
Evidence and Uncertainties on Lipoprotein(a) as a Marker of Cardiovascular Health Risk in Children and Adolescents

 ,
, {kind=link}
{kind=link}
Abstract
1. Introduction
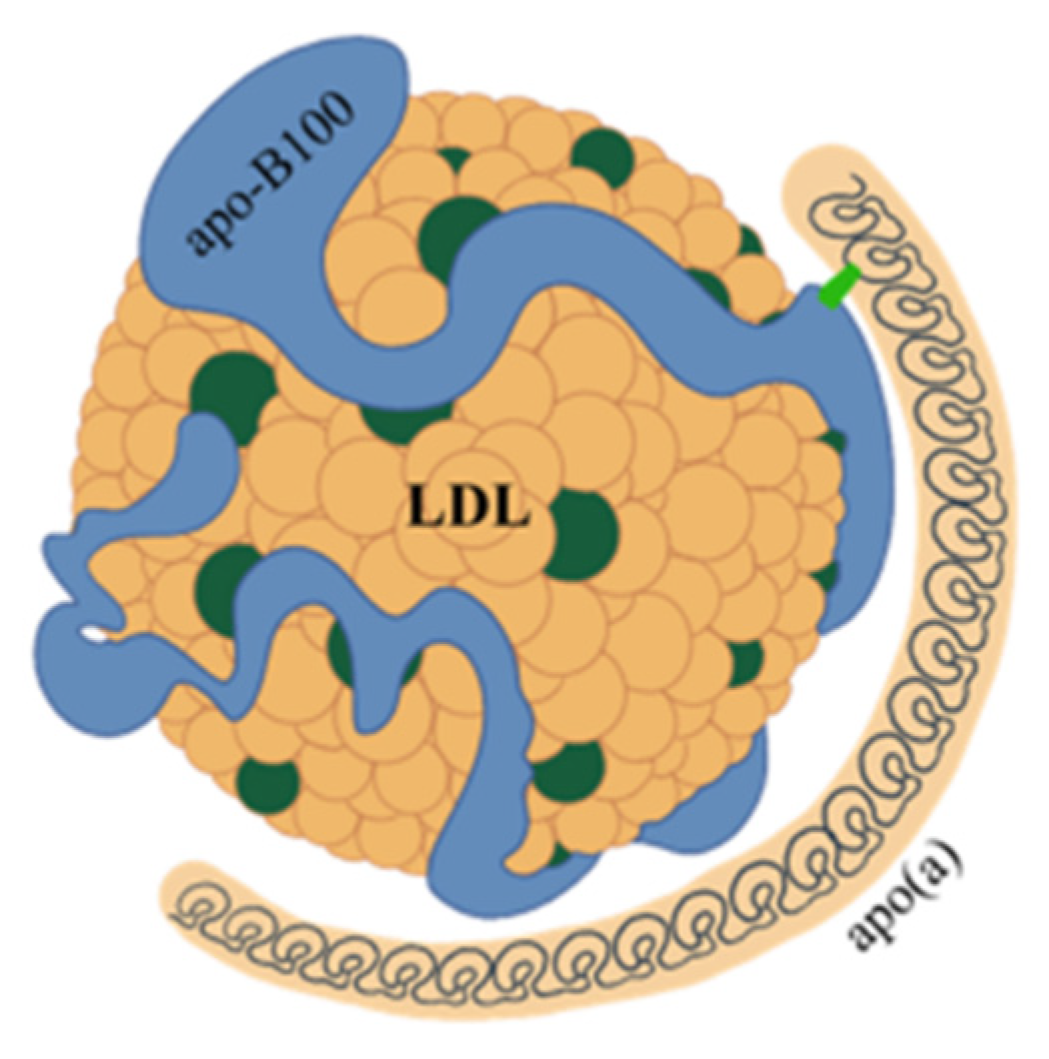
2. Structure and Features of Lp(a)
3. Lp(a) and Cardiovascular Risk
4. Lp(a) in Adults
5. Lp(a) in Children and Adolescents
5.1. Changes in Lp(a) Values in Childhood
5.2. Correlation between Pediatric Lp(a) Values and Clinical Data
6. Role of Lp(a) in Defining Pediatric Cardiovascular Risk and Preventive Activity
7. Conclusions and Research Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berg, K. A new serum type system in man—The lp system. Acta Pathol. Microbiol. Scand. 1963, 59, 369–382. [Google Scholar] [CrossRef] [PubMed]
- McLean, J.W.; Tomlinson, J.E.; Kuang, W.J.; Eaton, D.L.; Chen, E.Y.; Fless, G.M.; Scanu, A.M.; Lawn, R.M. CDNA Sequence of Human Apolipoprotein(a) Is Homologous to Plasminogen. Nature 1987, 330, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Erqou, S.; Thompson, A.; Di Angelantonio, E.; Saleheen, D.; Kaptoge, S.; Marcovina, S.; Danesh, J. Apolipoprotein(a) Isoforms and the Risk of Vascular Disease: Systematic Review of 40 Studies Involving 58,000 Participants. J. Am. Coll. Cardiol. 2010, 55, 2160–2167. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, P.R.; Tybjaerg-Hansen, A.; Steffensen, R.; Nordestgaard, B.G. Genetically Elevated Lipoprotein(a) and Increased Risk of Myocardial Infarction. JAMA 2009, 301, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Thanassoulis, G.; Campbell, C.Y.; Owens, D.S.; Smith, J.G.; Smith, A.V.; Peloso, G.M.; Kerr, K.F.; Pechlivanis, S.; Budoff, M.J.; Harris, T.B.; et al. Genetic Associations with Valvular Calcification and Aortic Stenosis. N. Engl. J. Med. 2013, 368, 503–512. [Google Scholar] [CrossRef]
- Langsted, A.; Nordestgaard, B.G.; Kamstrup, P.R. Elevated Lipoprotein(a) and Risk of Ischemic Stroke. J. Am. Coll. Cardiol. 2019, 74, 54–66. [Google Scholar] [CrossRef]
- Koschinsky, M.L.; Kronenberg, F. The Long Journey of Lipoprotein(a) from Cardiovascular Curiosity to Therapeutic Target. Atherosclerosis 2022, 349, 1–6. [Google Scholar] [CrossRef]
- van der Hoek, Y.Y.; Wittekoek, M.E.; Beisiegel, U.; Kastelein, J.J.; Koschinsky, M.L. The Apolipoprotein(a) Kringle IV Repeats Which Differ from the Major Repeat Kringle Are Present in Variably-Sized Isoforms. Hum. Mol. Genet. 1993, 2, 361–366. [Google Scholar] [CrossRef]
- Santonastaso, A.; Maggi, M.; De Jonge, H.; Scotti, C. High Resolution Structure of Human Apolipoprotein (a) Kringle IV Type 2: Beyond the Lysine Binding Site. J. Lipid Res. 2020, 61, 1687–1696. [Google Scholar] [CrossRef]
- Garner, B.; Merry, A.H.; Royle, L.; Harvey, D.J.; Rudd, P.M.; Thillet, J. Structural Elucidation of the N- and O-Glycans of Human Apolipoprotein(a): Role of o-Glycans in Conferring Protease Resistance. J. Biol. Chem. 2001, 276, 22200–22208. [Google Scholar] [CrossRef]
- Kostner, G.M. Lp(a) Biochemistry, Composition, and Structure. In Lipoprotein(a); Kostner, K., Kostner, G.M., Toth, P.P., Eds.; Contemporary Cardiology; Springer International Publishing: Cham, Switzerland, 2023; pp. 39–54. ISBN 978-3-031-24575-6. [Google Scholar]
- Zheng, K.H.; Tsimikas, S.; Pawade, T.; Kroon, J.; Jenkins, W.S.A.; Doris, M.K.; White, A.C.; Timmers, N.K.L.M.; Hjortnaes, J.; Rogers, M.A.; et al. Lipoprotein(a) and Oxidized Phospholipids Promote Valve Calcification in Patients with Aortic Stenosis. J. Am. Coll. Cardiol. 2019, 73, 2150–2162. [Google Scholar] [CrossRef] [PubMed]
