
ASL mRNA-LNP Therapeutic for the Treatment of Argininosuccinic Aciduria Enables Survival Benefit in a Mouse Model
 , , ,
, , ,
Abstract

1. Introduction
2. Materials and Methods
2.1. Manufacturing of ASL mRNA-LNP
2.2. In Vitro Cell Culture and Transfections of LNP-mRNA
2.3. In Vivo Delivery to Wild-Type and ASLNeo/Neo Mouse Model of ASLD
2.4. Western Blots, Jess, and Mass Spectroscopy Quantification
2.5. Cytokine and Chemokine Assays
2.6. Statistical Analysis
3. Results
3.1. mRNA Therapeutic for the Treatment of ASA
3.2. Robust Protein Expression of ASL mRNA Payloads In Vitro
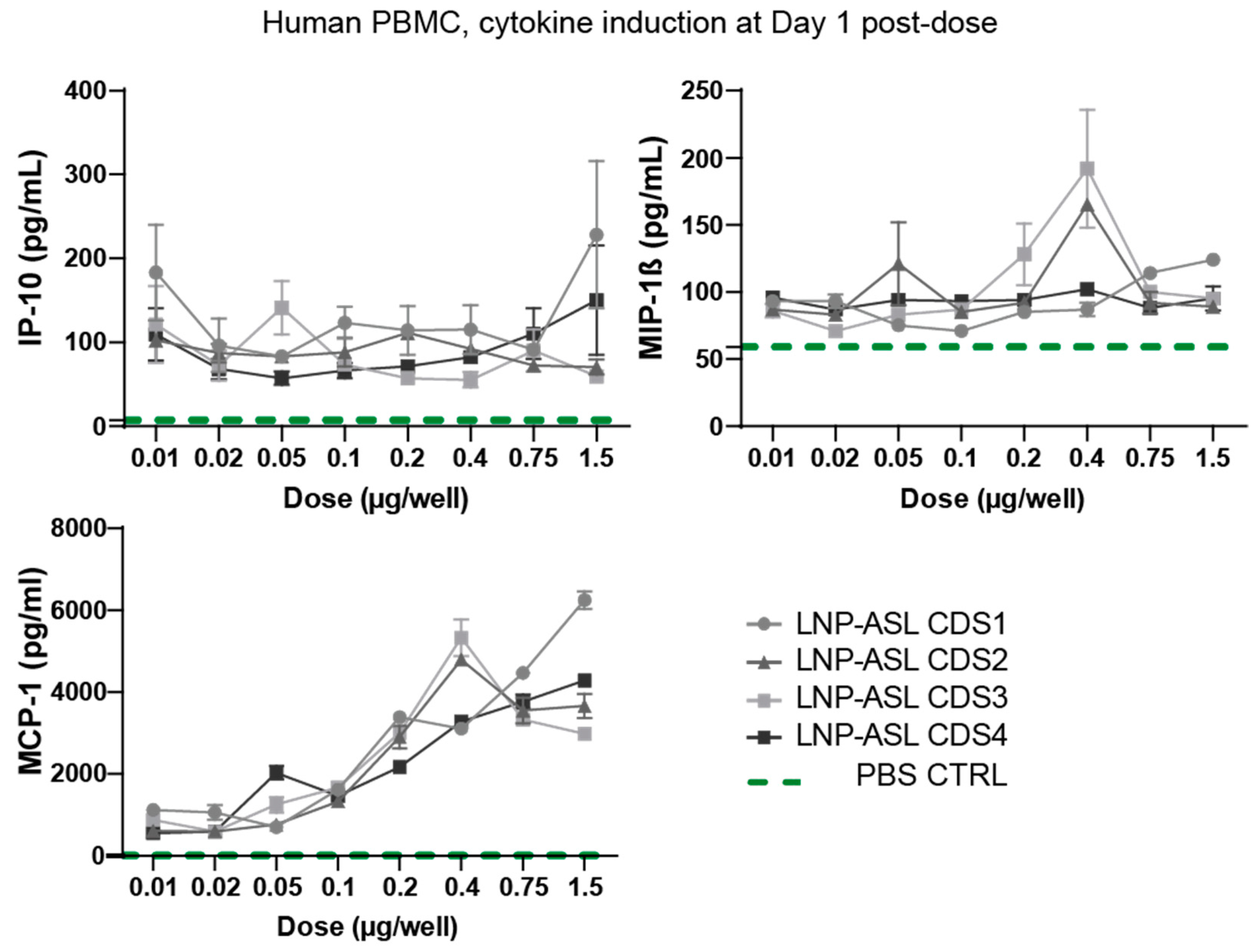
3.3. Low Cytokine Induction Post-mRNA-LNP Administration in Human Peripheral Blood Mononuclear Cells
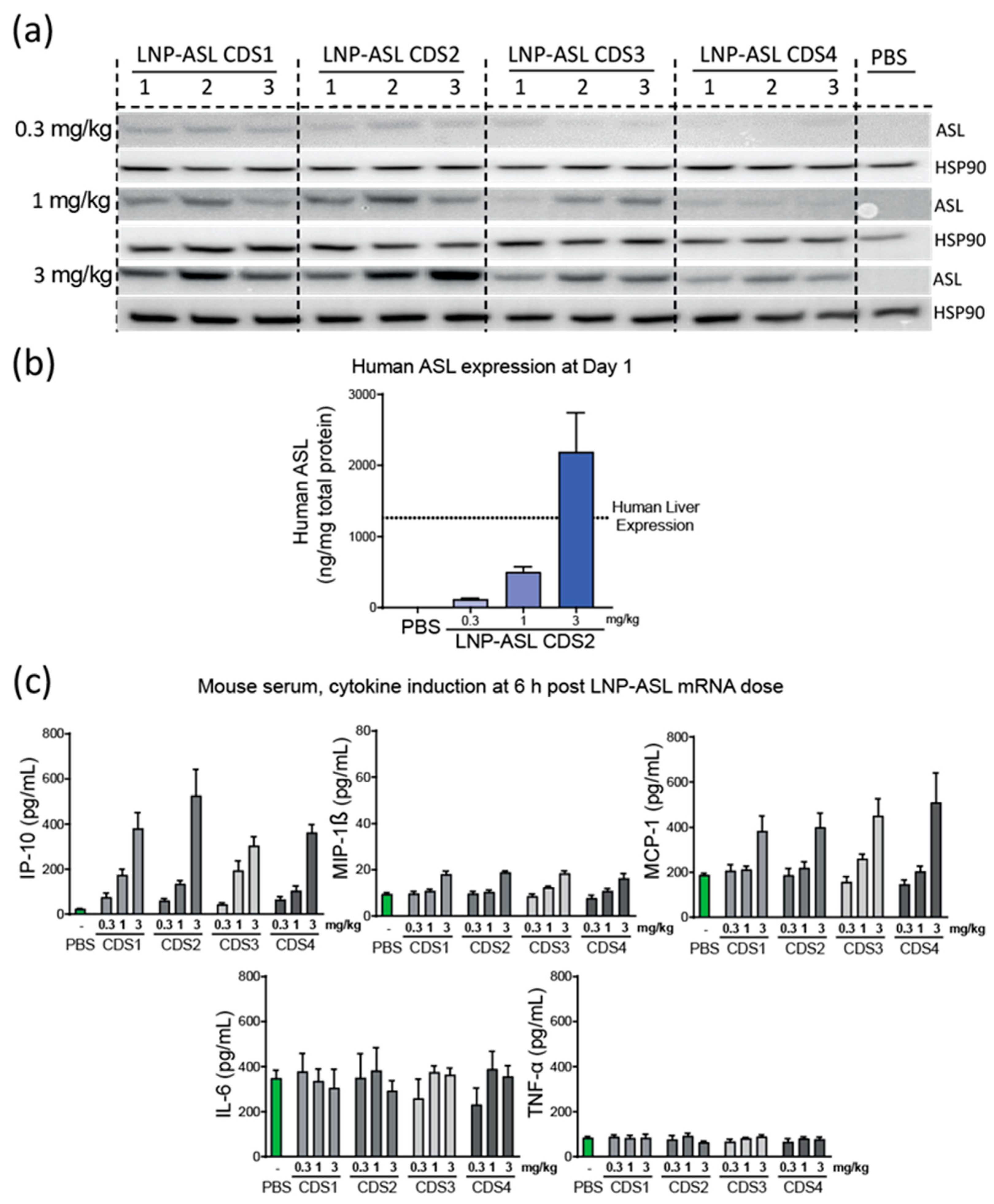
3.4. Dose Response in Wild-Type Mice
3.5. Repeat-Dose Efficacy of mRNA-LNP in ASLD Mouse Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Summar, M.L.; Koelker, S.; Freedenberg, D.; Le Mons, C.; Haberle, J.; Lee, H.-S.; Kirmse, B. The incidence of urea cycle disorders. Mol. Genet. Metab. 2013, 110, 179–180. [Google Scholar] [CrossRef]
- O’Brien, W.E.; McInnes, R.; Kalumuck, K.; Adcock, M. Cloning and sequence analysis of cDNA for human argininosuccinate lyase. Proc. Natl. Acad. Sci. USA 1986, 83, 7211–7215. [Google Scholar] [CrossRef] [PubMed]
- Erez, A. Argininosuccinic aciduria: From a monogenic to a complex disorder. Genet. Med. 2013, 15, 251–257. [Google Scholar] [CrossRef]
- Erez, A.; Nagamani, S.C.S.; Shchelochkov, O.A.; Premkumar, M.H.; Campeau, P.M.; Chen, Y.; Garg, H.K.; Li, L.; Mian, A.; Bertin, T.K.; et al. Requirement of argininosuccinate lyase for systemic nitric oxide production. Nat. Med. 2011, 17, 1619–1626. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Gotoh, T. Arginine metabolic enzymes, nitric oxide and infection. J. Nutr. 2004, 134, 2820S–2825S, discussion 2853S. [Google Scholar] [CrossRef] [PubMed]
- Baruteau, J.; Perocheau, D.P.; Hanley, J.; Lorvellec, M.; Rocha-Ferreira, E.; Karda, R.; Ng, J.; Suff, N.; Diaz, J.A.; Rahim, A.A.; et al. Argininosuccinic aciduria fosters neuronal nitrosative stress reversed by Asl gene transfer. Nat. Commun. 2018, 9, 3505. [Google Scholar] [CrossRef]
