
Clinicopathological Outlines of Post-COVID-19 Pulmonary Fibrosis Compared with Idiopathic Pulmonary Fibrosis
 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Materials and Methods
3. Risk Factors
4. Clinical Aspects
5. Pulmonary Function Tests
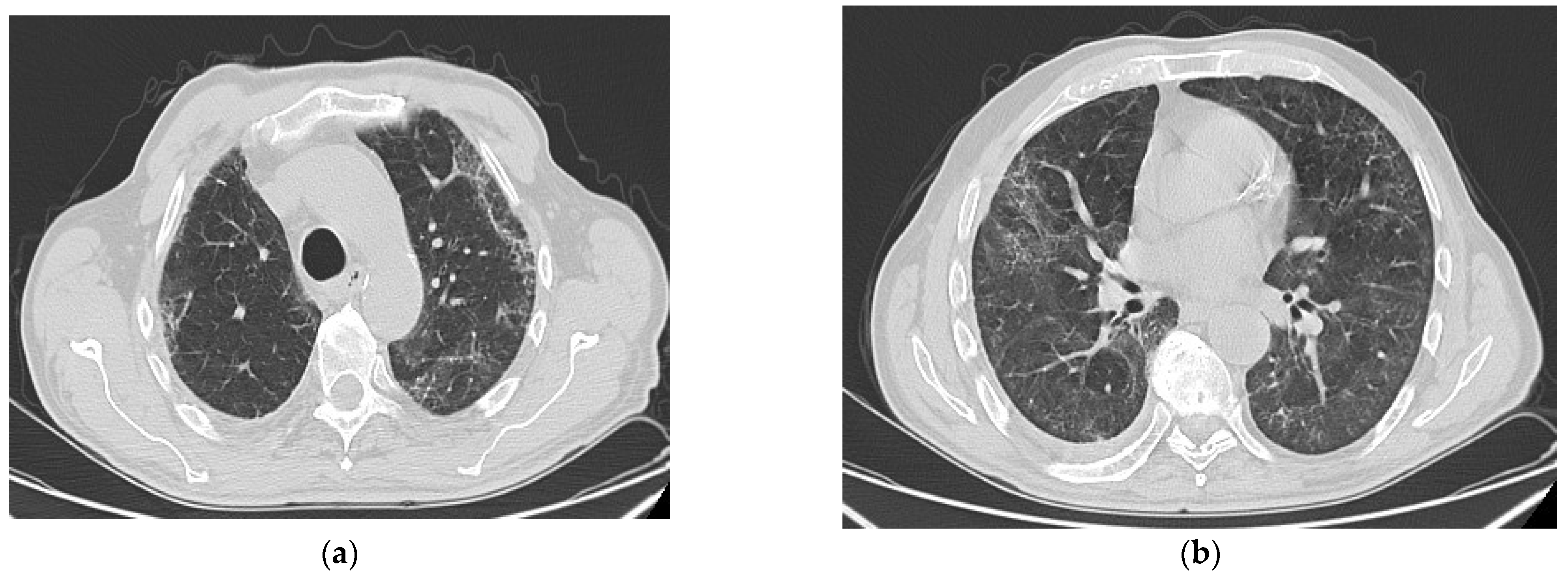
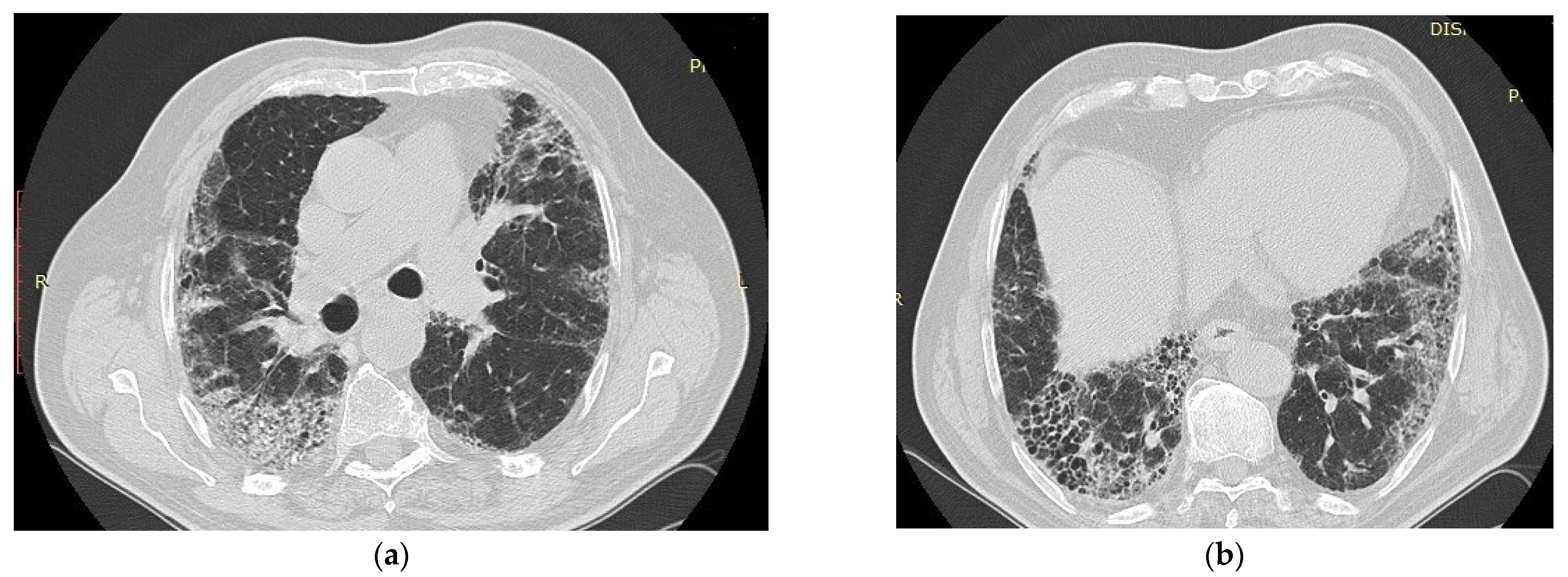
6. Radiologic Aspects
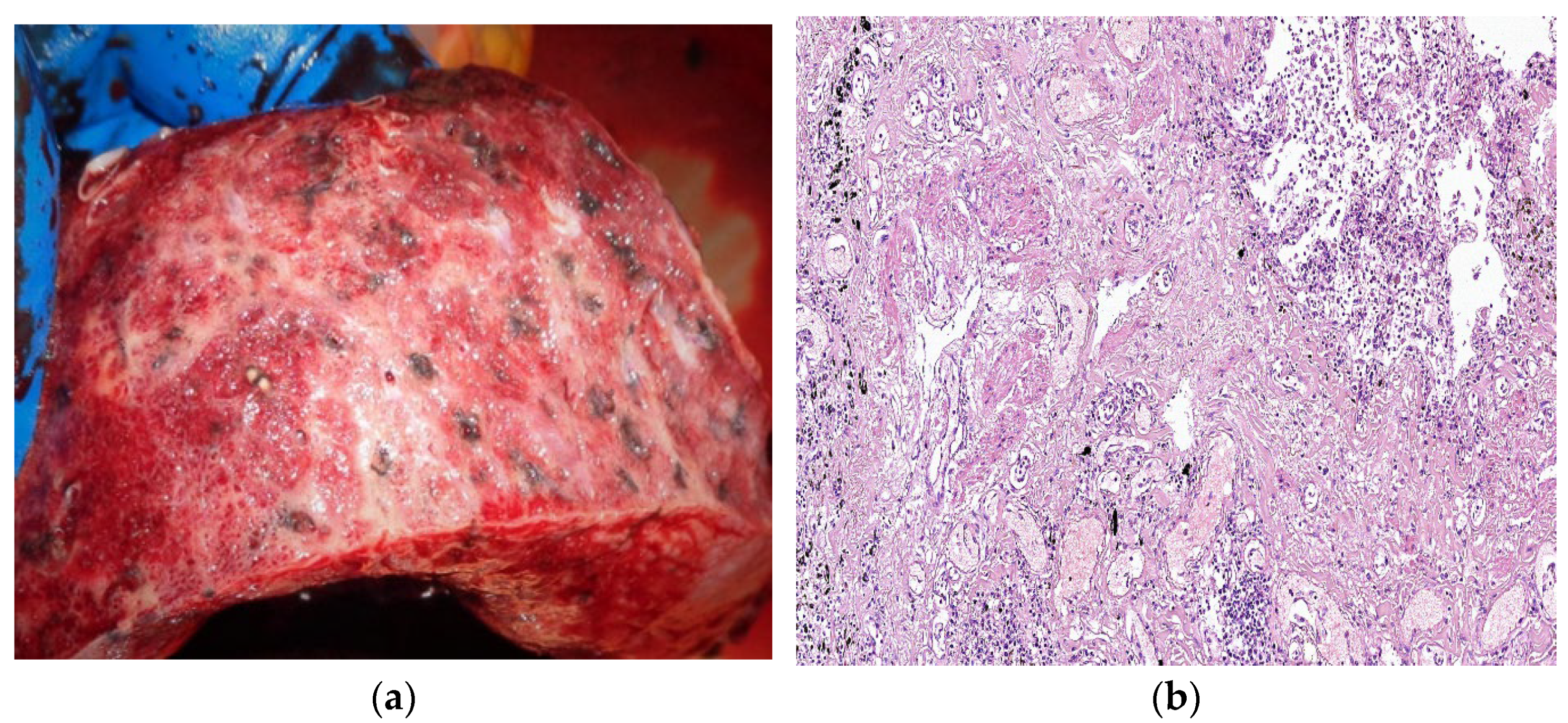
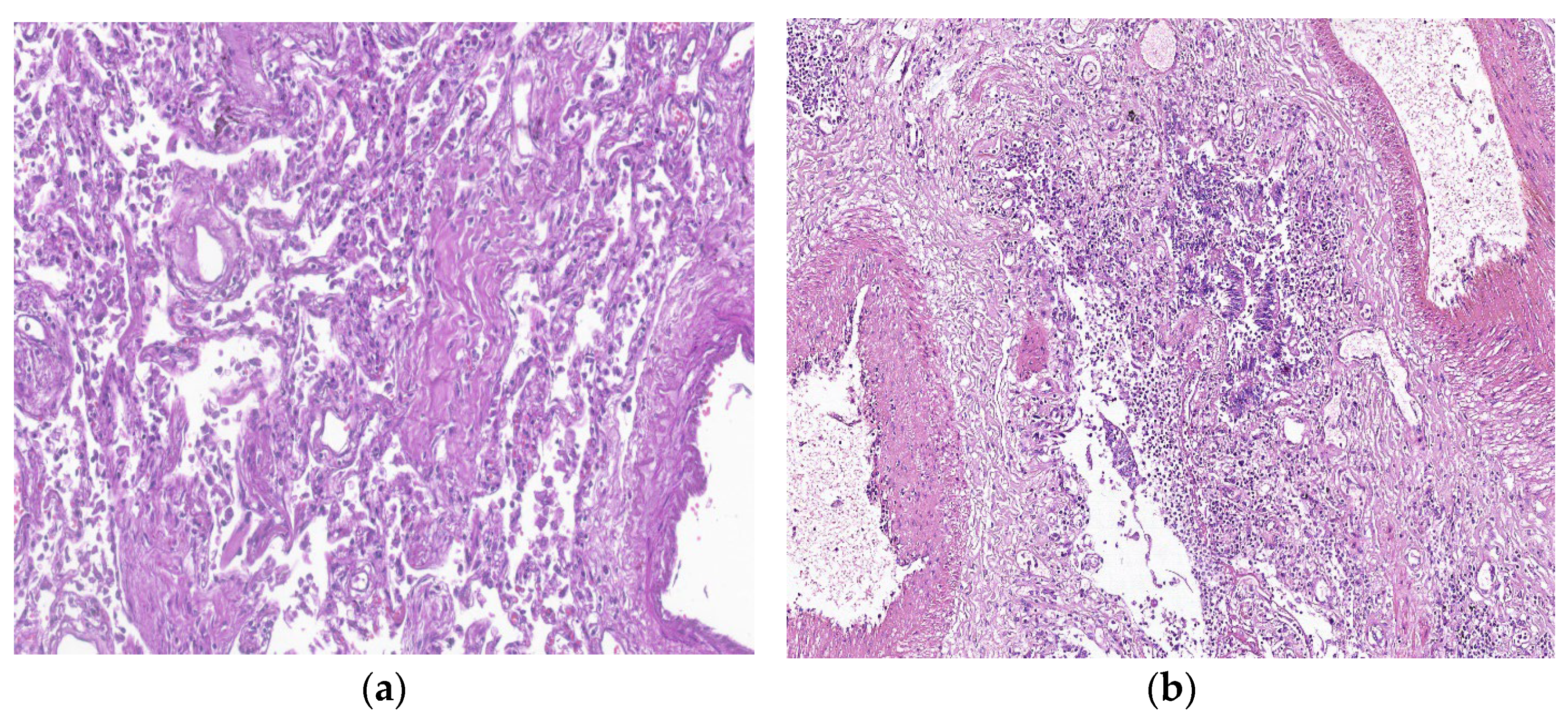
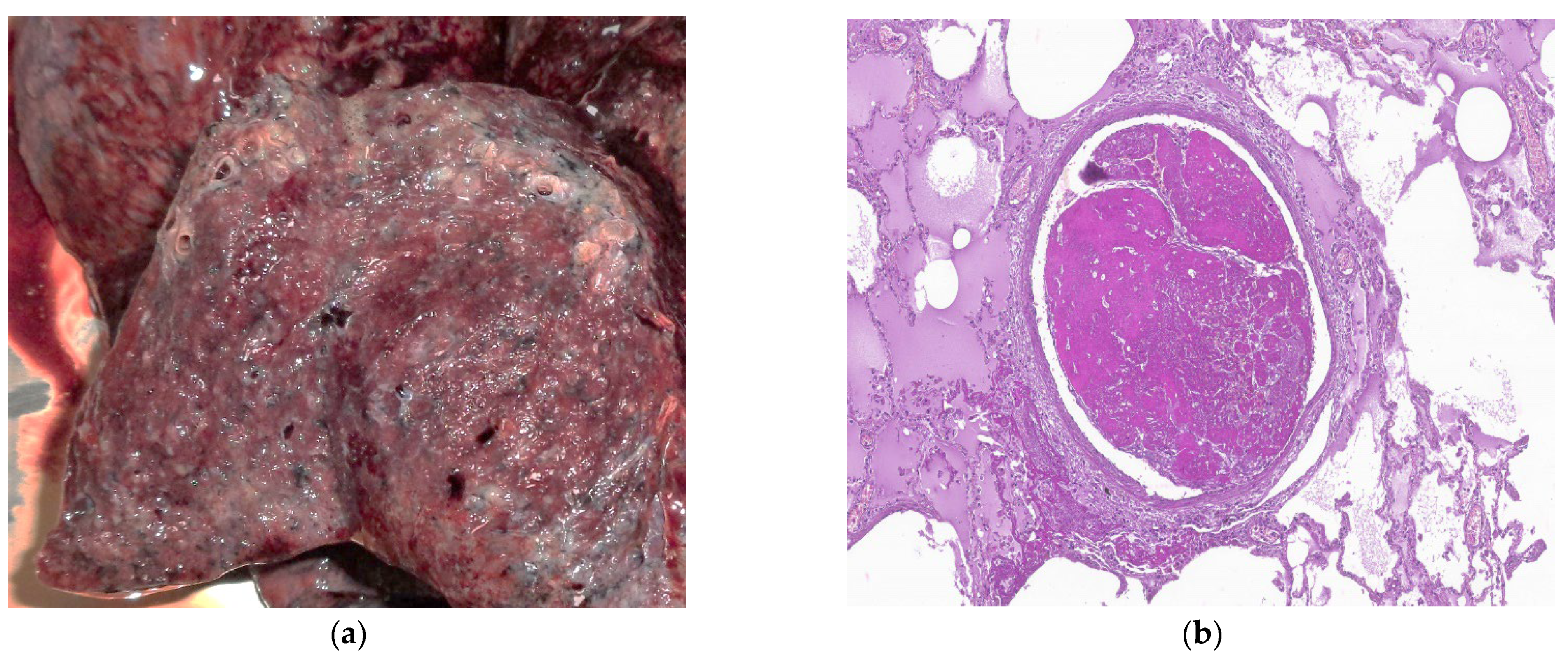
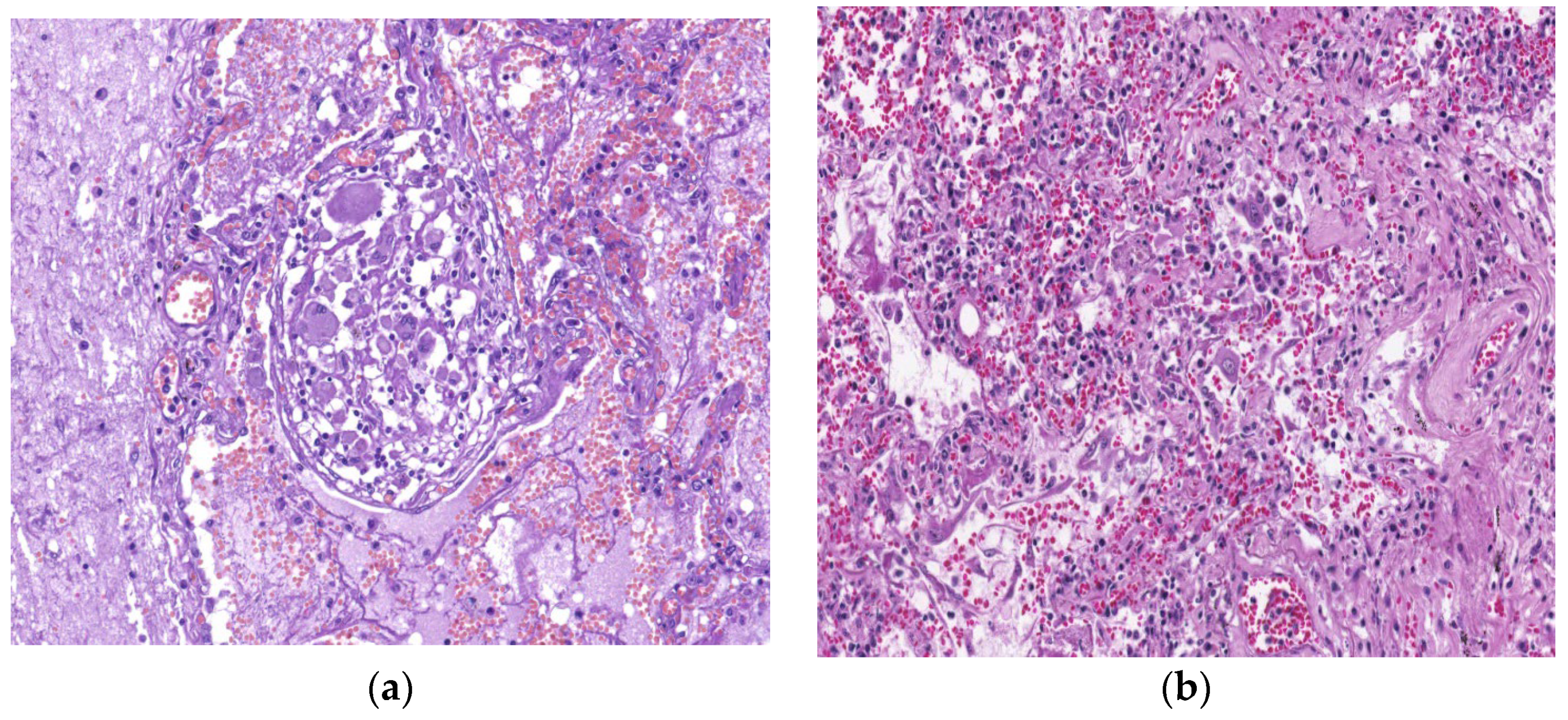
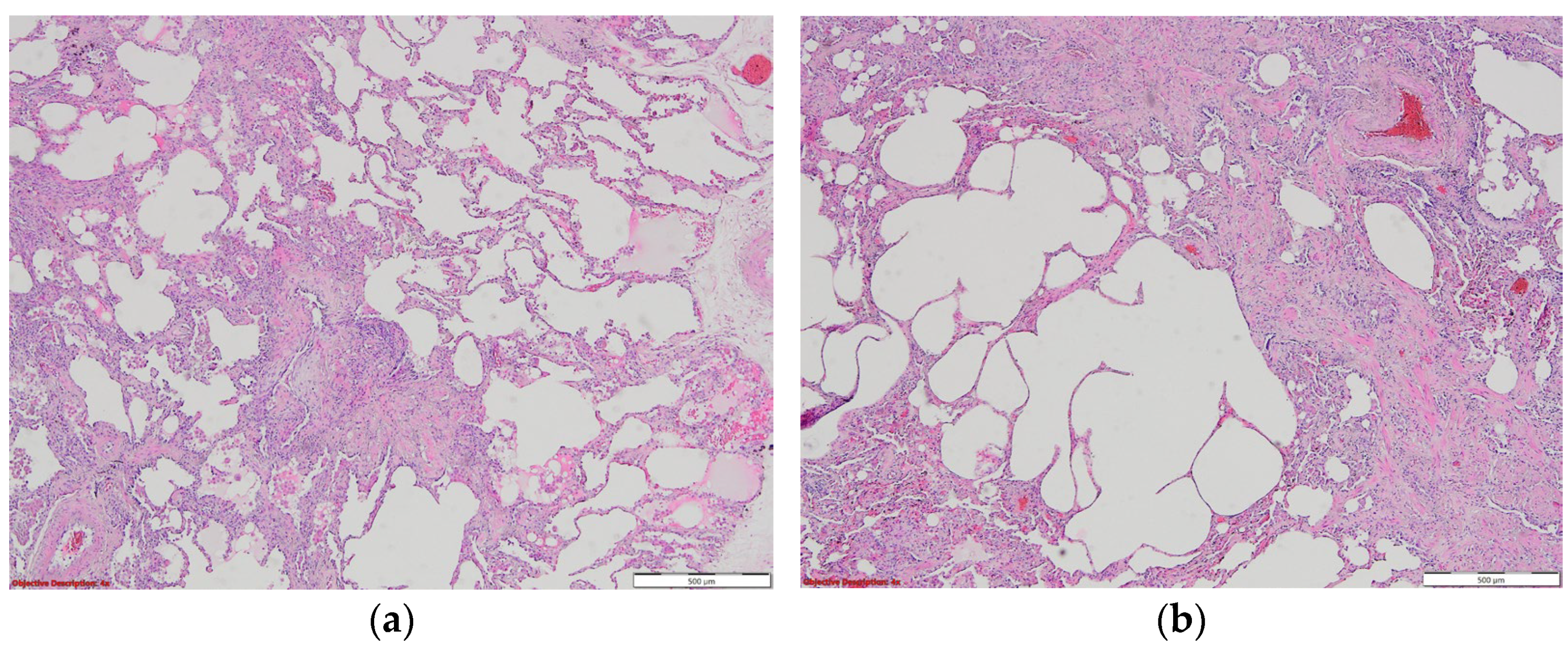
7. Histopathologic Characterization
8. Therapeutic Perspectives
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A.D.; et al. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J. Pathol. 2020, 251, 228–248. [Google Scholar] [CrossRef]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Ioannidis, J.P.A. Infection fatality rate of COVID-19 inferred from seroprevalence data. Bull. World Health Organ. 2021, 99, 19–33F. [Google Scholar] [CrossRef]
- Rai, D.K.; Sharma, P.; Kumar, R. Post COVID-19 pulmonary fibrosis. Is it real threat? Indian J. Tuberc. 2021, 68, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Cao, Y.; Jiang, N.; Chen, Y.; Alwalid, O.; Zhang, X.; Gu, J.; Dai, M.; Liu, J.; Zhu, W.; et al. Novel Coronavirus Disease 2019 (COVID-19) Pneumonia Progression Course in 17 Discharged Patients: Comparison of Clinical and Thin-Section Computed Tomography Features During Recovery. Clin. Infect. Dis. 2020, 71, 723–731. [Google Scholar] [CrossRef]
- Wang, Y.; Dong, C.; Hu, Y.; Li, C.; Ren, Q.; Zhang, X.; Shi, H.; Zhou, M. Temporal Changes of CT Findings in 90 Patients with COVID-19 Pneumonia: A Longitudinal Study. Radiology 2020, 296, E55–E64. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, W.; Pan, F.; Li, L.; Yang, L.; Zheng, D.; Wang, J.; Liang, B. The pulmonary sequelae in discharged patients with COVID-19: A short-term observational study. Respir. Res. 2020, 21, 125. [Google Scholar] [CrossRef] [PubMed]
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: A two-centre descriptive study. Lancet Infect. Dis. 2020, 20, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Lechowicz, K.; Drozdzal, S.; Machaj, F.; Rosik, J.; Szosta, B.; Zegan-Baranska, M.; Biernawska, J.; Dabrowski, W.; Rotter, I.; Kotfis, K. COVID-19: The potential treatment of pulmonary fibrosis associated with SARS-CoV-2 infection. J. Clin. Med. 2020, 9, 1917. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Fan, Y.; Alwalid, O.; Li, N.; Jia, X.; Yuan, M.; Li, Y.; Cao, Y.; Gu, J.; Wu, H.; et al. Six-month Follow-up Chest CT Findings after Severe COVID-19 Pneumonia. Radiology 2021, 299, E177–E186. [Google Scholar] [CrossRef]
- Krishna, R.; Chapman, K.; Ullah, S. Idiopathic Pulmonary Fibrosis; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Kawabata, Y. Pathology of IPF. In Idiopathic Pulmonary Fibrosis; Nakamura, H., Aoshiba, K., Eds.; Springer: Tokyo, Japan, 2016. [Google Scholar]
- Barratt, S.L.; Creamer, A.; Hayton, C.; Chaudhuri, N. Idiopathic Pulmonary Fibrosis (IPF): An Overview. J. Clin. Med. 2018, 7, 201. [Google Scholar] [CrossRef] [PubMed]
- Fadista, J.; Kraven, L.M.; Karjalainen, J.; Andrews, S.J.; Geller, F. Shared genetic aetiology between idiopathic pulmonary fibrosis and COVID-19 severity. EBioMedicine 2021, 65, 103277. [Google Scholar] [CrossRef] [PubMed]
- Marvisi, M.; Ferrozzi, F.; Balzarini, L.; Mancini, C.; Ramponi, S.; Uccelli, M. First report on clinical and radiological features of COVID-19 pneumonitis in a Caucasian population: Factors predicting fibrotic evolution. Int. J. Infect. Dis. 2020, 99, 485–488. [Google Scholar] [CrossRef]
- Ojo, A.S.; Balogun, S.A.; Williams, O.T.; Ojo, O.S. Pulmonary Fibrosis in COVID-19 Survivors: Predictive Factors and Risk Reduction Strategies. Pulm. Med. 2020, 2020, 1–10. [Google Scholar] [CrossRef]