- Chemello, K.; Chan, D.C.; Lambert, G.; Watts, G.F. Recent Advances in Demystifying the Metabolism of Lipoprotein(a). Atherosclerosis 2022, 349, 82–91. [Google Scholar] [CrossRef]
- Koschinsky, M.L.; Marcovina, S.M. Structure-Function Relationships in Apolipoprotein(a): Insights into Lipoprotein(a) Assembly and Pathogenicity. Curr. Opin. Lipidol. 2004, 15, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Becker, L.; Nesheim, M.E.; Koschinsky, M.L. Catalysis of Covalent Lp(a) Assembly: Evidence for an Extracellular Enzyme Activity That Enhances Disulfide Bond Formation. Biochemistry 2006, 45, 9919–9928. [Google Scholar] [CrossRef]
- Mooser, V.; Marcovina, S.M.; White, A.L.; Hobbs, H.H. Kringle-Containing Fragments of Apolipoprotein(a) Circulate in Human Plasma and Are Excreted into the Urine. J. Clin. Investig. 1996, 98, 2414–2424. [Google Scholar] [CrossRef]
- Borrelli, M.J.; Youssef, A.; Boffa, M.B.; Koschinsky, M.L. New Frontiers in Lp(a)-Targeted Therapies. Trends Pharmacol. Sci. 2019, 40, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Pagnan, A.; Kostner, G.; Braggion, M.; Ziron, L. Relationship between “sinking Pre-Beta-Lipoprotein” (Lp(a) Lipoprotein) and Age in a Family Kindred. Gerontology 1982, 28, 381–385. [Google Scholar] [CrossRef]
- Reyes-Soffer, G.; Westerterp, M. Beyond Lipoprotein(a) Plasma Measurements: Lipoprotein(a) and Inflammation. Pharmacol. Res. 2021, 169, 105689. [Google Scholar] [CrossRef]
- Kronenberg, F. Causes and Consequences of Lipoprotein(a) Abnormalities in Kidney Disease. Clin. Exp. Nephrol. 2014, 18, 234–237. [Google Scholar] [CrossRef]
- Barbagelata, L.; Masson, W.; Corral, P.; Lavalle-Cobo, A.; Nogueira, J.P.; Rosa Diez, G. Relationship between Lipoprotein(a) Levels, Cardiovascular Outcomes and Death in Patients with Chronic Kidney Disease: A Systematic Review of Prospective Studies. J. Nephrol. 2023. [Google Scholar] [CrossRef]
- Chan, D.C.; Watts, G.F.; Coll, B.; Wasserman, S.M.; Marcovina, S.M.; Barrett, P.H.R. Lipoprotein(a) Particle Production as a Determinant of Plasma Lipoprotein(a) Concentration Across Varying Apolipoprotein(a) Isoform Sizes and Background Cholesterol-Lowering Therapy. J. Am. Heart Assoc. 2019, 8, e011781. [Google Scholar] [CrossRef] [PubMed]
- Boffa, M.B.; Koschinsky, M.L. Understanding the Ins and Outs of Lipoprotein (a) Metabolism. Curr. Opin. Lipidol. 2022, 33, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Coassin, S.; Kronenberg, F. Lipoprotein(a) beyond the Kringle IV Repeat Polymorphism: The Complexity of Genetic Variation in the LPA Gene. Atherosclerosis 2022, 349, 17–35. [Google Scholar] [CrossRef]
- Noureen, A.; Fresser, F.; Utermann, G.; Schmidt, K. Sequence Variation within the KIV-2 Copy Number Polymorphism of the Human LPA Gene in African, Asian, and European Populations. PLoS ONE 2015, 10, e0121582. [Google Scholar] [CrossRef] [PubMed]
- McCormick, S.P.A.; Schneider, W.J. Lipoprotein(a) Catabolism: A Case of Multiple Receptors. Pathology 2019, 51, 155–164. [Google Scholar] [CrossRef]
- Cegla, J.; France, M.; Marcovina, S.M.; Neely, R.D.G. Lp(a): When and How to Measure It. Ann. Clin. Biochem. 2021, 58, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, F. Lipoprotein(a) Measurement Issues: Are We Making a Mountain out of a Molehill? Atherosclerosis 2022, 349, 123–135. [Google Scholar] [CrossRef]