- McNaughton, L.; Puttagunta, L.; Martinez-Cuesta, M.A.; Kneteman, N.; Mayers, I.; Moqbel, R.; Hamid, Q.; Radomski, M.W. Distribution of nitric oxide synthase in normal and cirrhotic human liver. Proc. Natl. Acad. Sci. USA 2002, 99, 17161–17166. [Google Scholar] [CrossRef]
- Häussinger, D.; Lamers, W.H.; Moorman, A.F. Hepatocyte heterogeneity in the metabolism of amino acids and ammonia. Enzyme 1992, 46, 72–93. [Google Scholar] [CrossRef]
- Mian, A.; Lee, B. Urea-cycle disorders as a paradigm for inborn errors of hepatocyte metabolism. Trends Mol. Med. 2002, 8, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Häberle, J.; Burlina, A.; Chakrapani, A.; Dixon, M.; Karall, D.; Lindner, M.; Mandel, H.; Martinelli, D.; Pintos-Morell, G.; Santer, R.; et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J. Inherit. Metab. Dis. 2019, 42, 1192–1230. [Google Scholar] [CrossRef] [PubMed]
- Osawa, Y.; Wada, A.; Ohtsu, Y.; Yamada, K.; Takizawa, T. Late-onset argininosuccinic aciduria associated with hyperammonemia triggered by influenza infection in an adolescent: A case report. Mol. Genet. Metab. Rep. 2020, 24, 100605. [Google Scholar] [CrossRef]
- Häberle, J.; Boddaert, N.; Burlina, A.; Chakrapani, A.; Dixon, M.; Huemer, M.; Karall, D.; Martinelli, D.; Crespo, P.S.; Santer, R.; et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J. Rare Dis. 2012, 7, 32. [Google Scholar] [CrossRef]
- Newnham, T.; Hardikar, W.; Allen, K.; Wellard, R.M.; Hamilton, C.; Angus, P.; Jones, R.; Boneh, A. Liver transplantation for argininosuccinic aciduria: Clinical, biochemical, and metabolic outcome. Liver Transpl. 2008, 14, 41–45. [Google Scholar] [CrossRef]
- Yankol, Y.; Mecit, N.; Kanmaz, T.; Acarli, K.; Kalayoglu, M. Argininosuccinic Aciduria-A Rare Indication for Liver Transplant: Report of Two Cases. Exp. Clin. Transplant. 2017, 15, 581–584. [Google Scholar] [CrossRef]
- Ziogas, I.A.; Wu, W.K.; Matsuoka, L.K.; Pai, A.K.; Hafberg, E.T.; Gillis, L.A.; Morgan, T.M.; Alexopoulos, S.P. Liver Transplantation in Children with Urea Cycle Disorders: The Importance of Minimizing Waiting Time. Liver Transpl. 2021, 27, 1799–1810. [Google Scholar] [CrossRef]
