requires additional studies.
7. Conclusions
NAFLD and AATD are hepatic diseases characterized by increased hepatic lipid content and consequently the intracellular accumulation of lipid droplets. To maintain liver homeostasis, hepatocytes in NAFLD eliminate the excess of lipids by exporting them to the bloodstream as lipoproteins, which results in an increased risk of cardiovascular disease in NAFLD patients. Conversely, in patients with AATD, the intrahepatic accumulation of misfolded AAT protein lowers lipid secretion and thus risk for cardiovascular disease. The use of patient-derived liver organoids as new cellular models is of great value, especially for the development of new personalized therapies, as well as for studying the underlying molecular mechanisms in NAFLD and AATD.
Author Contributions
Conceptualization, S.P.-L. and S.J.; resources, S.J., N.M. and G.G.-M.; writing—original draft preparation, S.P.-L. and S.J.; review and editing, N.M., G.G.-M., S.J. and B.M.-D.; supervision, B.M.-D. and S.J.; funding acquisition, B.M.-D. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from Instituto de Salud Carlos III (ISCIII): AESI PI20CIII/00015 and PISCIIIBB-PT20CIII/00009, and by Polish National Science Centre Grants 2015/17/B/NZ5/01370 and 2018/29/B/NZ5/02346.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
This study did not report any data.
Acknowledgments
We thank colleagues from the Institute of Rare Diseases Research (IIER), particularly from the Genetic Diagnosis and the Molecular Genetics Unit, and the BioNER, Biobanco Nacional de Enfermedades Raras for their help.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonalcoholic Fatty Liver Disease-Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatol. Baltim. Md. 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Teng, T.; Qiu, S.; Zhao, Y.; Zhao, S.; Sun, D.; Hou, L.; Li, Y.; Zhou, K.; Yu, X.; Yang, C.; et al. Pathogenesis and Therapeutic Strategies Related to Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 7841. [Google Scholar] [CrossRef] [PubMed]
- Liebe, R.; Esposito, I.; Bock, H.H.; Vom Dahl, S.; Stindt, J.; Baumann, U.; Luedde, T.; Keitel, V. Diagnosis and Management of Secondary Causes of Steatohepatitis. J. Hepatol. 2021, 74, 1455–1471. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.-F.; Schattenberg, J.M.; et al. A New Definition for Metabolic Dysfunction-Associated Fatty Liver Disease: An International Expert Consensus Statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Sanyal, A.J.; George, J. International Consensus Panel MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Rinella, M.E.; Sanyal, A.J.; Harrison, S.A.; Brunt, E.M.; Goodman, Z.; Cohen, D.E.; Loomba, R. From NAFLD to MAFLD: Implications of a Premature Change in Terminology. Hepatol. Baltim. Md. 2021, 73, 1194–1198. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Hadizadeh, F.; Faghihimani, E.; Adibi, P. Nonalcoholic Fatty Liver Disease: Diagnostic Biomarkers. World J. Gastrointest. Pathophysiol. 2017, 8, 11–26. [Google Scholar] [CrossRef]
- Targher, G.; Byrne, C.D.; Tilg, H. NAFLD and Increased Risk of Cardiovascular Disease: Clinical Associations, Pathophysiological Mechanisms and Pharmacological Implications. Gut 2020, 69, 1691–1705. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The Multiple-Hit Pathogenesis of Non-Alcoholic Fatty Liver Disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Harte, A.L.; da Silva, N.F.; Creely, S.J.; McGee, K.C.; Billyard, T.; Youssef-Elabd, E.M.; Tripathi, G.; Ashour, E.; Abdalla, M.S.; Sharada, H.M.; et al. Elevated Endotoxin Levels in Non-Alcoholic Fatty Liver Disease. J. Inflamm. 2010, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Pendyala, S.; Walker, J.M.; Holt, P.R. A High-Fat Diet Is Associated with Endotoxemia That Originates from the Gut. Gastroenterology 2012, 142, 1100–1101.e2. [Google Scholar] [CrossRef] [PubMed]
- Kessoku, T.; Kobayashi, T.; Imajo, K.; Tanaka, K.; Yamamoto, A.; Takahashi, K.; Kasai, Y.; Ozaki, A.; Iwaki, M.; Nogami, A.; et al. Endotoxins and Non-Alcoholic Fatty Liver Disease. Front. Endocrinol. 2021, 12, 770986. [Google Scholar] [CrossRef] [PubMed]