- Huang, W.; Wu, Q.; Chen, Z.; Xiong, Z.; Wang, K.; Tian, J.; Zhang, S. The potential indicators for pulmonary fibrosis in survivors of severe COVID-19. J. Infect. 2020, 82, e5–e7. [Google Scholar] [CrossRef]
- Yu, M.; Liu, Y.; Xu, D.; Zhang, R.; Lan, L.; Xu, H. Prediction of the Development of Pulmonary Fibrosis Using Serial Thin-Section CT and Clinical Features in Patients Discharged after Treatment for COVID-19 Pneumonia. Korean J. Radiol. 2020, 21, 746–755. [Google Scholar] [CrossRef]
- Liu, W.; Tao, Z.W.; Wang, L.; Yuan, M.L.; Liu, K.; Zhou, L.; Wei, S.; Deng, Y.; Liu, J.; Liu, H.G.; et al. Analysis of factors associated with disease outcomes in hospitalized patients with 2019 novel coronavirus disease. Chinese Med. J. 2020, 133, 1032e1038. [Google Scholar] [CrossRef]
- Vardavas, C.I.; Nikitara, K. COVID-19 and smoking: A systematic review of the evidence. Tob. Induc. Dis. 2020, 18, 20. [Google Scholar] [CrossRef]
- Jensen, K.; Nizamutdinov, D.; Guerrier, M.; Afroze, S.; Dostal, D.; Glaser, S. General mechanisms of nicotine-induced fibrogenesis. FASEB J. 2012, 26, 4778–4787. [Google Scholar] [CrossRef] [PubMed]
- Camelo, A.; Dunmore, R.; Sleeman, M.A.; Clarke, D.L. The epithelium in idiopathic pulmonary fibrosis: Breaking the barrier. Front. Pharmacol. 2014, 4, 173. [Google Scholar] [CrossRef]
- Vasarmidi, E.; Tsitoura, E.; Spandidos, D.A.; Tzanakis, N.; Antoniou, K.M. Pulmonary fibrosis in the aftermath of the Covid-19 era (Review). Exp. Ther. Med. 2020, 20, 2557–2560. [Google Scholar] [CrossRef] [PubMed]
- Guler, S.A.; Ebner, L.; Beigelman, C.; Bridevaux, P.O.; Brutsche, M.; Clarenbach, C.; Garzoni, C.; Geiser, T.K.; Lenoir, A.; Mancinetti, M.; et al. Pulmonary function and radiological features four months after COVID-19: First results from the national prospective observational Swiss COVID-19 lung study. Eur. Respir. J. 2021, 57, 2003690. [Google Scholar] [CrossRef]
- McGroder, C.F.; Zhang, D.; Choudhury, M.A.; Salvatore, M.M.; D’Souza, B.M.; Hoffman, E.A.; Wei, Y.; Baldwin, M.R.; Garcia, C.K. Pulmonary fibrosis four months after COVID-19 is associated with severity of illness and blood leucocyte telomere length. Thorax 2021, 76, 1242–1245. [Google Scholar] [CrossRef] [PubMed]
- Rogliani, P.; Calzetta, L.; Coppola, A.; Puxeddu, E.; Sergiacomi, G.; D’Amato, D.; Orlacchio, A. Are there pulmonary sequelae in patients recovering from COVID-19? Respir. Res. 2020, 21, 286. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Zhang, Q.; Li, Q.; Wu, T.; Huang, Y.-Z. Correlation analysis of the severity and clinical prognosis of 32 cases of patients with COVID-19. Respir. Med. 2020, 167, 105981. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.J.; Xu, J.; Yin, J.M.; Li, L.; Hou, W.; Zhang, L.L.; Zhou, Z.; Yu, Y.Z.; Li, H.J.; Feng, Y.M.; et al. Lower Circulating Interferon-Gamma Is a Risk Factor for Lung Fibrosis in COVID-19 Patients. Front. Immunol. 2020, 11, 585647. [Google Scholar] [CrossRef]
- Zaman, T.; Lee, J.S. Risk Factors for the Development of Idiopathic Pulmonary Fibrosis: A Review. Curr. Pulmonol. Rep. 2018, 7, 118–125. [Google Scholar] [CrossRef]
- Reyfman, P.A.; Gottardi, C.J. IPF and lung cancer: Finding similarities within differences. Am. J. Respir. Cell Mol. Biol. 2019, 61, 667–668. [Google Scholar] [CrossRef]
- Álvarez, D.; Cárdenes, N.; Sellarés, J.; Bueno, M.; Corey, C.; Hanumanthu, V.S.; Peng, Y.; D’cunha, H.; Sembrat, J.; Nouraie, M.; et al. IPF lung fibroblasts have a senescent phenotype. Am. J. Physiol. Cell. Mol. Physiol. 2017, 313, L1164–L1173. [Google Scholar] [CrossRef]
- Leung, J.; Cho, Y.; Lockey, R.F.; Kolliputi, N. The Role of Aging in Idiopathic Pulmonary Fibrosis. Lung 2015, 193, 605–610. [Google Scholar] [CrossRef]
- Jo, H.E.; Glaspole, I.; Grainge, C.; Goh, N.; Hopkins, P.M.; Moodley, Y.; Reynolds, P.N.; Chapman, S.; Walters, E.H.; Zappala, C.; et al. Baseline characteristics of idiopathic pulmonary fibrosis: Analysis from the Australian Idiopathic Pulmonary Fibrosis Registry. Eur. Respir. J. 2017, 49, 1601592. [Google Scholar] [CrossRef]