- Ashley, S.N.; Nordin, J.M.L.; Buza, E.L.; Greig, J.A.; Wilson, J.M. Adeno-associated viral gene therapy corrects a mouse model of argininosuccinic aciduria. Mol. Genet. Metab. 2018, 125, 241–250. [Google Scholar] [CrossRef]
- Chandler, R.J.; LaFave, M.C.; Varshney, G.K.; Burgess, S.M.; Venditti, C.P. Genotoxicity in Mice Following AAV Gene Delivery: A Safety Concern for Human Gene Therapy? Mol. Ther. 2016, 24, 198–201. [Google Scholar] [CrossRef]
- Chandler, R.; LaFave, M.; Varshney, G.K.; Trivedi, N.S.; Carrillo, N.; Senac, J.S.; Wu, W.; Hoffmann, V.; Elkahloun, A.G.; Burgess, S.; et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J. Clin. Investig. 2015, 125, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Chandler, R.J.; Sands, M.S.; Venditti, C.P. Recombinant Adeno-Associated Viral Integration and Genotoxicity: Insights from Animal Models. Hum. Gene Ther. 2017, 28, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Donsante, A.; Miller, D.G.; Li, Y.; Vogler, C.; Brunt, E.M.; Russell, D.W.; Sands, M.S. AAV vector integration sites in mouse hepatocellular carcinoma. Science 2007, 317, 477. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Beck, J.D.; Reidenbach, D.; Salomon, N.; Sahin, U.; Türeci, Ö.; Vormehr, M.; Kranz, L.M. mRNA therapeutics in cancer immunotherapy. Mol. Cancer. 2021, 20, 69. [Google Scholar] [CrossRef]
- Vlatkovic, I. Non-Immunotherapy Application of LNP-mRNA: Maximizing Efficacy and Safety. Biomedicines 2021, 9, 530. [Google Scholar] [CrossRef]
- Szabó, G.T.; Mahiny, A.J.; Vlatkovic, I. COVID-19 mRNA vaccines: Platforms and current developments. Mol. Ther. 2022, 30, 1850–1868. [Google Scholar] [CrossRef]
- von Niessen, A.G.O.; Poleganov, M.A.; Rechner, C.; Plaschke, A.; Kranz, L.M.; Fesser, S.; Diken, M.; Löwer, M.; Vallazza, B.; Beissert, T.; et al. Improving mRNA-Based Therapeutic Gene Delivery by Expression-Augmenting 3’ UTRs Identified by Cellular Library Screening. Mol. Ther. 2019, 27, 824–836. [Google Scholar] [CrossRef]