- Kinpara, K.; Okada, H.; Yoneyama, A.; Okubo, M.; Murase, T. Lipoprotein(a)-Cholesterol: A Significant Component of Serum Cholesterol. Clin. Chim. Acta 2011, 412, 1783–1787. [Google Scholar] [CrossRef]
- van der Valk, F.M.; Bekkering, S.; Kroon, J.; Yeang, C.; Van den Bossche, J.; van Buul, J.D.; Ravandi, A.; Nederveen, A.J.; Verberne, H.J.; Scipione, C.; et al. Oxidized Phospholipids on Lipoprotein(a) Elicit Arterial Wall Inflammation and an Inflammatory Monocyte Response in Humans. Circulation 2016, 134, 611–624. [Google Scholar] [CrossRef]
- Garg, P.K.; Guan, W.; Karger, A.B.; Steffen, B.T.; Budoff, M.; Tsai, M.Y. Lipoprotein (a) and Risk for Calcification of the Coronary Arteries, Mitral Valve, and Thoracic Aorta: The Multi-Ethnic Study of Atherosclerosis. J. Cardiovasc. Comput. Tomogr. 2021, 15, 154–160. [Google Scholar] [CrossRef]
- Kaiser, Y.; Daghem, M.; Tzolos, E.; Meah, M.N.; Doris, M.K.; Moss, A.J.; Kwiecinski, J.; Kroon, J.; Nurmohamed, N.S.; van der Harst, P.; et al. Association of Lipoprotein(a) With Atherosclerotic Plaque Progression. J. Am. Coll. Cardiol. 2022, 79, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Leibundgut, G.; Scipione, C.; Yin, H.; Schneider, M.; Boffa, M.B.; Green, S.; Yang, X.; Dennis, E.; Witztum, J.L.; Koschinsky, M.L.; et al. Determinants of Binding of Oxidized Phospholipids on Apolipoprotein (a) and Lipoprotein (a). J. Lipid Res. 2013, 54, 2815–2830. [Google Scholar] [CrossRef] [PubMed]
- Koschinsky, M.L.; Boffa, M.B. Oxidized Phospholipid Modification of Lipoprotein(a): Epidemiology, Biochemistry and Pathophysiology. Atherosclerosis 2022, 349, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Dzobo, K.E.; Kraaijenhof, J.M.; Stroes, E.S.G.; Nurmohamed, N.S.; Kroon, J. Lipoprotein(a): An Underestimated Inflammatory Mastermind. Atherosclerosis 2022, 349, 101–109. [Google Scholar] [CrossRef]
- Boffa, M.B.; Koschinsky, M.L. Lipoprotein (a): Truly a Direct Prothrombotic Factor in Cardiovascular Disease? J. Lipid Res. 2016, 57, 745–757. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Langsted, A. Lipoprotein (a) as a Cause of Cardiovascular Disease: Insights from Epidemiology, Genetics, and Biology. J. Lipid Res. 2016, 57, 1953–1975. [Google Scholar] [CrossRef]
- Kaiser, Y.; Singh, S.S.; Zheng, K.H.; Verbeek, R.; Kavousi, M.; Pinto, S.-J.; Vernooij, M.W.; Sijbrands, E.J.G.; Boekholdt, S.M.; de Rijke, Y.B.; et al. Lipoprotein(a) Is Robustly Associated with Aortic Valve Calcium. Heart 2021, 107, 1422–1428. [Google Scholar] [CrossRef]
- Kaltoft, M.; Sigvardsen, P.E.; Afzal, S.; Langsted, A.; Fuchs, A.; Kühl, J.T.; Køber, L.; Kamstrup, P.R.; Kofoed, K.F.; Nordestgaard, B.G. Elevated Lipoprotein(a) in Mitral and Aortic Valve Calcification and Disease: The Copenhagen General Population Study. Atherosclerosis 2022, 349, 166–174. [Google Scholar] [CrossRef]
- Tsimikas, S.; Gordts, P.L.S.M.; Nora, C.; Yeang, C.; Witztum, J.L. Statin Therapy Increases Lipoprotein(a) Levels. Eur. Heart J. 2020, 41, 2275–2284. [Google Scholar] [CrossRef]
- Awad, K.; Mikhailidis, D.P.; Katsiki, N.; Muntner, P.; Banach, M. Lipid and Blood Pressure Meta-Analysis Collaboration (LBPMC) Group Effect of Ezetimibe Monotherapy on Plasma Lipoprotein(a) Concentrations in Patients with Primary Hypercholesterolemia: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Drugs 2018, 78, 453–462. [Google Scholar] [CrossRef]