- Ali, R.M.M.; Ghonimy, M.B.I. Post-COVID-19 pneumonia lung fibrosis: A worrisome sequelae in surviving patients. Egypt. J. Radiol. Nucl. Med. 2021, 52, 1–8. [Google Scholar] [CrossRef]
- Voltz, J.W.; Card, J.W.; Carey, M.A.; DeGraff, L.M.; Ferguson, C.D.; Flake, G.P.; Bonner, J.C.; Korach, K.S.; Zeldin, D.C. Male Sex Hormones Exacerbate Lung Function Impairment after Bleomycin-Induced Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2008, 39, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Lekgabe, E.D.; Royce, S.G.; Hewitson, T.D.; Tang, M.L.K.; Zhao, C.; Moore, X.L.; Tregear, G.W.; Bathgate, R.A.D.; Du, X.-J.; Samuel, C.S. The Effects of Relaxin and Estrogen Deficiency on Collagen Deposition and Hypertrophy of Nonreproductive Organs. Endocrinology 2006, 147, 5575–5583. [Google Scholar] [CrossRef]
- Baumgartner, K.B.; Samet, J.M.; Stidley, C.A.; Colby, T.V.; Waldron, J.A. Cigarette smoking: A risk factor for idiopathic pul-monary fibrosis. Am. J. Respir. Crit. Care Med. 1997, 155, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Taskar, V.S.; Coultas, D.B. Is idiopathic pulmonary fibrosis an environmental disease? Proc. Am. Thorac. Soc. 2006, 3, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Velavan, T.P.; Pallerla, S.R.; Rüter, J.; Augustin, Y.; Kremsner, P.G.; Krishna, S.; Meyer, C.G. Host genetic factors determining COVID-19 susceptibility and severity. Ebiomedicine 2021, 72, 103629. [Google Scholar] [CrossRef]
- Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernández, J.; Prati, D.; Baselli, G.; Asselta, R.; et al. Severe COVID-19 GWAS Group. Genome-wide Association Study of Severe COVID-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar]
- COVID-19 Host Genetics Initiative. COVID-19 Host Genetics Initiative Mapping the human genetic architecture of COVID-19. Nature 2021, 600, 472–477. [Google Scholar] [CrossRef]
- Wein, A.N.; McMaster, S.R.; Takamura, S.; Dunbar, P.R.; Cartwright, E.K.; Hayward, S.L.; McManus, D.T.; Shimaoka, T.; Ueha, S.; Tsukui, T.; et al. CXCR6 regulates localisation of tissue-resident memory CD8 T cells to the airways. J. Exp. Med. 2019, 216, 2748–2762. [Google Scholar] [CrossRef]
- Kropski, J.A.; Blackwell, T.S.; Loyd, J.E. The genetic basis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1717–1727. [Google Scholar] [CrossRef]
- Kaur, A.; Mathai, S.K.; Schwartz, D.A. Genetics in Idiopathic Pulmonary Fibrosis Pathogenesis, Prognosis, and Treatment. Front. Med. 2017, 4, 154. [Google Scholar] [CrossRef]
- van Moorsel, C.H.M.; van der Vis, J.J.; Benschop, C.; Ruven, H.J.T.; Quanjel, M.; Grutters, J.C. THE MUC5B promotor poly-morphism associates with severe COVID-19. Front. Med. 2021, 8, 668024. [Google Scholar] [CrossRef]
- Molyneaux, P.L.; Cox, M.; Willis-Owen, S.A.; Mallia, P.; Russell, K.E.; Russell, A.M.; Murphy, E.; Johnston, S.L.; Schwartz, D.A.; Wells, A.U.; et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 906–913. [Google Scholar] [CrossRef]
- Han, M.K.; Zhou, Y.; Murray, S.; Tayob, N.; Noth, I.; Lama, V.N.; Moore, B.B.; White, E.S.; Flaherty, K.R.; Huffnagle, G.B.; et al. Lung microbiome and disease progression in idiopathic pulmonary fibrosis: An analysis of the COMET study. Lancet Respir. Med. 2014, 2, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Nasir, N.; Rehman, F.; Omair, S.F. Risk factors for bacterial infections in patients with moderate to severe COVID-19: A case-control study. J. Med. Virol. 2021, 93, 4564–4569. [Google Scholar] [CrossRef] [PubMed]