- Sahin, U.; Muik, A.; Derhovanessian, E.; Vogler, I.; Kranz, L.M.; Vormehr, M.; Baum, A.; Pascal, K.; Quandt, J.; Maurus, D.; et al. COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature 2020, 586, 594–599. [Google Scholar] [CrossRef]
- Baiersdörfer, M.; Boros, G.; Muramatsu, H.; Mahiny, A.; Vlatkovic, I.; Sahin, U.; Karikó, K. A Facile Method for the Removal of dsRNA Contaminant from In Vitro-Transcribed mRNA. Mol. Ther. Nucleic Acids 2019, 15, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Prieve, M.G.; Harvie, P.; Monahan, S.D.; Roy, D.; Li, A.G.; Blevins, T.L.; Paschal, A.E.; Waldheim, M.; Bell, E.C.; Galperin, A.; et al. Targeted mRNA Therapy for Ornithine Transcarbamylase Deficiency. Mol. Ther. 2018, 26, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Maclachlan, I.; Jeffs, L.B.; Yaworski, E.; Lam, K. Systems and Methods for Manufacturing Liposomes. U.S. Patent 9,005,654, 27 July 2006. [Google Scholar]
- Heyes, J.; Hall, K.; Tailor, V.; Lenz, R.; MacLachlan, I. Synthesis and characterization of novel poly(ethylene glycol)-lipid conjugates suitable for use in drug delivery. J. Control Release 2006, 112, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.; Schreiner, P.; Leung, A.; Stainton, P.; Reid, S.; Yaworski, E.; Lutwyche, P.; Heyes, J. Optimizing Lipid Nanoparticles for Delivery in Primates. Adv. Mater. 2023, e2211420. [Google Scholar] [CrossRef]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Hinnebusch, A.G.; Ivanov, I.P.; Sonenberg, N. Translational control by 5’-untranslated regions of eukaryotic mRNAs. Science 2016, 352, 1413–1416. [Google Scholar] [CrossRef] [PubMed]
- Gallie, D.R. The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes. Dev. 1991, 5, 2108–2116. [Google Scholar] [CrossRef]
- Karikó, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef]
- Daniel, S.; Kis, Z.; Kontoravdi, C.; Shah, N. Quality by Design for enabling RNA platform production processes. Trends Biotechnol. 2022, 40, 1213–1228. [Google Scholar] [CrossRef] [PubMed]