- Jaeger, B.R.; Richter, Y.; Nagel, D.; Heigl, F.; Vogt, A.; Roeseler, E.; Parhofer, K.; Ramlow, W.; Koch, M.; Utermann, G.; et al. Longitudinal cohort study on the effectiveness of lipid apheresis treatment to reduce high lipoprotein(a) levels and prevent major adverse coronary events. Nat. Clin. Pract. Cardiovasc. Med. 2009, 6, 229–239. [Google Scholar] [CrossRef] [PubMed]
- AIM-HIGH Investigators; Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in Patients with Low HDL Cholesterol Levels Receiving Intensive Statin Therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.L.; Fazio, S.; Giugliano, R.P.; Stroes, E.S.G.; Kanevsky, E.; Gouni-Berthold, I.; Im, K.; Lira Pineda, A.; Wasserman, S.M.; Češka, R.; et al. Lipoprotein(a), PCSK9 Inhibition, and Cardiovascular Risk. Circulation 2019, 139, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Tsushima, T.; Tsushima, Y.; Sullivan, C.; Hatipoglu, B. Lipoprotein(a) and Atherosclerotic Cardiovascular Disease, the Impact of Available Lipid-Lowering Medications on Lipoprotein(a): An Update on New Therapies. Endocr. Pract. 2022, S1530-891X(22)00901-6. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.; Stroes, E.S.G. Therapy of Elevated Lipoprotein(a). In Lipoprotein(a); Kostner, K., Kostner, G.M., Toth, P.P., Eds.; Contemporary Cardiology; Springer International Publishing: Cham, Switzerland, 2023; pp. 347–357. ISBN 978-3-031-24575-6. [Google Scholar]
- Mehta, A.; Jain, V.; Saeed, A.; Saseen, J.J.; Gulati, M.; Ballantyne, C.M.; Virani, S.S. Lipoprotein(a) and Ethnicities. Atherosclerosis 2022, 349, 42–52. [Google Scholar] [CrossRef]
- Patel, A.P.; Wang, M.; Pirruccello, J.P.; Ellinor, P.T.; Ng, K.; Kathiresan, S.; Khera, A.V. Lp(a) (Lipoprotein[a]) Concentrations and Incident Atherosclerotic Cardiovascular Disease: New Insights from a Large National Biobank. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 465–474. [Google Scholar] [CrossRef]
- Virani, S.S.; Brautbar, A.; Davis, B.C.; Nambi, V.; Hoogeveen, R.C.; Sharrett, A.R.; Coresh, J.; Mosley, T.H.; Morrisett, J.D.; Catellier, D.J.; et al. Associations between Lipoprotein(a) Levels and Cardiovascular Outcomes in Black and White Subjects: The Atherosclerosis Risk in Communities (ARIC) Study. Circulation 2012, 125, 241–249. [Google Scholar] [CrossRef]
- Tsimikas, S.; Clopton, P.; Brilakis, E.S.; Marcovina, S.M.; Khera, A.; Miller, E.R.; de Lemos, J.A.; Witztum, J.L. Relationship of Oxidized Phospholipids on Apolipoprotein B-100 Particles to Race/Ethnicity, Apolipoprotein(a) Isoform Size, and Cardiovascular Risk Factors: Results from the Dallas Heart Study. Circulation 2009, 119, 1711–1719. [Google Scholar] [CrossRef]
- Deo, R.C.; Wilson, J.G.; Xing, C.; Lawson, K.; Kao, W.H.L.; Reich, D.; Tandon, A.; Akylbekova, E.; Patterson, N.; Mosley, T.H.; et al. Single-Nucleotide Polymorphisms in LPA Explain Most of the Ancestry-Specific Variation in Lp(a) Levels in African Americans. PLoS ONE 2011, 6, e14581. [Google Scholar] [CrossRef]
- Paré, G.; Çaku, A.; McQueen, M.; Anand, S.S.; Enas, E.; Clarke, R.; Boffa, M.B.; Koschinsky, M.; Wang, X.; Yusuf, S.; et al. Lipoprotein(a) Levels and the Risk of Myocardial Infarction Among 7 Ethnic Groups. Circulation 2019, 139, 1472–1482. [Google Scholar] [CrossRef]
- Varvel, S.; McConnell, J.P.; Tsimikas, S. Prevalence of Elevated Lp(a) Mass Levels and Patient Thresholds in 532 359 Patients in the United States. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2239–2245. [Google Scholar] [CrossRef] [PubMed]