- Langford, B.J.; So, M.; Raybardhan, S.; Leung, V.; Westwood, D.; MacFadden, D.R.; Soucy, J.R.; Daneman, N. Bacterial co-infection and secondary infection in patients with COVID-19: A living rapid review and meta-analysis. Clin. Microbiol. Infect. 2020, 26, 1622–1629. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.M.; Ata, F.; Islam, A.M.; Bint, I.B.A.; Salih, A.A.; Yousaf, Z. Post COVID-19 fibrosis, an emerging complication of SARS-CoV-2 infection. IDCases 2020, 23, e01041. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.A.; Sverzellati, N.; Travis, W.D.; Brown, K.K.; Colby, T.V.; Galvin, J.R.; Goldin, J.G.; Hansell, D.M.; Inoue, Y.; Johkoh, T.; et al. Diagnostic criteria for idiopathic pulmonary fibrosis: A Fleischner Society White Paper. Lancet Respir. Med. 2018, 6, 138–153. [Google Scholar] [CrossRef] [PubMed]
- Farghaly, S.; Badedi, M.; Ibrahim, R.; Sadhan, M.H.; Alamoudi, A.; Alnami, A.; Muhajir, A. Clinical characteristics and out-comes of post-COVID-19 pulmonary fibrosis: A case-control study. Medicine 2022, 101, e28639. [Google Scholar] [CrossRef]
- Kamal, M.; Abo Omirah, M.; Hussein, A.; Saeed, H. Assessment and characterisation of post-COVID-19 manifestations. Int. J. Clin. Pract. 2021, 75, e13746. [Google Scholar] [CrossRef]
- Nolan, C.M.; Patel, S.; Barker, R.E.; George, P.; Maddocks, M.M.; Cullinan, P.; Maher, T.M.; Man, W.D.C. Anxiety and depression in idiopathic pulmonary fibrosis (IPF): Prevalence and clinical correlates. Eur. Respir. J. 2017, 50, PA848. [Google Scholar]
- Lechtzin, N.; Hilliard, M.E.; Horton, M.R. Validation of the Cough Quality-of-Life Questionnaire in Patients with Idiopathic Pulmonary Fibrosis. Chest 2013, 143, 1745–1749. [Google Scholar] [CrossRef]
- Araki, T.; Katsura, H.; Sawabe, M.; Kida, K. A Clinical Study of Idiopathic Pulmonary Fibrosis Based on Autopsy Studies in Elderly Patients. Intern. Med. 2003, 42, 483–489. [Google Scholar] [CrossRef]
- Carvajalino, S.; Reigada, C.; Johnson, M.J.; Dzingina, M.; Bajwah, S. Symptom prevalence of patients with fibrotic interstitial lung disease: A systematic literature review. BMC Pulm. Med. 2018, 18, 78. [Google Scholar] [CrossRef] [PubMed]
- Faverio, P.; Luppi, F.; Rebora, P.; Busnelli, S.; Stainer, A.; Catalano, M.; Parachini, L.; Monzani, A.; Galimberti, S.; Bini, F.; et al. Six-Month Pulmonary Impairment after Severe COVID-19: A Prospective, Multicentre Follow-Up Study. Respiration 2021, 100, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Kanematsu, T.; Kitaichi, M.; Nishimura, K.; Nagai, S.; Izumi, T. Clubbing of the Fingers and Smooth-Muscle Proliferation in Fibrotic Changes in the Lung in Patients With Idiopathic Pulmonary Fibrosis. Chest 1994, 105, 339–342. [Google Scholar] [CrossRef]
- Bonella, F.; di Marco, F.; Spagnolo, P. Pulmonary Function Tests in Idiopathic Pulmonary Fibrosis. In Idiopathic Pulmonary Fibrosis. Respiratory Medicine; Meyer, K., Nathan, S., Eds.; Humana Press: Cham, Switzerland, 2019. [Google Scholar]
- Torres, C.R.; Vasconcello, C.L.; Alsina, R.L.; Solis, N.F.; Burgos, H.; Puppo, J.V. Respiratory function in patients post-infection by COVID-19: A systematic review and meta-analysis. Pulmonology 2021, 27, 328–337. [Google Scholar] [CrossRef]
- British Thoracic Society. British Thoracic Society Guidance on Respiratory Follow-Up of Patients with a Clinico-Radiological Diagnosis of COVID-19 Pneumonia [Internet]. 2020. Available online: https://www.brit-thoracic.org.uk/document-library/quality-improvement/COVID-19/resp-follow-up-guidance-post-COVID-pneumonia/ (accessed on 2 July 2022).
- Cherrez-Ojeda, I.; Robles-Velasco, K.; Osorio, M.F.; Cottin, V.; Centeno, J.V.; Felix, M. Follow-up of two cases of suspected interstitial lung disease following severe COVID-19 infection shows persistent changes in imaging and lung function. Clin. Case Rep. 2021, 9, e04918. [Google Scholar] [CrossRef] [PubMed]
- Cabo-Gambin, R.; Benítez, I.D.; Carmona, P.; Santiesteve, S.; Mínguez, O.; Vaca, R.; Moncusí-Moix, A.; Gort-Paniello, C.; García-Hidalgo, M.C.; de Gonzalo-Calvo, D.; et al. Three to Six Months Evolution of Pulmonary Function and Radiological Features in Critical COVID-19 Patients: A Prospective Cohort. Arch. Bronconeumol. 2021, 58, 59–62. [Google Scholar] [CrossRef]