- Leroux, A.; Ferrere, G.; Godie, V.; Cailleux, F.; Renoud, M.-L.; Gaudin, F.; Naveau, S.; Prévot, S.; Makhzami, S.; Perlemuter, G.; et al. Toxic Lipids Stored by Kupffer Cells Correlates with Their Pro-Inflammatory Phenotype at an Early Stage of Steatohepatitis. J. Hepatol. 2012, 57, 141–149. [Google Scholar] [CrossRef]
- Walther, T.C.; Farese, R.V. Lipid Droplets And Cellular Lipid Metabolism. Annu. Rev. Biochem. 2012, 81, 687–714. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Carvalho, P. Dynamics and Functions of Lipid Droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Mashek, D.G. Hepatic Lipid Droplets: A Balancing Act between Energy Storage and Metabolic Dysfunction in NAFLD. Mol. Metab. 2021, 50, 101115. [Google Scholar] [CrossRef]
- Yang, H.; Liu, J. Chapter 11-Structure and Function of Lipid Droplets. In Biochemistry of Lipids, Lipoproteins and Membranes, 7th ed.; Ridgway, N.D., McLeod, R.S., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 357–394. ISBN 978-0-12-824048-9. [Google Scholar]
- Schmidt, A.F.; Joshi, R.; Gordillo-Marañón, M.; Drenos, F.; Charoen, P.; Giambartolomei, C.; Bis, J.C.; Gaunt, T.R.; Hughes, A.D.; Lawlor, D.A.; et al. Biomedical Consequences of Elevated Cholesterol-Containing Lipoproteins and Apolipoproteins on Cardiovascular and Non-Cardiovascular Outcomes. Commun. Med. 2023, 3, 9. [Google Scholar] [CrossRef]
- Janciauskiene, S.M.; Bals, R.; Koczulla, R.; Vogelmeier, C.; Köhnlein, T.; Welte, T. The Discovery of A1-Antitrypsin and Its Role in Health and Disease. Respir. Med. 2011, 105, 1129–1139. [Google Scholar] [CrossRef]
- Seixas, S.; Marques, P.I. Known Mutations at the Cause of Alpha-1 Antitrypsin Deficiency an Updated Overview of SERPINA1 Variation Spectrum. Appl. Clin. Genet. 2021, 14, 173–194. [Google Scholar] [CrossRef]
- Ogushi, F.; Fells, G.A.; Hubbard, R.C.; Straus, S.D.; Crystal, R.G. Z-Type Alpha 1-Antitrypsin Is Less Competent than M1-Type Alpha 1-Antitrypsin as an Inhibitor of Neutrophil Elastase. J. Clin. Investig. 1987, 80, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- de Serres, F.J.; Blanco, I. Prevalence of A1-Antitrypsin Deficiency Alleles PI*S and PI*Z Worldwide and Effective Screening for Each of the Five Phenotypic Classes PI*MS, PI*MZ, PI*SS, PI*SZ, and PI*ZZ: A Comprehensive Review. Ther. Adv. Respir. Dis. 2012, 6, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Carlson, J.A.; Rogers, B.B.; Sifers, R.N.; Hawkins, H.K.; Finegold, M.J.; Woo, S.L. Multiple Tissues Express Alpha 1-Antitrypsin in Transgenic Mice and Man. J. Clin. Investig. 1988, 82, 26–36. [Google Scholar] [CrossRef]
- Sinden, N.J.; Baker, M.J.; Smith, D.J.; Kreft, J.-U.; Dafforn, T.R.; Stockley, R.A. α-1-Antitrypsin Variants and the Proteinase/Antiproteinase Imbalance in Chronic Obstructive Pulmonary Disease. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2015, 308, L179–L190. [Google Scholar] [CrossRef] [PubMed]
- Blanco, I.; Lipsker, D.; Lara, B.; Janciauskiene, S. Neutrophilic Panniculitis Associated with Alpha-1-Antitrypsin Deficiency: An Update. Br. J. Dermatol. 2016, 174, 753–762. [Google Scholar] [CrossRef]
- Sun, R.; Xu, Z.; Zhu, C.; Chen, T.; Muñoz, L.E.; Dai, L.; Zhao, Y. Alpha-1 Antitrypsin in Autoimmune Diseases: Roles and Therapeutic Prospects. Int. Immunopharmacol. 2022, 110, 109001. [Google Scholar] [CrossRef]
- Lomas, D.A.; Evans, D.L.; Finch, J.T.; Carrell, R.W. The Mechanism of Z Alpha 1-Antitrypsin Accumulation in the Liver. Nature 1992, 357, 605–607. [Google Scholar] [CrossRef]
- Stoller, J.K.; Aboussouan, L.S. A Review of A1-Antitrypsin Deficiency. Am. J. Respir. Crit. Care Med. 2012, 185, 246–259. [Google Scholar] [CrossRef]
- Santos, G.; Turner, A.M. Alpha-1 Antitrypsin Deficiency: An Update on Clinical Aspects of Diagnosis and Management. Fac. Rev. 2020, 9, 1. [Google Scholar] [CrossRef]