- Wisse, E.; Jacobs, F.; Topal, B.; Frederik, P.; de Geest, B. The size of endothelial fenestrae in human liver sinusoids: Implications for hepatocyte-directed gene transfer. Gene Ther. 2008, 15, 1193–1199. [Google Scholar] [CrossRef]
- Verbeke, R.; Hogan, M.J.; Loré, K.; Pardi, N. Innate immune mechanisms of mRNA vaccines. Immunity 2022, 55, 1993–2005. [Google Scholar] [CrossRef] [PubMed]
- Wuebben, C.; Bartok, E.; Hartmann, G. Innate sensing of mRNA vaccines. Curr. Opin. Immunol. 2022, 79, 102249. [Google Scholar] [CrossRef]
- Samaridou, E.; Heyes, J.; Lutwyche, P. Lipid nanoparticles for nucleic acid delivery: Current perspectives. Adv. Drug Deliv. Rev. 2020, 154–155, 37–63. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef]
- Abrams, M.J.; Heyes, J.; Holland, R.J.; Lam, K.M.; Wood, M. Cationic Lipids Containing Silicon. Patent WO-2020097520-A1, 14 May 2020. [Google Scholar]
- Wetzel, M.A.; Steele, A.D.; Eisenstein, T.K.; Adler, M.W.; Henderson, E.E.; Rogers, T.J. Mu-opioid induction of monocyte chemoattractant protein-1, RANTES, and IFN-gamma-inducible protein-10 expression in human peripheral blood mononuclear cells. J. Immunol. 2000, 165, 6519–6524. [Google Scholar] [CrossRef]
- Barkovich, E.; Gropman, A.L. Late Onset Ornithine Transcarbamylase Deficiency Triggered by an Acute Increase in Protein Intake: A Review of 10 Cases Reported in the Literature. Case Rep. Genet. 2020, 2020, 7024735. [Google Scholar] [CrossRef]
- Thrane, V.R.; Thrane, A.S.; Wang, F.; Cotrina, M.L.; Smith, N.A.; Chen, M.; Xu, Q.; Kang, N.; Fujita, T.; Nagelhus, E.A.; et al. Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering. Nat. Med. 2013, 19, 1643–1648. [Google Scholar] [CrossRef] [PubMed]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. Methods Clin. Dev. 2018, 8, 87–104. [Google Scholar] [CrossRef]
- Imoto, K.; Tanaka, M.; Goya, T.; Aoyagi, T.; Takahashi, M.; Kurokawa, M.; Tashiro, S.; Kato, M.; Kohjima, M.; Ogawa, Y. Corticosteroid suppresses urea-cycle-related gene expressions in ornithine transcarbamylase deficiency. BMC Gastroenterol. 2022, 22, 144. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef]