- Fidler, L.; Shapera, S.; Mittoo, S.; Marras, T.K. Diagnostic disparity of previous and revised American Thoracic Society guide-lines for idiopathic pulmonary fibrosis. Can. Respir. J. 2015, 22, 86–90. [Google Scholar] [CrossRef]
- Collard, H.R.; King, T.E.; Bartelson, B.B.; Vourlekis, J.S.; Schwarz, M.I.; Brown, K.K. Changes in clinical and physiologic var-iables predict survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 538–542. [Google Scholar] [CrossRef]
- Nathan, S.D.; Shlobin, O.A.; Weir, N.; Ahmad, S.; Kaldjob, J.M.; Battle, E.; Sheridan, M.J.; du Bois, R.M. Long-term course and prognosis of idiopathic pulmonary fibrosis in the new millennium. Chest 2011, 140, 221–229. [Google Scholar] [CrossRef]
- Tanni, S.E.; Fabro, A.T.; de Albuquerque, A.; Ferreira, E.V.M.; Verrastro, C.G.Y.; Sawamura, M.V.Y.; Ribeiro, S.M.; Baldi, B.G. Pulmonary fibrosis secondary to COVID-19: A narrative review. Exp. Rev. Respir. Med. 2021, 15, 791–803. [Google Scholar] [CrossRef]
- Atabati, E.; Dehghani-Samani, A.; Mortazavimoghaddam, S.G. Association of COVID-19 and other viral infections with interstitial lung diseases, pulmonary fibrosis, and pulmonary hypertension: A narrative review. Can. J. Respir. Ther. 2020, 56, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Wie, J.; Yang, H.; Lei, P.; Fan, B.; Qiu, Y.; Zeng, B. Analysis of thin-section CT in patients with coronavirus disease (COVID-19) after hospital discharge. J. X-ray Sci. Technol. 2020, 28, 383–389. [Google Scholar]
- Francone, M.; Iafrate, F.; Masci, G.M.; Coco, S.; Cilia, F.; Manganaro, L.; Panebianco, V.; Andreoli, C.; Colaiacomo, M.C.; Zingaropoli, M.A.; et al. Chest CT score in COVID-19 patients: Correlation with disease severity and short-term prognosis. Eur. Radiol. 2020, 30, 6808–6817. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Xi, X.; Min, X.; Feng, Z.; Li, B.; Cai, W.; Fan, C.; Wang, L.; Xia, L. Long-term chest CT follow-up in COVID-19 Survivors: 102–361 days after onset. Ann. Transl. Med. 2021, 9, 1231. [Google Scholar] [CrossRef]
- Vijayakumar, B.; Tonkin, J.; Devaraj, A.; Philip, K.E.J.; Orton, C.M.; Desai, S.R.; Shah, P.L. CT Lung Abnormalities after COVID-19 at 3 Months and 1 Year after Hospital Discharge. Radiology 2022, 303, 444–454. [Google Scholar] [CrossRef]
- Camiciottoli, G.; Orlandi, I.; Bartolucci, M.; Meoni, E.; Nacci, F.; Diciotti, S.; Barcaroli, C.; Conforti, M.L.; Pistolesi, M.; Matucci-Cerinic, M.; et al. Lung CT densitometry in systemic sclerosis: Correlation with lung function, exercise testing, and quality of life. Chest 2007, 131, 672–681. [Google Scholar] [CrossRef]
- Combet, M.; Pavot, A.; Savale, L.; Humbert, M.; Monnet, X. Rapid onset honeycombing fibrosis in spontaneously breathing patient with COVID-19. Eur. Respir. J. 2020, 56, 2001808. [Google Scholar] [CrossRef] [PubMed]
- Balestro, E.; Cocconcelli, E.; Giraudo, C.; Polverosi, R.; Biondini, D.; Lacedonia, D.; Bazzan, E.; Mazzai, L.; Rizzon, G.; Lococo, S.; et al. High-Resolution CT Change over Time in Patients with Idiopathic Pulmonary Fibrosis on Anti-fibrotic Treatment. J. Clin. Med. 2019, 8, 1469. [Google Scholar] [CrossRef] [PubMed]
- Ley, B.; Elicker, B.M.; Hartman, T.E.; Ryerson, C.J.; Vittinghoff, E.; Ryu, J.H.; Lee, J.S.; Jones, K.D.; Richeldi, L.; King, T.E.; et al. Idiopathic Pulmonary Fibrosis: CT and Risk of Death. Radiology 2014, 273, 570–579. [Google Scholar] [CrossRef]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-based Guidelines for Diagnosis and Management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef]
- Adachi, T.; Chong, J.M.; Nakajima, N.; Sano, M.; Yamazaki, J.; Miyamoto, I.; Nishioka, H.; Akita, H.; Sato, Y.; Kataoka, M.; et al. Clinicopathologic and Immunohistochemical Findings from Autopsy of Patient with COVID-19, Japan. Emerg. Infect. Dis. 2020, 26, 2157–2161. [Google Scholar] [CrossRef] [PubMed]