- Trinder, M.; Paruchuri, K.; Haidermota, S.; Bernardo, R.; Zekavat, S.M.; Gilliland, T.; Januzzi, J.; Natarajan, P. Repeat Measures of Lipoprotein(a) Molar Concentration and Cardiovascular Risk. J. Am. Coll. Cardiol. 2022, 79, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Jenner, J.L.; Ordovas, J.M.; Lamon-Fava, S.; Schaefer, M.M.; Wilson, P.W.; Castelli, W.P.; Schaefer, E.J. Effects of Age, Sex, and Menopausal Status on Plasma Lipoprotein(a) Levels. The Framingham Offspring Study. Circulation 1993, 87, 1135–1141. [Google Scholar] [CrossRef] [PubMed]
- Derby, C.A.; Crawford, S.L.; Pasternak, R.C.; Sowers, M.; Sternfeld, B.; Matthews, K.A. Lipid Changes during the Menopause Transition in Relation to Age and Weight: The Study of Women’s Health Across the Nation. Am. J. Epidemiol. 2009, 169, 1352–1361. [Google Scholar] [CrossRef] [PubMed]
- Anagnostis, P.; Antza, C.; Trakatelli, C.; Lambrinoudaki, I.; Goulis, D.G.; Kotsis, V. The Effect of Menopause on Lipoprotein (a) Concentrations: A Systematic Review and Meta-Analysis. Maturitas 2023, 167, 39–45. [Google Scholar] [CrossRef]
- Simony, S.B.; Mortensen, M.B.; Langsted, A.; Afzal, S.; Kamstrup, P.R.; Nordestgaard, B.G. Sex Differences of Lipoprotein(a) Levels and Associated Risk of Morbidity and Mortality by Age: The Copenhagen General Population Study. Atherosclerosis 2022, 355, 76–82. [Google Scholar] [CrossRef]
- Nowak-Göttl, U.; Sträter, R.; Heinecke, A.; Junker, R.; Koch, H.G.; Schuierer, G.; von Eckardstein, A. Lipoprotein (a) and Genetic Polymorphisms of Clotting Factor V, Prothrombin, and Methylenetetrahydrofolate Reductase Are Risk Factors of Spontaneous Ischemic Stroke in Childhood. Blood 1999, 94, 3678–3682. [Google Scholar] [CrossRef]
- Nowak-Göttl, U.; Debus, O.; Findeisen, M.; Kassenböhmer, R.; Koch, H.G.; Pollmann, H.; Postler, C.; Weber, P.; Vielhaber, H. Lipoprotein (a): Its Role in Childhood Thromboembolism. Pediatrics 1997, 99, E11. [Google Scholar] [CrossRef]
- Nowak-Göttl, U.; Sträter, R.; Dübbers, A.; Oleszuk-Raschke, K.; Vielhaber, H. Ischaemic Stroke in Infancy and Childhood: Role of the Arg506 to Gln Mutation in the Factor V Gene. Blood Coagul. Fibrinolysis 1996, 7, 684–688. [Google Scholar] [CrossRef]
- Peynet, J.; Beaudeux, J.-L.; Woimant, F.; Flourié, F.; Giraudeaux, V.; Vicaut, E.; Launay, J.-M. Apolipoprotein(a) Size Polymorphism in Young Adults with Ischemic Stroke. Atherosclerosis 1999, 142, 233–239. [Google Scholar] [CrossRef]
- Chan, A.; Iorio, A.; Kenet, G. Impact of Thrombophilia on Risk of Arterial Ischemic Stroke or Cerebral Sinovenous Thrombosis in Neonates and Children: A Systematic Review and Meta-Analysis of Observational Studies. Circulation 2010, 121, 1838–1847. [Google Scholar]
- Strobl, W.; Widhalm, K.; Kostner, G.; Pollak, A. Serum Apolipoproteins and Lipoprotein (a) during the First Week of Life. Acta Paediatr. Scand. 1983, 72, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Van Biervliet, J.P.; Labeur, C.; Michiels, G.; Usher, D.C.; Rosseneu, M. Lipoprotein(a) Profiles and Evolution in Newborns. Atherosclerosis 1991, 86, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Rifai, N.; Heiss, G.; Doetsch, K. Lipoprotein(a) at Birth, in Blacks and Whites. Atherosclerosis 1992, 92, 123–129. [Google Scholar] [CrossRef]