- Andries, O.; Mc Cafferty, S.; de Smedt, S.C.; Weiss, R.; Sanders, N.N.; Kitada, T. N(1)-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J. Control Release 2015, 217, 337–344. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name (LNP-ASL mRNA) | mRNA Length (nt) | Codon Opt. | Bioanalyzer Integrity (%) | dsRNA per 1 µg mRNA (pg) | Capping % | Z-Avg (nm) | PDI | % Encap. |
|---|---|---|---|---|---|---|---|---|
| CDS1 | 1862 | Wild-type | 89 | <1 | 84 | 73 | 0.06 | 88 |
| CDS2 | 1862 | GC-rich | 92 | <1 | 80 | 76 | 0.07 | 87 |
| CDS3 | 1862 | Non-GC-rich | 93 | <1 | 87 | 77 | 0.05 | 89 |
| CDS4 | 1885 | GC-rich | 73 | Pass | 99 | 72 | 0.09 | 93 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daly, O.; Mahiny, A.J.; Majeski, S.; McClintock, K.; Reichert, J.; Boros, G.; Szabó, G.T.; Reinholz, J.; Schreiner, P.; Reid, S.; et al. ASL mRNA-LNP Therapeutic for the Treatment of Argininosuccinic Aciduria Enables Survival Benefit in a Mouse Model. Biomedicines 2023, 11, 1735. https://doi.org/10.3390/biomedicines11061735
Daly O, Mahiny AJ, Majeski S, McClintock K, Reichert J, Boros G, Szabó GT, Reinholz J, Schreiner P, Reid S, et al. ASL mRNA-LNP Therapeutic for the Treatment of Argininosuccinic Aciduria Enables Survival Benefit in a Mouse Model. Biomedicines. 2023; 11(6):1735. https://doi.org/10.3390/biomedicines11061735
Chicago/Turabian StyleDaly, Owen, Azita Josefine Mahiny, Sara Majeski, Kevin McClintock, Julia Reichert, Gábor Boros, Gábor Tamás Szabó, Jonas Reinholz, Petra Schreiner, Steve Reid, and et al. 2023. "ASL mRNA-LNP Therapeutic for the Treatment of Argininosuccinic Aciduria Enables Survival Benefit in a Mouse Model" Biomedicines 11, no. 6: 1735. https://doi.org/10.3390/biomedicines11061735
APA StyleDaly, O., Mahiny, A. J., Majeski, S., McClintock, K., Reichert, J., Boros, G., Szabó, G. T., Reinholz, J., Schreiner, P., Reid, S., Lam, K., Lepper, M., Adler, M., Meffen, T., Heyes, J., Karikó, K., Lutwyche, P., & Vlatkovic, I. (2023). ASL mRNA-LNP Therapeutic for the Treatment of Argininosuccinic Aciduria Enables Survival Benefit in a Mouse Model. Biomedicines, 11(6), 1735. https://doi.org/10.3390/biomedicines11061735