- Vishwajit, D.; Rohini, M.; Ashutosh, K.; Chiman, K.; Khursheed, R. Histopathological observations in COVID-19: A systematic review. J. Clin. Pathol. 2021, 74, 76–83. [Google Scholar]
- Martines, R.B.; Ritter, J.M.; Matkovic, E.; Gary, J.; Bollweg, B.C.; Bullock, H.; Goldsmith, C.S.; Silva-Flannery, L.; Seixas, J.N.; Reagan-Steiner, S.; et al. COVID-19 pathology working group. Pathology and pathogenesis of SARS-CoV-2 associated with fatal coronavirus disease, United States. Emerg. Infect. Dis. 2020, 26, 2005–2015. [Google Scholar] [CrossRef]
- Sauter, J.L.; Baine, M.K.; Butnor, K.J.; Buonocore, D.J.; Chang, J.C.; Jungbluth, A.A.; Szabolcs, M.J.; Morjaria, S.; Mount, S.L.; Rekhtman, N.; et al. Insights into pathogenesis of fatal COVID-19 pneumonia from histopathology with immunohistochemical and viral RNA studies. Histopathology 2020, 77, 915–925. [Google Scholar] [CrossRef]
- Chen, J.Y.; Qiao, K.; Liu, F.; Wu, B.; Xu, X.; Jiao, G.Q.; Lu, R.G.; Li, H.X.; Zhao, J.; Huang, J.; et al. Lung transplantation as therapeutic option in acute respiratory distress syndrome for coronavirus disease 2019-related pulmonary fibrosis. Chin. Med. J. 2020, 133, 1390–1396. [Google Scholar] [CrossRef]
- Tian, S.; Xiong, Y.; Liu, H.; Niu, L.; Guo, J.; Liao, M.; Xiao, S.Y. Pathological study of the 2019 novel coronavirus disease (COVID-19) through post-mortem core biopsies. Mod. Pathol. 2020, 33, 1007–1014. [Google Scholar] [CrossRef]
- Bernard, G.R.; Artigas, A.; Brigham, K.L.; Carlet, J.; Falke, K.; Hudson, L.; Lamy, M.; Legall, J.R.; Morris, A.; Spragg, R. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am. J. Respir. Crit. Care Med. 1994, 149, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Beasley, M.B.; Franks, T.J.; Galvin, J.R.; Gochuico, B.; Travis, W.D. Acute fibrinous and organising pneumonia: A histologic pattern of lung injury and possible variant of diffuse alveolar damage. Arch. Pathol. Lab. Med. 2002, 126, 1064–1070. [Google Scholar] [CrossRef]
- Tomashefski, J.F., Jr. Pulmonary Pathology of Acute Respiratory Distress Syndrome. Clin. Chest Med. 2000, 21, 435–466. [Google Scholar] [CrossRef]
- Sapru, A.; Wiemels, J.L.; Witte, J.S.; Ware, L.B.; Matthay, M.A. Acute lung injury and the coagulation pathway: Potential role of gene polymorphisms in the protein C and fibrinolytic pathways. Intensiv. Care Med. 2006, 32, 1293–1303. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, J.; Wang, S.; Li, X.; Zhou, J.; Huang, B.; Luo, D.; Cao, Q.; Chen, Y.; Chen, S.; et al. Progression to fibrosing diffuse alveolar damage in a series of 30 minimally invasive autopsies with COVID-19 pneumonia in Wuhan, China. Histopathology 2020, 78, 542–555. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, P.; Wei, Y.; Yue, H.; Wang, Y.; Hu, M.; Zhang, S.; Cao, T.; Yang, C.; Li, M.; et al. Histopathologic Changes and SARS-CoV-2 immunostaining in the lung of a patient with COVID-19. Ann. Intern. Med. 2020, 172, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Kory, P.; Jp, K. SARS-CoV-2 organising pneumonia: Has there been a widespread failure to identify and treat this prevalent condition in COVID-19? BMJ Open Respir. Res. 2020, 7, e000724. [Google Scholar] [CrossRef]
- Kommoss, F.K.; Schwab, C.; Tavernar, L.; Schreck, J.; Wagner, W.L.; Merle, U.; Jonigk, D.; Schirmacher, P.; Longerich, T. The Pathology of Severe COVID-19-Related Lung Damage. Deutsches Ärzteblatt Int. 2020, 117, 500–506. [Google Scholar] [CrossRef]
- Hanley, B.; Naresh, K.N.; Roufosse, C.; Nicholson, A.G.; Weir, J.; Cooke, G.S.; Thursz, M.; Manousou, P.; Corbett, R.; Goldin, R.; et al. Histopathological findings and viral tropism in UK patients with severe fatal COVID-19: A post-mortem study. Lancet Microbe 2020, 1, e245–e253. [Google Scholar] [CrossRef]
- Copin, M.C.; Parmentier, E.; Duburcq, T.; Poissy, J.; Mathieu, D.; The Lille COVID-19 ICU and Anatomopathology Group. Time to consider histologic pattern of lung injury to treat critically ill patients with COVID-19 infection. Intensiv. Care Med. 2020, 46, 1124–1126. [Google Scholar] [CrossRef]