- Bouchecareilh, M. Alpha-1 Antitrypsin Deficiency-Mediated Liver Toxicity: Why Do Some Patients Do Poorly? What Do We Know So Far? Chronic Obstr. Pulm. Dis. J. COPD Found. 2020, 7, 172–181. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, L.R.; Lee, J.; Mohammad, N.S.; Aranyos, A.M.; Gould, C.; Khodayari, N.; Oshins, R.A.; Moneypenny, C.G.; Brantly, M.L. The Unfolded Protein Response to PI*Z Alpha-1 Antitrypsin in Human Hepatocellular and Murine Models. Hepatol. Commun. 2022, 6, 2354–2367. [Google Scholar] [CrossRef] [PubMed]
- Mela, M.; Smeeton, W.; Davies, S.E.; Miranda, E.; Scarpini, C.; Coleman, N.; Alexander, G.J.M. The Alpha-1 Antitrypsin Polymer Load Correlates With Hepatocyte Senescence, Fibrosis Stage and Liver-Related Mortality. Chronic Obstr. Pulm. Dis. Miami Fla 2020, 7, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Stoller, J.K.; Hupertz, V.; Aboussouan, L.S. Alpha-1 Antitrypsin Deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Topic, A.; Alempijevic, T.; Milutinovic, A.S.; Kovacevic, N. Alpha-1-Antitrypsin Phenotypes in Adult Liver Disease Patients. Ups. J. Med. Sci. 2009, 114, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Hamesch, K.; Mandorfer, M.; Pereira, V.M.; Moeller, L.S.; Pons, M.; Dolman, G.E.; Reichert, M.C.; Schneider, C.V.; Woditsch, V.; Voss, J.; et al. Liver Fibrosis and Metabolic Alterations in Adults With Alpha-1-Antitrypsin Deficiency Caused by the Pi*ZZ Mutation. Gastroenterology 2019, 157, 705–719.e18. [Google Scholar] [CrossRef] [PubMed]
- Khodayari, N.; Wang, R.L.; Oshins, R.; Lu, Y.; Millett, M.; Aranyos, A.M.; Mostofizadeh, S.; Scindia, Y.; Flagg, T.O.; Brantly, M. The Mechanism of Mitochondrial Injury in Alpha-1 Antitrypsin Deficiency Mediated Liver Disease. Int. J. Mol. Sci. 2021, 22, 13255. [Google Scholar] [CrossRef]
- Winther, S.V.; Ahmed, D.; Al-Shuweli, S.; Landt, E.M.; Nordestgaard, B.G.; Seersholm, N.; Dahl, M. Severe A1-Antitrypsin Deficiency Associated with Lower Blood Pressure and Reduced Risk of Ischemic Heart Disease: A Cohort Study of 91,540 Individuals and a Meta-Analysis. Respir. Res. 2022, 23, 55. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and Metabolic Disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Greenberg, A.S.; Obin, M.S. Obesity and the Role of Adipose Tissue in Inflammation and Metabolism. Am. J. Clin. Nutr. 2006, 83, 461S–465S. [Google Scholar] [CrossRef]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 Contributes to Macrophage Infiltration into Adipose Tissue, Insulin Resistance, and Hepatic Steatosis in Obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef]
- Weisberg, S.P.; Hunter, D.; Huber, R.; Lemieux, J.; Slaymaker, S.; Vaddi, K.; Charo, I.; Leibel, R.L.; Ferrante, A.W. CCR2 Modulates Inflammatory and Metabolic Effects of High-Fat Feeding. J. Clin. Investig. 2006, 116, 115–124. [Google Scholar] [CrossRef]
- Chakarov, S.; Lim, H.Y.; Tan, L.; Lim, S.Y.; See, P.; Lum, J.; Zhang, X.-M.; Foo, S.; Nakamizo, S.; Duan, K.; et al. Two Distinct Interstitial Macrophage Populations Coexist across Tissues in Specific Subtissular Niches. Science 2019, 363, eaau0964. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xu, M.M.; Wang, K.; Adler, A.J.; Vella, A.T.; Zhou, B. Macrophage Polarization and Metainflammation. Transl. Res. J. Lab. Clin. Med. 2018, 191, 29–44. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and Pathogenic Functions of Macrophage Subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Chawla, A. Alternative Macrophage Activation and Metabolism. Annu. Rev. Pathol. 2011, 6, 275–297. [Google Scholar] [CrossRef] [PubMed]
- Pradere, J.-P.; Kluwe, J.; De Minicis, S.; Jiao, J.-J.; Gwak, G.-Y.; Dapito, D.H.; Jang, M.-K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic Macrophages but Not Dendritic Cells Contribute to Liver Fibrosis by Promoting the Survival of Activated Hepatic Stellate Cells in Mice. Hepatol. Baltim. Md. 2013, 58, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Hammoutene, A.; Rautou, P.-E. Role of Liver Sinusoidal Endothelial Cells in Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2019, 70, 1278–1291. [Google Scholar] [CrossRef]
- Pigeolet, E.; Corbisier, P.; Houbion, A.; Lambert, D.; Michiels, C.; Raes, M.; Zachary, M.-D.; Remacle, J. Glutathione Peroxidase, Superoxide Dismutase, and Catalase Inactivation by Peroxides and Oxygen Derived Free Radicals. Mech. Ageing Dev. 1990, 51, 283–297. [Google Scholar] [CrossRef]