- Schumacher, M.; Kessler, A.; Meier, A.; Weigert, S.; Wood, W.G. Lipoprotein(a) Concentrations in Cord and Capillary Blood from Newborns and in Serum from in-Patient Children, Adolescents and Adults. Eur. J. Clin. Chem. Clin. Biochem. 1994, 32, 341–347. [Google Scholar] [CrossRef]
- Wood, W.G.; Schumacher, M.; Weigert, S. (Apo)Lipoprotein(a) Concentrations at Birth and in the First Days and Months of Life—Studies on the Distribution of Serum Levels and the Predictive Value of Measurements Made at This Time. Eur. J. Clin. Chem. Clin. Biochem. 1995, 33, 139–145. [Google Scholar] [CrossRef]
- Wilcken, D.E.; Wang, X.L.; Dudman, N.P. The Relationship between Infant and Parent Lp(a) Levels. Chem. Phys. Lipids 1994, 67–68, 299–304. [Google Scholar] [CrossRef]
- Routi, T.; Rönnemaa, T.; Viikari, J.S.; Leino, A.; Välimäki, I.A.; Simell, O.G. Tracking of Serum Lipoprotein (a) Concentration and Its Contribution to Serum Cholesterol Values in Children from 7 to 36 Months of Age in the STRIP Baby Study. Special Turku Coronary Risk Factor Intervention Project for Babies. Ann. Med. 1997, 29, 541–547. [Google Scholar] [CrossRef]
- Strandkjær, N.; Hansen, M.K.; Nielsen, S.T.; Frikke-Schmidt, R.; Tybjærg-Hansen, A.; Nordestgaard, B.G.; Tabor, A.; Bundgaard, H.; Iversen, K.; Kamstrup, P.R. Lipoprotein(a) Levels at Birth and in Early Childhood: The COMPARE Study. J. Clin. Endocrinol. Metab. 2022, 107, 324–335. [Google Scholar] [CrossRef]
- de Boer, L.M.; Hof, M.H.; Wiegman, A.; Stroobants, A.K.; Kastelein, J.J.P.; Hutten, B.A. Lipoprotein(a) Levels from Childhood to Adulthood: Data in Nearly 3000 Children Who Visited a Pediatric Lipid Clinic. Atherosclerosis 2022, 349, 227–232. [Google Scholar] [CrossRef]
- Choi, R.; Lee, S.G.; Lee, E.H. Lipoprotein(a) in the Korean Pediatric Population Visiting Local Clinics and Hospitals. Nutrients 2022, 14, 2820. [Google Scholar] [CrossRef] [PubMed]
- Qayum, O.; Alshami, N.; Ibezim, C.F.; Reid, K.J.; Noel-MacDonnell, J.R.; Raghuveer, G. Lipoprotein (a): Examination of Cardiovascular Risk in a Pediatric Referral Population. Pediatr. Cardiol. 2018, 39, 1540–1546. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou-Legbelou, K.; Triantafyllou, A.; Vampertzi, O.; Koletsos, N.; Douma, S.; Papadopoulou-Alataki, E. Similar Myocardial Perfusion and Vascular Stiffness in Children and Adolescents with High Lipoprotein (a) Levels, in Comparison with Healthy Controls. Pulse 2021, 9, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Pederiva, C.; Capra, M.E.; Biasucci, G.; Banderali, G.; Fabrizi, E.; Gazzotti, M.; Casula, M.; Catapano, A.L.; Lipigen Paediatric Group; Members of the Lipigen Steering Committee; et al. Lipoprotein(a) and Family History for Cardiovascular Disease in Paediatric Patients: A New Frontier in Cardiovascular Risk Stratification. Data from the LIPIGEN Paediatric Group. Atherosclerosis 2022, 349, 233–239. [Google Scholar] [CrossRef]
- Raitakari, O.; Kartiosuo, N.; Pahkala, K.; Hutri-Kähönen, N.; Bazzano, L.A.; Chen, W.; Urbina, E.M.; Jacobs, D.R.; Sinaiko, A.; Steinberger, J.; et al. Lipoprotein(a) in Youth and Prediction of Major Cardiovascular Outcomes in Adulthood. Circulation 2023, 147, 23–31. [Google Scholar] [CrossRef]
- de Boer, L.M.; Hutten, B.A.; Zwinderman, A.H.; Wiegman, A. Lipoprotein(a) Levels in Children with Suspected Familial Hypercholesterolaemia: A Cross-Sectional Study. Eur. Heart J. 2023, 44, 1421–1428. [Google Scholar] [CrossRef]
- Averna, M.R.; Cefalù, A.B. Lp(a): A Genetic Cause of Clinical FH in Children. Eur. Heart J. 2023, 44, 1429–1431. [Google Scholar] [CrossRef]