- Bharat, A.; Querrey, M.; Markov, N.S.; Kim, S.; Kurihara, C.; Garza-Castillon, R.; Manerikar, A.; Shilatifard, A.; Tomic, R.; Politanska, Y.; et al. Lung transplantation for patients with severe COVID-19. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- John, A.E.; Joseph, C.; Jenkins, G.; Tatler, A.L. COVID-19 and pulmonary fibrosis: A potential role for lung epithelial cells and fibroblasts. Immunol. Rev. 2021, 302, 228–240. [Google Scholar] [CrossRef]
- Buja, L.M.; Wolf, D.A.; Zhao, B.; Akkanti, B.; McDonald, M.; Lelenwa, L.; Reilly, N.; Ottaviani, G.; Elghetany, M.T.; Trujillo, D.O.; et al. The emerging spectrum of cardiopulmonary pathology of the coronavirus disease 2019 (COVID-19): Report of 3 autopsies from Houston, Texas, and review of autopsy findings from other United States cities. Cardiovasc. Pathol. 2020, 48, 107233. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in COVID-19. N Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Burel-Vandenbos, F.; Cardot-Leccia, N.; Passeron, T. Pulmonary Vascular Pathology in Covid-19. New Engl. J. Med. 2020, 383, 886–889. [Google Scholar] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef] [PubMed]
- Nienhold, R.; Ciani, Y.; Koelzer, V.H.; Tzankov, A.; Haslbauer, J.D.; Menter, T.; Schwab, N.; Henkel, M.; Frank, A.; Zsikla, V.; et al. Two distinct immunopathological profiles in autopsy lungs of COVID-19. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Parambil, J.G.; Myers, J.L.; Aubry, M.C.; Ryu, J.H. Causes and Prognosis of Diffuse Alveolar Damage Diagnosed on Surgical Lung Biopsy. Chest 2007, 132, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.; Nathan, S.D.; Hill, C.; Marshall, J.; Dejonckheere, F.; Thuresson, P.O.; Maher, T.M. Predicting Life Expectancy for Pirfenidone in Idiopathic Pulmonary Fibrosis. J. Manag. Care Spéc. Pharm. 2017, 23, S17–S24. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.J.; Ni, Z.Y.; Hu, Y. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020, 18, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Jolly, L.; Stavrou, A.; Vanderstoken, G. Influenza promotes collagen deposition via αvβ6 integrin-mediated transforming growth factor β activation. J. Biol. Chem. 2014, 289, 35246–35263. [Google Scholar] [CrossRef]
- Mackinnon, A.C.; Gibbons, M.A.; Farnworth, S.L. Regulation of transforming growth factor-β1-driven lung fibrosis by galec-tin-3. Am. J. Respir. Crit. Care Med. 2012, 185, 537–546. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.A. SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Raghu, G.; van den Blink, B.; Hamblin, M.J. Effect of recombinant human pentraxin 2 vs placebo on change in forced vital capacity in patients with idiopathic pulmonary fibrosis: A randomised clinical trial. JAMA 2018, 319, 2299–2307. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, F.; Xu, J. Anti-hypertensive Angiotensin II receptor blockers associated to mitigation of disease severity in elderly COVID-19 patients. Ann. Intern. Med. 2020, 172, 629–632. [Google Scholar]
- Skurikhin, E.; Nebolsin, V.; Widera, D.; Ermakova, N.; Pershina, O.; Pakhomova, A.; Krupin, V.; Pan, E.; Zhukova, M.; Novikov, F.; et al. Anti-fibrotic and regenerative effects of treamid in pulmonary fibrosis. Int. J. Mol. Sci. 2020, 21, 8380. [Google Scholar] [CrossRef]
- Pure Tech Company. A Phase 2 Randomised, Double-Blind, Placebo-Controlled Trial and Open Label Extension to Evaluate the Safety and Efficacy of Deupirfenidone (LYT-100) in Post-Acute COVID-19 Respiratory Disease; Pure Tech Company: Boston, MA, USA, 2020. [Google Scholar]
- Bazdyrev, E.; Rusina, P.; Panova, M.; Novikov, F.; Grishagin, I.; Nebolsin, V. Lung Fibrosis after COVID-19: Treatment Pro-spects. Pharmaceuticals 2021, 14, 807. [Google Scholar] [CrossRef] [PubMed]