- Clare, K.; Dillon, J.F.; Brennan, P.N. Reactive Oxygen Species and Oxidative Stress in the Pathogenesis of MAFLD. J. Clin. Transl. Hepatol. 2022, 10, 939–946. [Google Scholar] [CrossRef]
- Song, M.J.; Malhi, H. The Unfolded Protein Response and Hepatic Lipid Metabolism in Non Alcoholic Fatty Liver Disease. Pharmacol. Ther. 2019, 203, 107401. [Google Scholar] [CrossRef]
- Febbraio, M.; Guy, E.; Coburn, C.; Knapp, F.F.; Beets, A.L.; Abumrad, N.A.; Silverstein, R.L. The Impact of Overexpression and Deficiency of Fatty Acid Translocase (FAT)/CD36. Mol. Cell. Biochem. 2002, 239, 193–197. [Google Scholar] [CrossRef]
- Wilson, C.G.; Tran, J.L.; Erion, D.M.; Vera, N.B.; Febbraio, M.; Weiss, E.J. Hepatocyte-Specific Disruption of CD36 Attenuates Fatty Liver and Improves Insulin Sensitivity in HFD-Fed Mice. Endocrinology 2016, 157, 570–585. [Google Scholar] [CrossRef] [PubMed]
- Rey, E.; del Pozo-Maroto, E.; Marañón, P.; Beeler, B.; García-García, Y.; Landete, P.; Isaza, S.C.; Farré, R.; García-Monzón, C.; Almendros, I.; et al. Intrahepatic Expression of Fatty Acid Translocase CD36 Is Increased in Obstructive Sleep Apnea. Front. Med. 2020, 7, 450. [Google Scholar] [CrossRef] [PubMed]
- Carroll, T.P.; Greene, C.M.; O’Connor, C.A.; Nolan, A.M.; O’Neill, S.J.; McElvaney, N.G. Evidence for Unfolded Protein Response Activation in Monocytes from Individuals with Alpha-1 Antitrypsin Deficiency. J. Immunol. Baltim. Md 1950 2010, 184, 4538–4546. [Google Scholar] [CrossRef]
- Bazzan, E.; Tinè, M.; Biondini, D.; Benetti, R.; Baraldo, S.; Turato, G.; Fagiuoli, S.; Sonzogni, A.; Rigobello, C.; Rea, F.; et al. A1-Antitrypsin Polymerizes in Alveolar Macrophages of Smokers With and Without A1-Antitrypsin Deficiency. Chest 2018, 154, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Belchamber, K.B.R.; Walker, E.M.; Stockley, R.A.; Sapey, E. Monocytes and Macrophages in Alpha-1 Antitrypsin Deficiency. Int. J. Chron. Obstruct. Pulmon. Dis. 2020, 15, 3183–3192. [Google Scholar] [CrossRef] [PubMed]
- Khodayari, N.; Oshins, R.; Aranyos, A.M.; Duarte, S.; Mostofizadeh, S.; Lu, Y.; Brantly, M. Characterization of Hepatic Inflammatory Changes in a C57BL/6J Mouse Model of Alpha1-Antitrypsin Deficiency. Am. J. Physiol.-Gastrointest. Liver Physiol. 2022, 323, G594–G608. [Google Scholar] [CrossRef]
- Daemen, S.; Gainullina, A.; Kalugotla, G.; He, L.; Chan, M.M.; Beals, J.W.; Liss, K.H.; Klein, S.; Feldstein, A.E.; Finck, B.N.; et al. Dynamic Shifts in the Composition of Resident and Recruited Macrophages Influence Tissue Remodeling in NASH. Cell Rep. 2021, 34, 108626. [Google Scholar] [CrossRef]
- Pott, G.B.; Chan, E.D.; Dinarello, C.A.; Shapiro, L. Alpha-1-Antitrypsin Is an Endogenous Inhibitor of Proinflammatory Cytokine Production in Whole Blood. J. Leukoc. Biol. 2009, 85, 886–895. [Google Scholar] [CrossRef]
- Lee, J.; Mohammad, N.; Lu, Y.; Kang, K.; Han, K.; Brantly, M. Alu RNA Induces NLRP3 Expression through TLR7 Activation in α-1-Antitrypsin-Deficient Macrophages. JCI Insight 2022, 7, e158791. [Google Scholar] [CrossRef]
- Callea, F.; Francalanci, P.; Giovannoni, I. Hepatic and Extrahepatic Sources and Manifestations in Endoplasmic Reticulum Storage Diseases. Int. J. Mol. Sci. 2021, 22, 5778. [Google Scholar] [CrossRef]
- Anand, P.K. Lipids, Inflammasomes, Metabolism, and Disease. Immunol. Rev. 2020, 297, 108–122. [Google Scholar] [CrossRef]
- Krahmer, N.; Farese, R.V.; Walther, T.C. Balancing the Fat: Lipid Droplets and Human Disease. EMBO Mol. Med. 2013, 5, 905–915. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD). European Association for the Study of Obesity (EASO) EASL-EASD-EASO Clinical Practice Guidelines for the Management of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef] [PubMed]
- Tulenko, T. The Physiology of Lipoproteins. J. Nucl. Cardiol. 2002, 9, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Diffenderfer, M.R.; Schaefer, E.J. The Composition and Metabolism of Large and Small LDL. Curr. Opin. Lipidol. 2014, 25, 221–226. [Google Scholar] [CrossRef]