- Olmastroni, E.; Gazzotti, M.; Averna, M.; Arca, M.; Tarugi, P.; Calandra, S.; Bertolini, S.; Catapano, A.L.; Casula, M.; LIPIGEN Study Group. Lipoprotein(a) Genotype Influences the Clinical Diagnosis of Familial Hypercholesterolemia. J. Am. Heart Assoc. 2023, 12, e029223. [Google Scholar] [CrossRef]
- WHO Mortality Database—WHO. Available online: https://www.who.int/data/data-collection-tools/who-mortality-database (accessed on 28 February 2023).
- Global Health Estimates. Available online: https://www.who.int/data/global-health-estimates (accessed on 28 February 2023).
- Birger, M.; Kaldjian, A.S.; Roth, G.A.; Moran, A.E.; Dieleman, J.L.; Bellows, B.K. Spending on Cardiovascular Disease and Cardiovascular Risk Factors in the United States: 1996 to 2016. Circulation 2021, 144, 271–282. [Google Scholar] [CrossRef]
- Moran, A.E.; Forouzanfar, M.H.; Roth, G.A.; Mensah, G.A.; Ezzati, M.; Murray, C.J.L.; Naghavi, M. Temporal Trends in Ischemic Heart Disease Mortality in 21 World Regions, 1980 to 2010: The Global Burden of Disease 2010 Study. Circulation 2014, 129, 1483–1492. [Google Scholar] [CrossRef]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Takura, T.; Yokoi, H.; Tanaka, N.; Matsumoto, N.; Yoshida, E.; Nakata, T.; J-CONCIOUS Investigators. Health Economics-Based Verification of Functional Myocardial Ischemia Evaluation of Stable Coronary Artery Disease in Japan: A Long-Term Longitudinal Study Using Propensity Score Matching. J. Nucl. Cardiol. 2022, 29, 1356–1369. [Google Scholar] [CrossRef] [PubMed]
- Berenson, G.S.; Srinivasan, S.R.; Bao, W.; Newman, W.P.; Tracy, R.E.; Wattigney, W.A. Association between Multiple Cardiovascular Risk Factors and Atherosclerosis in Children and Young Adults. The Bogalusa Heart Study. N. Engl. J. Med. 1998, 338, 1650–1656. [Google Scholar] [CrossRef] [PubMed]
- Milei, J.; Ottaviani, G.; Lavezzi, A.M.; Grana, D.R.; Stella, I.; Matturri, L. Perinatal and Infant Early Atherosclerotic Coronary Lesions. Can. J. Cardiol. 2008, 24, 137–141. [Google Scholar] [CrossRef]
- Lechner, K.; von Schacky, C.; McKenzie, A.L.; Worm, N.; Nixdorff, U.; Lechner, B.; Kränkel, N.; Halle, M.; Krauss, R.M.; Scherr, J. Lifestyle Factors and High-Risk Atherosclerosis: Pathways and Mechanisms beyond Traditional Risk Factors. Eur. J. Prev. Cardiol. 2020, 27, 394–406. [Google Scholar] [CrossRef]
- Adar, S.D.; Sheppard, L.; Vedal, S.; Polak, J.F.; Sampson, P.D.; Diez Roux, A.V.; Budoff, M.; Jacobs, D.R.; Barr, R.G.; Watson, K.; et al. Fine Particulate Air Pollution and the Progression of Carotid Intima-Medial Thickness: A Prospective Cohort Study from the Multi-Ethnic Study of Atherosclerosis and Air Pollution. PLoS Med. 2013, 10, e1001430. [Google Scholar] [CrossRef]
- Kronenberg, F.; Mora, S.; Stroes, E.S.G.; Ference, B.A.; Arsenault, B.J.; Berglund, L.; Dweck, M.R.; Koschinsky, M.; Lambert, G.; Mach, F.; et al. Lipoprotein(a) in Atherosclerotic Cardiovascular Disease and Aortic Stenosis: A European Atherosclerosis Society Consensus Statement. Eur. Heart J. 2022, 43, 3925–3946. [Google Scholar] [CrossRef]
- Wilson, D.P.; Jacobson, T.A.; Jones, P.H.; Koschinsky, M.L.; McNeal, C.J.; Nordestgaard, B.G.; Orringer, C.E. Use of Lipoprotein(a) in Clinical Practice: A Biomarker Whose Time Has Come. A Scientific Statement from the National Lipid Association. J. Clin. Lipidol. 2019, 13, 374–392. [Google Scholar] [CrossRef]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, e596–e646. [Google Scholar] [CrossRef]