- Echeverría-Esnal, D.; Martin-Ontiyuelo, C.; Navarrete-Rouco, M.E.; Cuscó, M.D.-A.; Ferrández, O.; Horcajada, J.P.; Grau, S. Azithromycin in the treatment of COVID-19: A review. Expert Rev. Anti-Infective Ther. 2020, 19, 147–163. [Google Scholar] [CrossRef] [PubMed]
- P, K.M.; Sivashanmugam, K.; Kandasamy, M.; Subbiah, R.; Ravikumar, V. Repurposing of histone deacetylase inhibitors: A promising strategy to combat pulmonary fibrosis promoted by TGF-β signalling in COVID-19 survivors. Life Sci. 2020, 266, 118883. [Google Scholar] [CrossRef] [PubMed]
- Reina-Gutiérrez, S.; Torres-Costoso, A.; Martínez-Vizcaíno, V.; de Arenas-Arroyo, S.N.; Fernández-Rodríguez, R.; Pozuelo-Carrascosa, D.P. Effectiveness of Pulmonary Rehabilitation in Interstitial Lung Disease, Including Coronavirus Diseases: A Systematic Review and Meta-analysis. Arch. Phys. Med. Rehabilit. 2021, 102, 1989–1997.e3. [Google Scholar] [CrossRef] [PubMed]
- Egan, J.J. Follow-up and nonpharmacological management of the idiopathic pulmonary fibrosis patient. Eur. Respir. Rev. 2011, 20, 114–117. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Risk Factor | PCPF | IPF |
|---|---|---|
| Gender-male | yes | yes |
| Advanced age | yes | yes |
| Smoking | yes | yes |
| Comorbidities | yes | yes |
| Toxic environmental exposure | no | yes |
| Severity of dyspnea | yes | yes |
| Prolonged hospital stay | yes | no |
| ICU admission | yes | yes |
| Mechanical ventilation | yes | yes |
| Lung microbiome | yes | yes |
| Gene | Mutation Consequence |
|---|---|
| SPTPC | Altered encoding of surfactant proteins |
| SPTPA1 | Altered encoding of surfactant proteins |
| SFTPA2 | Altered encoding of surfactant proteins |
| TERT | Telomere shortening |
| TERC | Telomere shortening |
| DKC1 | Telomere shortening |
| RTELI | Telomere shortening |
| PARN | Telomere shortening |
| DSP | The affection of the epithelial barrier |
| MUC5B | Host defense affection |
| TOLLIP | Host defense affection |
| Clinical Aspect | PCPF | IPF |
|---|---|---|
| Dyspnea | yes | yes |
| Cough | yes | yes |
| Fever | yes | no |
| Fatigue | yes | yes |
| Chest pain | yes | yes |
| Depression | yes | yes |
| Velcro crackles | yes | yes |
| Wheezing | yes | no |
| Clubbing fingers | no | yes |
| PFT | PCPF | IPF |
|---|---|---|
| Restrictive dysfunction | yes | yes |
| Obstructive dysfunction | yes | Coexistence of obstructive disease |
| Decreased DLCO | yes | yes |
| 6MWT desaturation | yes | yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cîrjaliu, R.-E.; Deacu, M.; Gherghișan, I.; Marghescu, A.-Ș.; Enciu, M.; Băltățescu, G.I.; Nicolau, A.A.; Tofolean, D.-E.; Arghir, O.C.; Fildan, A.-P. Clinicopathological Outlines of Post-COVID-19 Pulmonary Fibrosis Compared with Idiopathic Pulmonary Fibrosis. Biomedicines 2023, 11, 1739. https://doi.org/10.3390/biomedicines11061739
Cîrjaliu R-E, Deacu M, Gherghișan I, Marghescu A-Ș, Enciu M, Băltățescu GI, Nicolau AA, Tofolean D-E, Arghir OC, Fildan A-P. Clinicopathological Outlines of Post-COVID-19 Pulmonary Fibrosis Compared with Idiopathic Pulmonary Fibrosis. Biomedicines. 2023; 11(6):1739. https://doi.org/10.3390/biomedicines11061739
Chicago/Turabian StyleCîrjaliu, Roxana-Elena, Mariana Deacu, Ioana Gherghișan, Angela-Ștefania Marghescu, Manuela Enciu, Gabriela Izabela Băltățescu, Antonela Anca Nicolau, Doina-Ecaterina Tofolean, Oana Cristina Arghir, and Ariadna-Petronela Fildan. 2023. "Clinicopathological Outlines of Post-COVID-19 Pulmonary Fibrosis Compared with Idiopathic Pulmonary Fibrosis" Biomedicines 11, no. 6: 1739. https://doi.org/10.3390/biomedicines11061739
APA StyleCîrjaliu, R.-E., Deacu, M., Gherghișan, I., Marghescu, A.-Ș., Enciu, M., Băltățescu, G. I., Nicolau, A. A., Tofolean, D.-E., Arghir, O. C., & Fildan, A.-P. (2023). Clinicopathological Outlines of Post-COVID-19 Pulmonary Fibrosis Compared with Idiopathic Pulmonary Fibrosis. Biomedicines, 11(6), 1739. https://doi.org/10.3390/biomedicines11061739