- Olofsson, S.O.; Stillemark-Billton, P.; Asp, L. Intracellular Assembly of VLDL: Two Major Steps in Separate Cell Compartments. Trends Cardiovasc. Med. 2000, 10, 338–345. [Google Scholar] [CrossRef]
- Stillemark-Billton, P.; Beck, C.; Borén, J.; Olofsson, S.-O. Relation of the Size and Intracellular Sorting of ApoB to the Formation of VLDL 1 and VLDL 2. J. Lipid Res. 2005, 46, 104–114. [Google Scholar] [CrossRef]
- Björkegren, J.; Beigneux, A.; Bergo, M.O.; Maher, J.J.; Young, S.G. Blocking the Secretion of Hepatic Very Low Density Lipoproteins Renders the Liver More Susceptible to Toxin-Induced Injury. J. Biol. Chem. 2002, 277, 5476–5483. [Google Scholar] [CrossRef]
- Adiels, M.; Olofsson, S.-O.; Taskinen, M.-R.; Borén, J. Overproduction of Very Low-Density Lipoproteins Is the Hallmark of the Dyslipidemia in the Metabolic Syndrome. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1225–1236. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Seth, D.; Day, C.P. Genetic Factors That Affect Risk of Alcoholic and Nonalcoholic Fatty Liver Disease. Gastroenterology 2016, 150, 1728–1744.e7. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic Variation in PNPLA3 Confers Susceptibility to Nonalcoholic Fatty Liver Disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Borén, J.; Adiels, M.; Björnson, E.; Matikainen, N.; Söderlund, S.; Rämö, J.; Ståhlman, M.; Ripatti, P.; Ripatti, S.; Palotie, A.; et al. Effects of TM6SF2 E167K on Hepatic Lipid and Very Low-Density Lipoprotein Metabolism in Humans. JCI Insight 2020, 5, e144079. [Google Scholar] [CrossRef] [PubMed]
- Borén, J.; Adiels, M.; Björnson, E.; Matikainen, N.; Söderlund, S.; Rämö, J.; Henricsson, M.; Ripatti, P.; Ripatti, S.; Palotie, A.; et al. Effects of PNPLA3 I148M on Hepatic Lipid and Very-Low-Density Lipoprotein Metabolism in Humans. J. Intern. Med. 2022, 291, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Prill, S.; Caddeo, A.; Baselli, G.; Jamialahmadi, O.; Dongiovanni, P.; Rametta, R.; Kanebratt, K.P.; Pujia, A.; Pingitore, P.; Mancina, R.M.; et al. The TM6SF2 E167K Genetic Variant Induces Lipid Biosynthesis and Reduces Apolipoprotein B Secretion in Human Hepatic 3D Spheroids. Sci. Rep. 2019, 9, 11585. [Google Scholar] [CrossRef] [PubMed]
- Obermayer, G.; Afonyushkin, T.; Binder, C.J. Oxidized Low-Density Lipoprotein in Inflammation-Driven Thrombosis. J. Thromb. Haemost. 2018, 16, 418–428. [Google Scholar] [CrossRef]
- Kunjathoor, V.V.; Febbraio, M.; Podrez, E.A.; Moore, K.J.; Andersson, L.; Koehn, S.; Rhee, J.S.; Silverstein, R.; Hoff, H.F.; Freeman, M.W. Scavenger Receptors Class A-I/II and CD36 Are the Principal Receptors Responsible for the Uptake of Modified Low Density Lipoprotein Leading to Lipid Loading in Macrophages. J. Biol. Chem. 2002, 277, 49982–49988. [Google Scholar] [CrossRef]
- Florance, I.; Ramasubbu, S. Current Understanding on the Role of Lipids in Macrophages and Associated Diseases. Int. J. Mol. Sci. 2022, 24, 589. [Google Scholar] [CrossRef]
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R.; et al. Platelet CD36 Links Hyperlipidemia, Oxidant Stress and a Prothrombotic Phenotype. Nat. Med. 2007, 13, 1086–1095. [Google Scholar] [CrossRef]
- He, J.; Lee, J.H.; Febbraio, M.; Xie, W. The Emerging Roles of Fatty Acid Translocase/CD36 and the Aryl Hydrocarbon Receptor in Fatty Liver Disease. Exp. Biol. Med. Maywood NJ 2011, 236, 1116–1121. [Google Scholar] [CrossRef]
- Bonen, A.; Chabowski, A.; Luiken, J.J.F.P.; Glatz, J.F.C. Is Membrane Transport of FFA Mediated by Lipid, Protein, or Both? Mechanisms and Regulation of Protein-Mediated Cellular Fatty Acid Uptake: Molecular, Biochemical, and Physiological Evidence. Physiol. Bethesda Md 2007, 22, 15–29. [Google Scholar] [CrossRef]
- Devereux, C.J.; Bayliss, J.; Keenan, S.N.; Montgomery, M.K.; Watt, M.J. Investigating Dual Inhibition of ACC and CD36 for the Treatment of Non-Alcoholic Fatty Liver Disease in Mice. Am. J. Physiol. Endocrinol. Metab. 2023, 324, E187–E198. [Google Scholar] [CrossRef] [PubMed]
- Samovski, D.; Abumrad, N.A. Regulation of Lipophagy in NAFLD by Cellular Metabolism and CD36. J. Lipid Res. 2019, 60, 755–757. [Google Scholar] [CrossRef]