- Jellinger, P.S.; Handelsman, Y.; Rosenblit, P.D.; Bloomgarden, Z.T.; Fonseca, V.A.; Garber, A.J.; Grunberger, G.; Guerin, C.K.; Bell, D.S.H.; Mechanick, J.I.; et al. American association of clinical endocrinologists and american college of endocrinology guidelines for management of dyslipidemia and prevention of cardiovascular disease. Endocr. Pract. 2017, 23, 1–87. [Google Scholar] [CrossRef]
- McNeal, C.J. Lipoprotein(a): Its Relevance to the Pediatric Population. J. Clin. Lipidol. 2015, 9, S57–S66. [Google Scholar] [CrossRef] [PubMed]
- Kohn, B.; Ashraf, A.P.; Wilson, D.P. Should Lipoprotein(a) Be Measured in Youth? J. Pediatr. 2021, 228, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Simon, G.R.; Baker, C.; Barden, G.A.; Brown, O.W.; Hardin, A.; Lessin, H.R.; Meade, K.; Moore, S.; Rodgers, C.T.; Committee on Practice and Ambulatory Medicine; et al. 2014 Recommendations for Pediatric Preventive Health Care. Pediatrics 2014, 133, 568–570. [Google Scholar] [CrossRef]
- Daniels, S.R.; Gidding, S.S.; de Ferranti, S.D. Pediatric Aspects of Familial Hypercholesterolemias: Recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J. Clin. Lipidol. 2011, 5, S30–S37. [Google Scholar] [CrossRef] [PubMed]
- Descamps, O.S.; Tenoutasse, S.; Stephenne, X.; Gies, I.; Beauloye, V.; Lebrethon, M.-C.; De Beaufort, C.; De Waele, K.; Scheen, A.; Rietzschel, E.; et al. Management of Familial Hypercholesterolemia in Children and Young Adults: Consensus Paper Developed by a Panel of Lipidologists, Cardiologists, Paediatricians, Nutritionists, Gastroenterologists, General Practitioners and a Patient Organization. Atherosclerosis 2011, 218, 272–280. [Google Scholar] [CrossRef]
- Watts, G.F.; Gidding, S.; Wierzbicki, A.S.; Toth, P.P.; Alonso, R.; Brown, W.V.; Bruckert, E.; Defesche, J.; Lin, K.K.; Livingston, M.; et al. Integrated Guidance on the Care of Familial Hypercholesterolaemia from the International FH Foundation. Int. J. Cardiol. 2014, 171, 309–325. [Google Scholar] [CrossRef]
- Koutsogianni, A.; Liamis, G.; Liberopoulos, E.; Adamidis, P.S.; Florentin, M. Effects of Lipid-Modifying and Other Drugs on Lipoprotein(a) Levels—Potent Clinical Implications. Pharmaceuticals 2023, 16, 750. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Genovesi, S.; Giussani, M.; Lieti, G.; Orlando, A.; Patti, I.; Parati, G. Evidence and Uncertainties on Lipoprotein(a) as a Marker of Cardiovascular Health Risk in Children and Adolescents. Biomedicines 2023, 11, 1661. https://doi.org/10.3390/biomedicines11061661
Genovesi S, Giussani M, Lieti G, Orlando A, Patti I, Parati G. Evidence and Uncertainties on Lipoprotein(a) as a Marker of Cardiovascular Health Risk in Children and Adolescents. Biomedicines. 2023; 11(6):1661. https://doi.org/10.3390/biomedicines11061661
Chicago/Turabian StyleGenovesi, Simonetta, Marco Giussani, Giulia Lieti, Antonina Orlando, Ilenia Patti, and Gianfranco Parati. 2023. "Evidence and Uncertainties on Lipoprotein(a) as a Marker of Cardiovascular Health Risk in Children and Adolescents" Biomedicines 11, no. 6: 1661. https://doi.org/10.3390/biomedicines11061661
APA StyleGenovesi, S., Giussani, M., Lieti, G., Orlando, A., Patti, I., & Parati, G. (2023). Evidence and Uncertainties on Lipoprotein(a) as a Marker of Cardiovascular Health Risk in Children and Adolescents. Biomedicines, 11(6), 1661. https://doi.org/10.3390/biomedicines11061661