- Feng, Y.; Sun, W.; Sun, F.; Yin, G.; Liang, P.; Chen, S.; Liu, X.; Jiang, T.; Zhang, F. Biological Mechanisms and Related Natural Inhibitors of CD36 in Nonalcoholic Fatty Liver. Drug Des. Devel. Ther. 2022, 16, 3829–3845. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, P.; Zhao, L.; Chen, Y.; Zhang, X.; Zeng, S.; Wei, L.; Varghese, Z.; Moorhead, J.F.; Chen, Y.; et al. CD36 Plays a Negative Role in the Regulation of Lipophagy in Hepatocytes through an AMPK-Dependent Pathway. J. Lipid Res. 2019, 60, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Ferrarotti, I.; Corsico, A.G.; Stolk, J.; Ottaviani, S.; Fumagalli, M.; Janciauskiene, S.; Iadarola, P. Advances in Identifying Urine/Serum Biomarkers in Alpha-1 Antitrypsin Deficiency for More Personalized Future Treatment Strategies. COPD 2017, 14, 56–65. [Google Scholar] [CrossRef]
- Siebers, K.; Fink, B.; Zakrzewicz, A.; Agné, A.; Richter, K.; Konzok, S.; Hecker, A.; Zukunft, S.; Küllmar, M.; Klein, J.; et al. Alpha-1 Antitrypsin Inhibits ATP-Mediated Release of Interleukin-1β via CD36 and Nicotinic Acetylcholine Receptors. Front. Immunol. 2018, 9, 877. [Google Scholar] [CrossRef]
- Nati, M.; Haddad, D.; Birkenfeld, A.L.; Koch, C.A.; Chavakis, T.; Chatzigeorgiou, A. The Role of Immune Cells in Metabolism-Related Liver Inflammation and Development of Non-Alcoholic Steatohepatitis (NASH). Rev. Endocr. Metab. Disord. 2016, 17, 29–39. [Google Scholar] [CrossRef]
- Moon, S.W.; Kim, S.Y.; Jung, J.Y.; Kang, Y.A.; Park, M.S.; Kim, Y.S.; Chang, J.; Ro, J.S.; Lee, Y.-H.; Lee, S.H. Relationship between Obstructive Lung Disease and Non-Alcoholic Fatty Liver Disease in the Korean Population: Korea National Health and Nutrition Examination Survey, 2007–2010. Int. J. Chron. Obstruct. Pulmon. Dis. 2018, 13, 2603–2611. [Google Scholar] [CrossRef]
- Viglino, D.; Plazanet, A.; Bailly, S.; Benmerad, M.; Jullian-Desayes, I.; Tamisier, R.; Leroy, V.; Zarski, J.-P.; Maignan, M.; Joyeux-Faure, M.; et al. Impact of Non-Alcoholic Fatty Liver Disease on Long-Term Cardiovascular Events and Death in Chronic Obstructive Pulmonary Disease. Sci. Rep. 2018, 8, 16559. [Google Scholar] [CrossRef]
- Viglino, D.; Jullian-Desayes, I.; Minoves, M.; Aron-Wisnewsky, J.; Leroy, V.; Zarski, J.-P.; Tamisier, R.; Joyeux-Faure, M.; Pépin, J.-L. Nonalcoholic Fatty Liver Disease in Chronic Obstructive Pulmonary Disease. Eur. Respir. J. 2017, 49, 1601923. [Google Scholar] [CrossRef]
- Rodríguez-Roisin, R.; Krowka, M.J. Hepatopulmonary Syndrome--a Liver-Induced Lung Vascular Disorder. N. Engl. J. Med. 2008, 358, 2378–2387. [Google Scholar] [CrossRef] [PubMed]
- Cheeney, G.; Pac, L.J.; Gopal, P.; Landis, C.S.; Konnick, E.Q.; Swanson, P.E.; Greene, D.N.; Lockwood, C.M.; Westerhoff, M. Increased Frequency of Heterozygous Alpha-1-Antitrypsin Deficiency in Liver Explants From Nonalcoholic Steatohepatitis Patients. Liver Transpl. 2020, 26, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Lockett, A.D.; Kimani, S.; Ddungu, G.; Wrenger, S.; Tuder, R.M.; Janciauskiene, S.M.; Petrache, I. A₁-Antitrypsin Modulates Lung Endothelial Cell Inflammatory Responses to TNF-α. Am. J. Respir. Cell Mol. Biol. 2013, 49, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Madyaningrana, K.; Vijayan, V.; Nikolin, C.; Aljabri, A.; Tumpara, S.; Korenbaum, E.; Shah, H.; Stankov, M.; Fuchs, H.; Janciauskiene, S.; et al. Alpha1-Antitrypsin Counteracts Heme-Induced Endothelial Cell Inflammatory Activation, Autophagy Dysfunction and Death. Redox Biol. 2021, 46, 102060. [Google Scholar] [CrossRef]
- Nakagiri, T.; Wrenger, S.; Sivaraman, K.; Ius, F.; Goecke, T.; Zardo, P.; Grau, V.; Welte, T.; Haverich, A.; Knöfel, A.-K.; et al. A1-Antitrypsin Attenuates Acute Rejection of Orthotopic Murine Lung Allografts. Respir. Res. 2021, 22, 295. [Google Scholar] [CrossRef]
- Jedicke, N.; Struever, N.; Aggrawal, N.; Welte, T.; Manns, M.P.; Malek, N.P.; Zender, L.; Janciauskiene, S.; Wuestefeld, T. α-1-Antitrypsin Inhibits Acute Liver Failure in Mice. Hepatol. Baltim. Md. 2014, 59, 2299–2308. [Google Scholar] [CrossRef]
- Grander, C.; Schaefer, B.; Schwärzler, J.; Grabherr, F.; de Graaf, D.M.; Enrich, B.; Oberhuber, G.; Mayr, L.; Sangineto, M.; Jaschke, N.; et al. Alpha-1 Antitrypsin Governs Alcohol-Related Liver Disease in Mice and Humans. Gut 2021, 70, 585–594. [Google Scholar] [CrossRef]
- Cho, J.-H.; Ryu, H.-M.; Oh, E.-J.; Yook, J.-M.; Ahn, J.-S.; Jung, H.-Y.; Choi, J.-Y.; Park, S.-H.; Kim, Y.-L.; Kwak, I.S.; et al. Alpha1-Antitrypsin Attenuates Renal Fibrosis by Inhibiting TGF-Β1-Induced Epithelial Mesenchymal Transition. PLoS ONE 2016, 11, e0162186. [Google Scholar] [CrossRef]
- Kalis, M.; Kumar, R.; Janciauskiene, S.; Salehi, A.; Cilio, C.M. α 1-Antitrypsin Enhances Insulin Secretion and Prevents Cytokine-Mediated Apoptosis in Pancreatic β-Cells. Islets 2010, 2, 185–189. [Google Scholar] [CrossRef]
- Abiru, S.; Migita, K.; Maeda, Y.; Daikoku, M.; Ito, M.; Ohata, K.; Nagaoka, S.; Matsumoto, T.; Takii, Y.; Kusumoto, K.; et al. Serum Cytokine and Soluble Cytokine Receptor Levels in Patients with Non-Alcoholic Steatohepatitis. Liver Int. Off. J. Int. Assoc. Study Liver 2006, 26, 39–45. [Google Scholar] [CrossRef]
- Schutgens, F.; Clevers, H. Human Organoids: Tools for Understanding Biology and Treating Diseases. Annu. Rev. Pathol. 2020, 15, 211–234. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Huch, M. Disease Modelling in Human Organoids. Dis. Model. Mech. 2019, 12, dmm039347. [Google Scholar] [CrossRef] [PubMed]
- Marsee, A.; Roos, F.J.M.; Verstegen, M.M.A.; Gehart, H.; de Koning, E.; Lemaigre, F.; Forbes, S.J.; Peng, W.C.; Huch, M.; Takebe, T.; et al. Building Consensus on Definition and Nomenclature of Hepatic, Pancreatic, and Biliary Organoids. Cell Stem Cell 2021, 28, 816–832. [Google Scholar] [CrossRef] [PubMed]
- Mayhew, C.N.; Singhania, R. A Review of Protocols for Brain Organoids and Applications for Disease Modeling. STAR Protoc. 2022, 4, 101860. [Google Scholar] [CrossRef] [PubMed]
- Yanakiev, M.; Soper, O.; Berg, D.A.; Kang, E. Modelling Alzheimer’s Disease Using Human Brain Organoids: Current Progress and Challenges. Expert Rev. Mol. Med. 2022, 25, e3. [Google Scholar] [CrossRef]
- Harada, K.; Sakamoto, N. Cancer Organoid Applications to Investigate Chemotherapy Resistance. Front. Mol. Biosci. 2022, 9, 1067207. [Google Scholar] [CrossRef]
- Riedel, N.C.; de Faria, F.W.; Alfert, A.; Bruder, J.M.; Kerl, K. Three-Dimensional Cell Culture Systems in Pediatric and Adult Brain Tumor Precision Medicine. Cancers 2022, 14, 5972. [Google Scholar] [CrossRef]
- Chen, J.; Na, F. Organoid Technology and Applications in Lung Diseases: Models, Mechanism Research and Therapy Opportunities. Front. Bioeng. Biotechnol. 2022, 10, 1066869. [Google Scholar] [CrossRef]
- Silva, I.A.L.; Laselva, O.; Lopes-Pacheco, M. Advances in Preclinical In Vitro Models for the Translation of Precision Medicine for Cystic Fibrosis. J. Pers. Med. 2022, 12, 1321. [Google Scholar] [CrossRef]
- Peng, L.; Gao, L.; Wu, X.; Fan, Y.; Liu, M.; Chen, J.; Song, J.; Kong, J.; Dong, Y.; Li, B.; et al. Lung Organoids as Model to Study SARS-CoV-2 Infection. Cells 2022, 11, 2758. [Google Scholar] [CrossRef]
- Nuciforo, S.; Heim, M.H. Organoids to Model Liver Disease. JHEP Rep. Innov. Hepatol. 2021, 3, 100198. [Google Scholar] [CrossRef]
- Ramli, M.N.B.; Lim, Y.S.; Koe, C.T.; Demircioglu, D.; Tng, W.; Gonzales, K.A.U.; Tan, C.P.; Szczerbinska, I.; Liang, H.; Soe, E.L.; et al. Human Pluripotent Stem Cell-Derived Organoids as Models of Liver Disease. Gastroenterology 2020, 159, 1471–1486.e12. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K.; Bowen, W.C.; Mulè, K.; Stolz, D.B. Histological Organization in Hepatocyte Organoid Cultures. Am. J. Pathol. 2001, 159, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Dorrell, C.; Boj, S.F.; van Es, J.H.; Li, V.S.W.; van de Wetering, M.; Sato, T.; Hamer, K.; Sasaki, N.; Finegold, M.J.; et al. In Vitro Expansion of Single Lgr5+ Liver Stem Cells Induced by Wnt-Driven Regeneration. Nature 2013, 494, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.-R.; Ueno, Y.; Zheng, Y.-W.; Koike, N.; et al. Vascularized and Functional Human Liver from an IPSC-Derived Organ Bud Transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef]
- Prior, N.; Inacio, P.; Huch, M. Liver Organoids: From Basic Research to Therapeutic Applications. Gut 2019, 68, 2228–2237. [Google Scholar] [CrossRef]
- Gómez-Mariano, G.; Matamala, N.; Martínez, S.; Justo, I.; Marcacuzco, A.; Jimenez, C.; Monzón, S.; Cuesta, I.; Garfia, C.; Martínez, M.T.; et al. Liver Organoids Reproduce Alpha-1 Antitrypsin Deficiency-Related Liver Disease. Hepatol. Int. 2020, 14, 127–137. [Google Scholar] [CrossRef]
- Huch, M.; Gehart, H.; van Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.A.; Ellis, E.; van Wenum, M.; Fuchs, S.A.; de Ligt, J.; et al. Long-Term Culture of Genome-Stable Bipotent Stem Cells from Adult Human Liver. Cell 2015, 160, 299–312. [Google Scholar] [CrossRef]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The Liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Deng, P.; Tao, T.; Liu, H.; Wu, S.; Chen, W.; Qin, J. Modeling Human Nonalcoholic Fatty Liver Disease (NAFLD) with an Organoids-on-a-Chip System. ACS Biomater. Sci. Eng. 2020, 6, 5734–5743. [Google Scholar] [CrossRef]
- Carter, J.K.; Friedman, S.L. Hepatic Stellate Cell-Immune Interactions in NASH. Front. Endocrinol. 2022, 13, 867940. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Yu, H.; Li, Q.-Y.; Wei, Y.-T.; Fu, J.; Dong, H.; Cao, D.; Guo, L.-N.; Chen, L.; Yang, Y.; et al. Hepatocyte-Derived VEGFA Accelerates the Progression of Non-Alcoholic Fatty Liver Disease to Hepatocellular Carcinoma via Activating Hepatic Stellate Cells. Acta Pharmacol. Sin. 2022, 43, 2917–2928. [Google Scholar] [CrossRef]
- Pingitore, P.; Sasidharan, K.; Ekstrand, M.; Prill, S.; Lindén, D.; Romeo, S. Human Multilineage 3D Spheroids as a Model of Liver Steatosis and Fibrosis. Int. J. Mol. Sci. 2019, 20, 1629. [Google Scholar] [CrossRef] [PubMed]
- Gwag, T.; Ma, E.; Zhou, C.; Wang, S. Anti-CD47 Antibody Treatment Attenuates Liver Inflammation and Fibrosis in Experimental Non-Alcoholic Steatohepatitis Models. Liver Int. Off. J. Int. Assoc. Study Liver 2022, 42, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Sendi, H.; Mead, I.; Wan, M.; Mehrab-Mohseni, M.; Koch, K.; Atala, A.; Bonkovsky, H.L.; Bishop, C.E. MiR-122 Inhibition in a Human Liver Organoid Model Leads to Liver Inflammation, Necrosis, Steatofibrosis and Dysregulated Insulin Signaling. PLoS ONE 2018, 13, e0200847. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, R.; Togo, S.; Kimura, M.; Shinozawa, T.; Koido, M.; Koike, H.; Thompson, W.; Karns, R.A.; Mayhew, C.N.; McGrath, P.S.; et al. Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids. Cell Metab. 2019, 30, 374–384.e6. [Google Scholar] [CrossRef] [PubMed]
- Collin de l’Hortet, A.; Takeishi, K.; Guzman-Lepe, J.; Morita, K.; Achreja, A.; Popovic, B.; Wang, Y.; Handa, K.; Mittal, A.; Meurs, N.; et al. Generation of Human Fatty Livers Using Custom-Engineered Induced Pluripotent Stem Cells with Modifiable SIRT1 Metabolism. Cell Metab. 2019, 30, 385–401.e9. [Google Scholar] [CrossRef]
- Uygun, B.E.; Soto-Gutierrez, A.; Yagi, H.; Izamis, M.-L.; Guzzardi, M.A.; Shulman, C.; Milwid, J.; Kobayashi, N.; Tilles, A.; Berthiaume, F.; et al. Organ Reengineering through Development of a Transplantable Recellularized Liver Graft Using Decellularized Liver Matrix. Nat. Med. 2010, 16, 814–820. [Google Scholar] [CrossRef]
- Park, Y.; Thadasina, D.; Bolujo, I.; Isidan, A.; Cross-Najafi, A.A.; Lopez, K.; Li, P.; Dahlem, A.M.; Kennedy, L.; Sato, K.; et al. Three-Dimensional Organoids as a Model to Study Nonalcoholic Fatty Liver Disease. Semin. Liver Dis. 2022, 42, 423–433. [Google Scholar] [CrossRef]
- Anant, S.; Davidson, N.O. Identification and Regulation of Protein Components of the Apolipoprotein B MRNA Editing Enzyme. A Complex Event. Trends Cardiovasc. Med. 2002, 12, 311–317. [Google Scholar] [CrossRef]
- Schneider, C.V.; Hamesch, K.; Gross, A.; Mandorfer, M.; Moeller, L.S.; Pereira, V.; Pons, M.; Kuca, P.; Reichert, M.C.; Benini, F.; et al. Liver Phenotypes of European Adults Heterozygous or Homozygous for Pi∗Z Variant of AAT (Pi∗MZ vs Pi∗ZZ Genotype) and Noncarriers. Gastroenterology 2020, 159, 534–548.e11. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).